

Abstract
New pH‐sensitive perylene bisimide indicator dyes were synthesized and used for fabrication of optical sensors. The highly photostable dyes show absorption/emission bands in the red/near‐infrared (NIR) region of the electromagnetic spectrum, high molar absorption coefficients (up to 100 000 m −1 cm−1), and fluorescence quantum yields close to unity. The absorption and emission spectra show strong bathochromic shifts upon deprotonation of the imidazole nitrogen atom, which makes the dyes promising as ratiometric fluorescent indicators. Physical entrapment of the indicators into a polyurethane hydrogel enables pH determination at alkaline pH values. It is also shown that a plastic carbon dioxide solid‐state sensor can be manufactured by immobilization of the pH indicator in a hydrophilic polymer, along with a quaternary ammonium base. The influences of the plasticizer, different lipophilic bases, and humidity on the sensitivity of the sensor material are systematically investigated. The disubstituted perylene, particularly, features two deprotonation equilibria, enabling sensing over a very broad pCO2 range of 0.5 to 1000 hPa.
Keywords: carbon dioxide, dyes/pigments, fluorescence, ratiometry, sensors
Introduction
pH determination is one of the most frequently performed analytical measurements in industry and numerous scientific disciplines, such as biotechnology,1, 2, 3 marine science,4, 5 and medical diagnostics6, 7 to mention only a few. pH is most commonly measured with a glass electrode, which provides information about the activity of hydronium ions in aqueous environment. Ion‐sensitive field‐effect transistors (ISFETs) belong to other very common tools for pH measurement. The easiness of use, fast response times, broad working range, and relatively low cost are advantages of such methods. On the other hand, disadvantages include challenging miniaturization, electrical interferences, interferences due to the reference electrode (e.g., from variation in salinity), and rigid design allowing only point measurements.
In the last decades, optical pH sensors (optodes) have been intensively investigated. pH optodes do not suffer from electromagnetic interferences, do not require reference sensors, are easy to miniaturize, and can be manufactured in a variety of formats, all of which enable high versatility of application. Particularly, planar optodes are excellent tools for imaging of pH distribution, fiber‐optic sensors and microsensors enable higher mechanical flexibility, and nanoparticles are of high interest for extra‐ and intracellular imaging.4, 8, 9, 10, 11, 12, 13, 14, 15, 16
In general, an optical pH sensor consists of a pH‐sensitive dye entrapped in a hydrophilic polymer matrix or a sol–gel. The protonated and deprotonated forms of the indicator dye feature different optical properties, most commonly either pH‐dependent excitation and/or emission spectra17, 18, 19 or fluorescence quenching through photoinduced electron transfer (PET) without any change in the spectral properties.20, 21, 22, 23, 24 PET indicators can be prepared with virtually any class of chromophores25 but require an additional luminescent reference for reliable measurements. On the contrary, ratiometric fluorescent indicators are essentially self‐referenced but are limited only to several dye classes. The most frequently used representatives are fluorescein derivatives,26, 27 hydroxycoumarins,28, 29 8‐hydroxypyrene‐1,3,6‐trisulfonic acid (HPTS),30, 31 and seminaphthorhodafluors (SNARFs).32 Recently, several other representatives have been reported.33, 34, 35, 36, 37, 38
The use of fluorescent dyes with absorption and emission in the red/near‐infrared (NIR) part of the spectrum has several advantages such as good light penetration in tissues essential for in vivo measurements, lower autofluorescence and light scattering from biological samples and optical components, and lower energy demand for the excitation, which is useful for autonomous sensing. The number of reported red/NIR‐emitting indicators is rather limited and only a few of them show ratiometric character.39 Moreover, high fluorescence brightness and high photostability are also highly desirable properties of a pH indicator.
Perylene tetracarboxylic acid bisimide dyes (PBIs) and pigments have gained substantial attention in academic and industrial research over the last decades. They are promising as n‐type semiconductors for electronics applications and as indicators for optical sensors.40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51 For sensing applications, excellent chemical and photochemical stability of the perylene dyes, their high molar absorption coefficients, and their high quantum yields close to unity are particularly attractive. Unfortunately, perylene dyes that are strongly emissive in the red part of the spectrum are rare. Often, synthetic modifications resulting in a bathochromic shift in absorption and emission are accompanied by a strong decrease in fluorescence brightness.52, 53, 54, 55 Imidazole‐substituted perylenes reported by Langhals et al. represent a notable exception.56
In this study, we present easily accessible core‐extended perylene bisimide indicators that show efficient absorption in the orange–red part of the spectrum and strong red–NIR fluorescence. We will demonstrate that these dyes are intrinsically pH sensitive, showing a strong bathochromic shift in the absorption and emission spectra upon deprotonation as a result of intramolecular charge transfer and thus enabling ratiometric sensing. The pK a values in the alkaline range enable several specific applications as well as the preparation of optical carbon dioxide sensors for different dynamic ranges.
Results and Discussion
Synthesis
Perylene bisimides are suitable for different synthetic modifications in the imide and bay regions of the chromophore.42, 45, 48, 57, 58 Lateral extension of the conjugated system in the bay region represents an interesting possibility to induce a bathochromic shift in the absorption and emission.56 We chose fluorescent dye 1 as a starting compound owing to its high solubility rendered by the diisopropylphenyl groups at the imide position and because of its commercial availability. The strong electron‐withdrawing character of the imide groups enabled its reaction with a strongly nucleophilic sodium amide and a dipolar aprotic solvent (i.e., benzonitrile) to yield 2‐phenylimidazole‐substituated perylene bisimides (Figure 1). Monosubstituted derivatives 2 a, 2 b, and 2 c were obtained from various benzonitriles, and doubly substituted 3 a was obtained in the case of benzonitrile. Notably, no doubly substituted products were formed upon using 4‐chlorobenzonitrile and 2,6‐dichlorobenzonitrile. Although the use of other benzonitriles is likely possible, we selected chloro‐substituted derivatives owing to the electron‐withdrawing effect of this halogen, and therefore, we expected a lowering of the pK a value (see below) of the perylene bisimide dyes. In the case of doubly substituted perylene 3 a, the formation of different isomers is possible and indeed was observed during the reaction. Isomer 3 a could be isolated in pure form with the position of the substituents confirmed by 1H NMR and 13C NMR spectroscopy (Figures S15–S31, Supporting Information).
Figure 1.
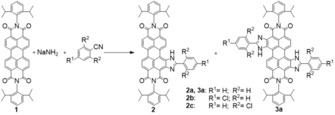
One‐pot synthesis of laterally extended perylene bisimide dyes. Double substitution with 2‐phenylimidazole groups was only observed for 3 a.
Photophysical properties
The introduction of 2‐phenylimidazole substituents in the bay position of the perylene chromophore results in a strong bathochromic shift in the absorption and emission spectra (Figure 2 a). Relative to the absorption spectrum of 1, the spectra of monosubstituted dyes 2 a, 2 b, and 2 c (neutral form) are shifted by Δλ≈55–65 nm, whereas the shift is as high as Δλ=115 nm for disubstituted 3 a (Table 1). The absorption maximum of 2 c is hypsochromically shifted by Δλ≈12 nm relative to those of 2 a and 2 b, and this may be due to the electron‐withdrawing character of the chlorine atoms and the decreased conjugation of the phenyl substituent because of steric hindrance. The molar absorption coefficients are rather high (Table 1) and are comparable to those of 1 (80 300 m −1 cm−1).59
Figure 2.
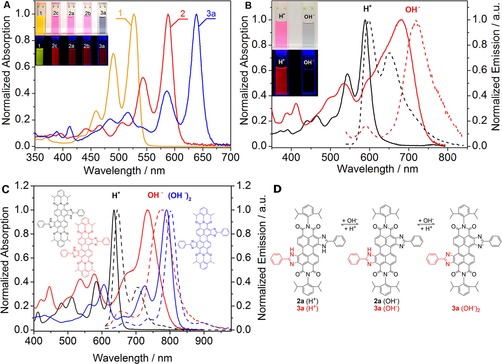
Spectral properties of the perylene dyes. a) absorption spectra of 1, 2, and 3 a in tetrahydrofuran. b) Representative absorption spectra (—) and emission spectra (‐ ‐ ‐ ‐) of the neutral form (black) and anionic form (red) of 2 a in tetrahydrofuran. The insets show photographic images of the dye solutions under daylight (top) and UV (bottom) illumination (λ=365 nm). c) Absorption spectra (—) and emission spectra (‐ ‐ ‐ ‐) of the neutral form (black), monoanionic form (red), and dianionic form (blue) of 3 a in tetrahydrofuran. d) Equilibria of the neutral and anionic forms of 2 a and 3 a.
Table 1.
Photophysical properties of perylene dyes in tetrahydrofuran. Absorption maxima (λ abs), molar absorption coefficients (ϵ), emission maxima (λ em), fluorescence quantum yields (Φ), and fluorescence lifetimes (τ) for the acidic and basic forms.
| Dye | λ abs [nm] | ϵ [m −1 cm−1] | λ em [nm] | Φ | τ [ns] | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| acidic | basic[a] | acidic | basic | acidic | basic[a] | acidic | basic[a] | acidic | basic[a] | |
| 2 a | 589 | 676 | 10 1300 | 55 500 | 597 | 735 | 0.99 | 0.10 | 5.3 | 3.2 |
| 2 b | 590 | 673 | 71 800 | 40 300 | 597 | 736 | 0.99 | 0.10 | 5.3 | 3.1 |
| 2 c | 578 | 662 | 75 900 | 47 300 | 584 | 713 | 0.98 | 0.15 | 5.3 | 4 |
| 3 a | 635 | 736[b]
789[c] |
96 600 | 48 400[b]
80 400[c] |
646 | 776[b]
802[c] |
0.85 | 0.25[b]
0.05[c] |
6.4 | 2.8[b]
3.1[c] |
[a] Basic form was generated by the addition of TOAOH to the solution. [b] Monoanionic form was generated by the addition of TOAOH to the solution and bubbling with CO2. [c] Dianionic form was generated by the addition of TOAOH to the solution.
All of the monosubstituted dyes in their neutral forms show very strong fluorescence with quantum yields approaching unity (Table 1) and small Stokes shifts of Δλ≈8 nm. The fluorescence quantum yield of 3 a is only slightly lower (0.85). Overall, the brightness of the dyes (defined as the product of the molar absorption coefficients and fluorescence quantum yields) is excellent. Additionally, an intense absorption band (ϵ≈40 000 m −1 cm−1) located at a wavelength that is approximately 50 nm shorter than the wavelength of the main band represents a very nice feature for practical applications, as it efficiently enhances the effective Stokes shift and, therefore, enables collection of the full emission spectrum with a simple optical setup.
Intrinsic pH sensitivity is the most striking property of the dyes. In basic media, deprotonation results in a strong bathochromic shift in the absorption and emission spectra of the monosubstituted dyes (Figure 2 b), and these shifts are approximately Δλ=85 and 130 nm, respectively (Table 1). Such behavior is typical for pH indicators based on an intramolecular charge transfer.26, 32 Compound 3 a, bearing two imidazole groups, shows the formation of the monoanionic and the dianionic forms, and the spectra of these forms are bathochromically shifted by approximately Δλ=100 and 150 nm, respectively, relative to the spectra of the neutral form of the dye (Figure 2 c). Notably, the absorption and emission spectra become significantly broader upon deprotonation. Broadening of the absorption spectra correlates well with a decrease in the molar absorption coefficients. Interestingly, the spectra of the dianionic form of 3 a are much less broadened than those of the neutral form, and the molar absorption coefficients are similar to those of the neutral form; this may be due to the symmetrical character of the formed species. The fluorescence quantum yields for the deprotonated form are approximately 10‐fold lower than those for the neutral form (Table 1). Overall, considering that the brightness of NIR dyes is generally significantly lower than that of UV/Vis chromophores, the new dyes in their deprotonated forms can be viewed as moderately strong NIR emitters.
The fluorescence lifetimes of the neutral forms are in a range of 5.3 to 6.4 ns (Table 1), which is typical for perylene fluorophores.60 The lifetimes of the negatively charged forms are 1.3–3.6 ns shorter. The introduction of a chlorine atom does not affect the lifetimes of the neutral forms. However, the increase in the conjugated system in 3 a results in a slight increase in the fluorescence lifetime.
Perylene dyes are known for their extraordinarily high photostability.43 Irradiation of the neutral forms of the dyes in toluene solution with a green high‐power LED array (λ max=528 nm, light intensity=353 mW cm−2) over 240 min did not result in any detectable bleaching (Figure 3). In contrast, irradiation of the basic form of 2 a in toluene solution with a red high‐power LED array (λ max=635 nm) of comparable intensity (377 mW cm−2) over 240 min showed degradation of approximately 6 % of the dye (Figure 3). The lower photostability of the basic form may be due to the more electron‐rich character of the core and, therefore, more favorable photooxidation. Nevertheless, even in the case of the anionic form the photostability is still sufficiently high considering the very high light intensities used.
Figure 3.
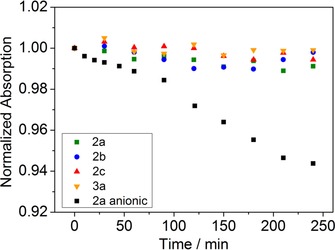
Photodegredation profiles of the neutral forms of 2 a, 2 b, 2 c, and 3 a and the anionic form of 2 a in toluene.
pH sensing properties
As mentioned above, all the dyes show pH‐dependent spectral properties. More detailed investigation conducted for THF/EtOH/aqueous buffered solution (3:2:2, v/v/v) revealed pK a values of 10.15–10.8. The pK a value of 2 b is slightly lower than those of 2 a, 2 c, and 3 a (Table 2). This effect can be explained by the electron‐withdrawing character of the chlorine atom in 2 b, which enhances the acidity of the imidazole. On the other hand, the steric hindrance introduced by the two chlorine atoms in the ortho positions in 2 c appears to compensate for the electron‐withdrawing effect.
Table 2.
pH sensing properties of the perylene dyes in solution and embedded in hydrogel D4.[a]
| THF/EtOH/buffer (3:2:2, v/v/v) | Hydrogel D4 | ||||
|---|---|---|---|---|---|
| Dye | pK a abs | λ abs (acidic/monoanionic) [nm] | pK a′ abs | λ em (acidic/monoanionic) [nm] | pK a′ em |
| 2 a | 10.63 | 596/689 | 10.88 | 608/728 | 10.14 |
| 2 b | 10.15 | 596/684 | 10.73 | 608/720 | 9.84 |
| 2 c | 10.78 | 586/664 | 11.45 | 595/708 | 10.56 |
| 3 a | 10.68 | 650/731 | 11.36 | 662/768 | 10.15 |
[a] Ionic strength (NaCl): 0.15 m, T=25 °C.
To enable continuous measurements of pH, it is essential to immobilize an indicator dye into a proton‐permeable polymer matrix. The two most common methods rely on covalent coupling of the dye to the polymer and noncovalent physical entrapment. Covalent coupling prevents leaching of the matrix and migration, but further synthetic steps are necessary to introduce functional groups for coupling, which may also negatively affect the photophysical properties of the indicator. Moreover, noncovalent immobilization is a simple method to obtain pH sensors provided that an adequate combination of the indicator and matrix is found. Planar solid‐state pH sensors were obtained by immobilization of the new perylenes into a commercially available polyurethane hydrogel (Hydromed D4) consisting of hydrophilic and hydrophobic domains and a water content in the swollen state of 50 % weight.61
As can be seen in Table 3, the pH‐sensing behavior of the immobilized dyes is very similar to that of the dyes in solution. Several isosbestic points are clearly visible in the absorption spectra, and this is indicative of an equilibrium between two species (neutral and anionic) (Figure 4 a). Thus, the new perylene dyes are suitable for colorimetric readout. The apparent pK a values determined from the absorption spectra are slightly higher for the immobilized dyes than for the dissolved ones (Table 2). This may be due to localization of the indicators in the more hydrophobic domains of the hydrogel, at which deprotonation (resulting in the formation of the charged form of the dye) is less favored. Similarly to the absorption spectra, the fluorescence spectra show the existence of two forms (Figure 4 b), which enables ratiometric readout. The apparent pK a′ values in hydrogel D4 determined from the fluorescence spectra are 0.7–1.2 pH units lower than those obtained from the absorption measurements (Table 2). This disparity of the pK a′ values can be explained by Förster resonance energy transfer (FRET) from the neutral form to the deprotonated form of the dye. In fact, FRET is favored by good overlap between the absorption spectrum of the deprotonated form and the emission spectrum of the neutral form (Figure 2 b) and by the comparably high concentration of the dye in the polymer, which results in a short distance between the donor and acceptor molecules. Evidently, because of the high pK a′ values, the new sensors are not suitable for conventional applications (e.g., biotechnology, biology, and medicine). On the other hand, the new materials are promising for pH measurements under alkaline conditions. Potential applications include the measurement of pH on the surface of concrete; this is known for extremely alkaline conditions, which, however, change owing to microbiological activity and interaction with acidic gases thus promoting corrosion of steel.62, 63 Here, optical pH sensors can be a promising alternative to pH electrodes because they do not suffer from alkali error, are easy to miniaturize, and can also be used for imaging of pH distribution. The new sensors may also be used in studies involving alkaliphilic microorganisms, which are of great interest for many biotechnological applications and in the detergent and textile industries.64, 65, 66
Table 3.
Composition of the carbon dioxide sensors.
| Sensor | Dye | EC/TBP [w/w] | Base[a] |
|---|---|---|---|
| CO2_1 | 2 a | 1:2 | TOAOH |
| CO2_2 | 2 b | 1:2 | TOAOH |
| CO2_3 | 2 c | 1:2 | TOAOH |
| CO2_4 | 2 c | 2:1 | TOAOH |
| CO2_5 | 2 c | 1:0 | TOAOH |
| CO2_6 | 2 c | 1:0 | TBAOH |
| CO2_7 | 2 c | 2:1 | TBAOH |
| CO2_8 | 3 a | 1:2 | TOAOH |
| CO2_9 | 3 a | 1:2 | TBAOH |
Figure 4.
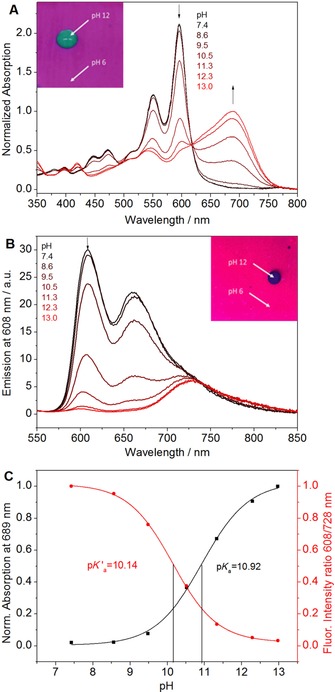
pH sensing properties of 2 a embedded into polyurethane hydrogel D4. a, b) pH dependence of the absorption and fluorescence spectra, respectively. c) Corresponding calibration curves. The insets in panels a and b show photographic images of the sensor foils under daylight and UV illumination (λ=365 nm), respectively.
The response of the sensors to dynamic pH changes is reversible (Figures S4 and S5) but fairly slow (t 90≈77 s) considering the comparably small thickness of the sensing layer (2.5 μm). Some hysteresis in the response is also observed. This behavior suggests localization of the dyes mostly in the hydrophobic domains of the hydrogel and some redistribution upon (de)protonation. Similar behavior was previously observed for other hydrophobic perylene dyes.49 It may be overcome by covalent immobilization of the dyes in polymeric matrixes52 or by increasing the hydrophilicity of the perylene (e.g., by employing other substituents in the imide position) and represents room for future improvements.
Carbon dioxide sensors
The high pK a′ values of the dyes imply their potential suitability for carbon dioxide sensing. Solid‐state or “plastic” optical carbon dioxide sensors rely on pH indicators embedded into polymers along with a lipophilic base, most commonly a quaternary ammonium base.67, 68, 69, 70 Several sensing materials based on ethyl cellulose (EC) as a matrix were manufactured to investigate systematically the influence of the indicator (i.e., 2 a, 2 b, 2 c, and 3 a), base [i.e., tetraoctylammonium hydroxide (TOAOH) and tetrabutylammonium hydroxide (TBAOH)], and plasticizer [i.e., tributyl phosphate (TBP)]. The sensor composition is schematically shown in Figure 5 a. A porous Teflon membrane was applied over the sensing layer to provide a homogenous white background and to eliminate the influence of ionic species in case of solution measurements.
Figure 5.
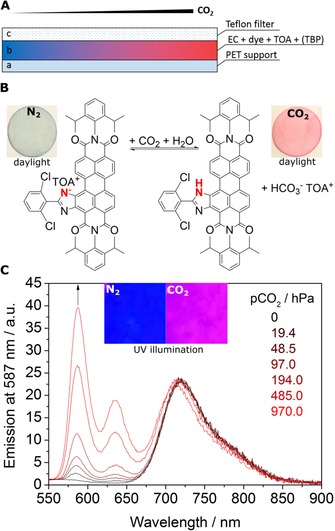
a) Cross section of the optical carbon dioxide sensor. b) Sensing mechanism and photographic images of the planar sensor foil based on 2 c in ethyl cellulose with tributyl phosphate as a plasticizer (1:2, w/w, material CO2_3) measured in water purged with nitrogen and 100 % carbon dioxide recorded under daylight. c) Corresponding emission spectra (λ ex=493 nm) for CO2_3 sensor measured in water at different pCO2 and photographic images of the sensor under UV illumination (λ=365 nm).
As can be seen (Figure 5 b), the sensors indeed respond to carbon dioxide. The color of the sensor changes from blue (in the absence of carbon dioxide) to red (in the presence of carbon dioxide), which enables colorimetric measurements of carbon dioxide. An increase in pCO2 also results in the appearance of strong red fluorescence. The corresponding emission spectra (Figure 5 c) show an increase in the fluorescence of the neutral form (λ max=587 nm) in the presence of carbon dioxide. In contrast, the fluorescence intensity at λ=720 nm corresponding to the deprotonated form is nearly constant. The likely reason is fairly efficient FRET from the neutral form to the deprotonated form of the dye. Thus, a strong increase in the absorption of the neutral form at the excitation wavelength (λ ex=493 nm) at high pCO2 results in higher fluorescence intensity of the neutral form but also for the deprotonated form (by FRET), which compensates for the decrease in the concentration of the deprotonated form. Referenced ratiometric measurements at two emission wavelengths are clearly possible.
The sensitivity of carbon dioxide sensors is known to be governed by the pK a value of the indicator and by many other reasons such as the presence of a plasticizer.71, 72 Figure 6 a shows that despite rather high pK a values, all of the materials show comparably low sensitivity. All sensors operate in the dynamic range of pCO2=10 to 970 hPa, which makes them interesting candidates for modified atmosphere packaging applications.73 In good agreement with the pK a values (Table 2), the sensitivity is the highest for 2 c (the highest pK a) and the lowest for 2 b (the lowest pK a), whereas 2 a occupies the intermediate position. The sensitivity of 3 a (first deprotonation step) is comparable to that of 2 c; however, steric reasons and the lipophilicity of the dye might be additional contributing factors. The less‐sensitive sensors based on 2 a and 2 b (CO2_1 and CO2_2, respectively) are promising for the measurement of carbon dioxide pressures >970 hPa, typical for the beverage industry,74, 75, 76 because they show high residual protonation capability relative to the sensors based on 2 c and 3 a (CO2_3 and CO2_8, respectively) at pCO2=970 hPa (Figure 6 a).
Figure 6.
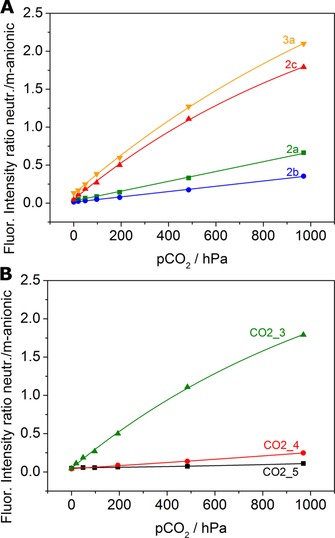
Carbon dioxide sensing properties of various materials measured in water at 25 °C a) Responses of sensors based on 2 a, 2 b, 2 c, and 3 a in ethyl cellulose with tributyl phosphate as a plasticizer (1:2, w/w, materials CO2_1, CO2_2, CO2_3, and CO2_8, respectively). b) Influence of plasticizer on the response of the sensor based on 2 c. Ethyl cellulose/tributyl phosphate=1:0 (CO2_5), 2:1 (CO2_4), 1:2 (CO2_3).
The sensitivity of the sensors is dramatically affected by the presence and concentration of a plasticizer (Figure 6 b). Whereas the sensor is almost insensitive in pure ethyl cellulose, a plasticizer/polymer ratio of 2:1 ensures optimal response. The plasticizer is likely to influence the diffusion and solubility of carbon dioxide but also to alter polarity of the environment and water uptake. Thus, adjustment of the ratio of the plasticizer to the polymer represents an excellent possibility to fine‐tune the sensitivity of the sensor. Further adjustment of the sensor sensitivity can be achieved by substitution of tetraoctylammonium hydroxide with tetrabutylammonium hydroxide. Shorter carbon chains of the latter are associated with higher sensitivity of the sensors most likely because of higher hydrophilicity of the dye environment (Figures S10–S12).
Humidity also plays an important role for response of the sensor material, and it has to be adjusted accurately. As the highest reproducibility of calibrations for the fabricated sensor materials could be achieved by using a flow‐through cell filled with water and bubbled with carbon dioxide, this setup was utilized for all measurements. Although the sensor also shows response in the gas phase, the sensitivity at 85 % relative humidity is significantly lower than that in water (Figures S13 and S14). Because of such strong cross‐talk, application of the new sensors in the gas phase might be problematic.
In contrast to monosubstituted dyes 2 a–c, disubstituted dye 3 a features two pH‐sensitive groups. This results in unique behavior that enables ratiometric sensing of carbon dioxide in two different dynamic ranges with a single indicator dye. Notably, all three forms of the indicator (neutral, monoanionic, and dianionic) are fluorescent (λ max=658, 776, and 802 nm, respectively) (Figure 7). The monoanionic form is converted into the neutral form at comparably high pCO2 (20–1000 hPa). In this dynamic range, the behavior of the sensor is similar to that of the materials based on 2 a–c. In contrast, the dianionic form is stable only at very low pCO2 (Figure 7 a). It is completely converted into the monoanionic form at approximately pCO2=2 hPa. Remarkably, pCO2 values well around atmospheric levels of 0.4 hPa can be resolved excellently. Thus, the sensor based on 3 a covers an extremely broad dynamic range of pCO2=0 to 1000 hPa, which is unprecedented in the literature.
Figure 7.
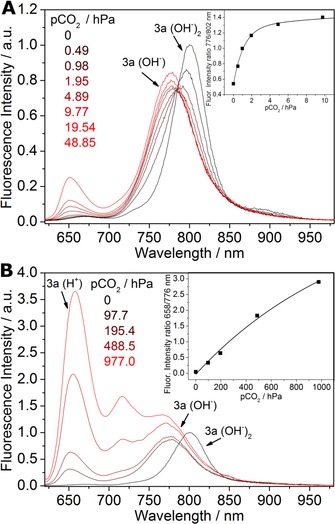
Response of the sensor based on 3 a (CO2_9, 25 °C, 85 % relative humidity, λ ex=590 nm) for a) pCO2=0 to 49 hPa and b) pCO2=98 to1000 hPa. The insets show the corresponding calibration curves. Equilibria for the neutral, monoanionic, and dianionic forms of 3 a are shown in Figure 2 d.
Conclusions
In summary, we introduced a new class of pH indicators based on bay‐modified perylene dyes that can be utilized for optical pH sensors as well as for optical carbon dioxide sensors. The dyes show high luminescence brightness, extraordinary good photostability, and versatility with respect to chemical modification. The indicators are accessible through a simple one‐step reaction starting from commercially available Lumogen Orange. The characteristic absorption and emission spectra of the neutral and deprotonated forms of the perylene bisimides enable colorimetric as well as ratiometric measurements of pH and carbon dioxide. The high pK a values enable preparation of optical pH sensors for measurements under alkaline conditions. The sensitivity of the carbon dioxide sensor can be adjusted by varying the dye, ratio of plasticizer to polymer, and nature of the quaternary ammonium base. The disubstituted perylene shows unique behavior with two protonation/deprotonation equilibria, enabling sensing of carbon dioxide over an unprecedentedly broad dynamic range. Whereas the new carbon dioxide sensors are likely to be promising for applications in food packaging and the beverage industry, the pH sensors with responses in alkaline media are of interest for research on alkaliphilic bacteria and monitoring of pH in concrete. Here, excellent chemical and photochemical stability of the new indicators is expected to be very valuable for long‐term deployment of the sensors.
Experimental Section
Materials
Anhydrous benzonitrile, 2,6‐dichlorobenzonitrile, ethyl cellulose (EC49, ethoxyl content 49 %), tetrabutylammonium hydroxide (TBAOH, 40 % in water), tetraoctylammonium hydroxide (TOAOH, 20 % in methanol), and tributyl phosphate (TBP) were purchased from Sigma–Aldrich (http://www.sigmaaldrich.com). 4‐Chlorobenzonitrile was obtained from TCI Europe (http://www.tcieurope.de). Lumogen Orange (1) was purchased from Kremer Pigmente (http://www.kremer-pigmente.de). Dichloromethane was received from Fisher Scientific (http://www.fishersci.com). Cyclohexane, ethyl acetate, tetrahydrofuran, toluene (synthesis grade), and hydrochloric acid (37 %) were purchased from VWR Chemicals (http://www.vwr.com). Polyurethane hydrogel (Hydromed D4) was obtained from AdvanSource biomaterials (http://www.advbiomaterials.com). Poly(ethylene terephthalate) (PET) support was received from Pütz (http://www.puetz-folien.com). Anhydrous sodium sulfate, anhydrous disodium phosphate, and the buffer salts CAPS, MOPS, and TRIS were obtained from Carl Roth (http://www.roth.de). Sodium amide (99 %) was purchased from Acros (http://www.acros.com). Deuterated chloroform (CDCl3) was purchased from Euriso‐top (http://www.eurisotop.com). All purchased chemicals were used without further purification. Nitrogen, 0.2 % carbon dioxide in nitrogen, 5 % carbon dioxide in nitrogen, and carbon dioxide (all 99.999 % purity) were received from Linde (http://www.linde-gas.at). Potassium chloride and silica gel 60 (0.063–0.200 mm) were obtained from Merck (http://www.merck.at). Polytetrafluoroethylene (PTFE) hydrophobic membranes (45 μm) were obtained from Millipore Merck (http://www.merckmillipore.com).
Syntheses
Compounds 2 a and 3 a: Compound 1 (400 mg, 0.563 mmol) and NaNH2 (99 %, 440 mg, 11.3 mmol) were dispersed in anhydrous benzonitrile (50 mL) under an inert atmosphere and were heated to 165 °C. The color changed from orange to green and afterwards to dark blue. Compressed air was bubbled through the mixture for 30 min. The mixture was cooled to RT, and a mixture of 1 m aq HCl/dichloromethane (1:1, v/v, 200 mL) was added. The organic phase was washed with 1 m aq HCl (3×100 mL) and dried (Na2SO4). The organic solvents were removed under reduced pressure. The purple crude products were purified by column chromatography (silica gel, 35–70 μm) with cyclohexane/ethyl acetate (93:7, v/v) as the mobile phase to elute 2 a (220 mg, 47 %) and cyclohexane/ethyl acetate (7:1, v/v) to elute 3 a (116 mg, 22 %). Data for 2 a: 1H NMR (300 MHz, CDCl3): δ=11.58 (s, 1 H), 11.01 (d, J=8.2 Hz, 1 H), 8.98 (d, J=8.2 Hz, 1 H), 8.91–8.78 (m, 4 H), 8.34 (dd, J=6.6, 3.0 Hz, 2 H), 7.70–7.62 (m, 3 H), 7.53 (dt, J=13.1, 7.9 Hz, 2 H), 7.39 (dd, J=12.6, 7.7 Hz, 4 H), 2.82 (h, J=6.9 Hz, 4 H), 1.21 ppm (dd, J=6.9, 4.6 Hz, 24 H); 13C NMR (126 MHz, CDCl3, 30 °C): δ=164.89, 163.84, 163.74, 163.68, 157.82, 145.82, 145.73, 144.18, 139.63, 136.00, 135.53, 135.41, 132.68, 132.36, 131.46, 131.37, 131.19, 130.79, 130.14, 129.86, 129.81, 129.59, 129.51, 128.12, 127.69, 127.21, 127.18, 125.83, 124.23, 124.08, 124.05, 123.29, 123.08, 122.70, 122.66, 121.59, 103.66, 29.29, 29.24, 24.09, 24.03, 24.02 ppm; MS (MALDI‐TOF): m/z: calcd for C55H46N4O4: 826.352 [M]+; found: 826.458; data for 3 a: 1H NMR (300 MHz, CDCl3): δ=11.69 (s, 2 H), 11.24 (d, J=8.2 Hz, 2 H), 9.02 (d, J=8.2 Hz, 2 H), 8.36 (dd, J=6.8, 2.9 Hz, 4 H), 7.66 (m, J=5.0, 2.1 Hz, 6 H), 7.48 (m, 6 H), 2.87 (h, J=7.0 Hz, 4 H), 1.09–0.67 ppm (m, 24 H); 13C NMR (126 MHz, CDCl3): δ=164.79, 163.92, 156.51, 145.92, 142.95, 139.43, 135.58, 131.90, 131.33, 130.97, 129.76, 129.44, 129.28, 128.42, 127.55, 127.44, 124.65, 124.02, 122.67, 121.52, 103.04, 31.90, 29.63, 29.33, 29.22, 24.08, 24.00, 22.66, 14.07 ppm; MS (MALDI‐TOF): m/z: calcd for C62H50N6O4: 942.389 [M]+; found: 942.485.
Compound 2 b: Compound 1 (105 mg, 0.148 mmol) and NaNH2 (99 %, 160 mg, 4.10 mmol) were quickly homogenized in a mortar and 4‐chlorobenzonitrile (7.16 g) was added. The powder was transferred into a Schlenk flask and was heated to 150 °C under an inert atmosphere. The color changed from orange to purple/dark red. At 160 °C, compressed air was bubbled through the mixture, which was stirred for 3 h under reflux conditions. The mixture was cooled to RT, a mixture of 1 m aq HCl/dichloromethane (1:1, v/v, 150 mL) was added, and the organic phase was washed with 1 m aq HCl (3×50 mL) (color change from green to purple) and dried (Na2SO4). The organic solvents were removed under reduced pressure. The crude product was purified by column chromatography (silica gel, 35–70 μm, cyclohexane/ethyl acetate 94:6, v/v) to yield 2 b (47 mg, 37 %): 1H NMR (300 MHz, CDCl3): δ=11.56 (s, 1 H), 10.96 (d, J=8.2 Hz, 1 H), 8.97 (d, J=8.2 Hz, 1 H), 8.93–8.76 (m, 4 H), 8.31–8.23 (m, 2 H), 7.67–7.61 (m, 2 H), 7.53 (dt, J=12.6, 8.0 Hz, 2 H), 7.39 (dd, J=12.1, 7.7 Hz, 4 H), 2.81 (m, 4 H), 1.23–1.08 ppm (m, 24 H); 13C NMR (126 MHz, CDCl3): δ=164.88, 163.79, 163.70, 163.65, 156.66, 145.80, 145.72, 143.98, 139.55, 138.73, 136.06, 135.41, 135.35, 132.66, 131.61, 131.40, 130.75, 130.07, 129.86, 129.80, 129.61, 128.88, 127.26, 127.19, 126.63, 126.02, 124.24, 124.11, 124.09, 123.35, 123.18, 122.77, 122.70, 121.68, 103.71, 29.29, 29.24, 24.08, 24.03, 24.01 ppm; MS (MALDI‐TOF): m/z: calcd for C55H45ClN4O4: 860.313 [M]+; found: 860.348.
Compound 2 c: Compound 2 c was prepared analogously to 2 b, except that 1 (120 mg, 0.169 mmol), NaNH2 (500 mg, 12.82 mmol), and 2,6‐dichlorobenzonitrile (7.05 g) were used. The mixture was heated in a Schlenk flask at 160 °C under an inert atmosphere. At 170 °C, compressed air was bubbled through the mixture, which was stirred for 1 h under reflux conditions. Chromatographic purification of the crude product was performed (silica gel, 35–70 μm, dichloromethane) to yield 2 c (101 mg, 67 %): 1H NMR (300 MHz, CDCl3): δ=11.40 (s, 1 H), 10.89 (d, J=8.2 Hz, 1 H), 9.00–8.70 (m, 5 H), 7.58 (d, J=7.0 Hz, 2 H), 7.54–7.45 (m, 3 H), 7.37 (dd, J=10.3, 7.7 Hz, 4 H), 2.81 (m, 4 H), 1.19 ppm (m, 24 H); 13C NMR (126 MHz, CDCl3): δ=164.49, 163.84, 163.70, 163.63, 152.69, 145.85, 145.71, 142.81, 138.24, 136.19, 135.90, 135.27, 135.20, 132.77, 132.40, 131.92, 131.58, 131.35, 130.74, 130.13, 129.80, 129.73, 129.58, 128.96, 128.35, 127.50, 127.16, 127.03, 124.18, 124.05, 123.97, 123.41, 123.30, 122.90, 122.78, 121.70, 104.43, 29.27, 29.23, 24.04, 24.00 ppm; MS (MALDI‐TOF): m/z: calcd for C55H44Cl2N4O4: 894.2740 [M]+; found: 894.2684.
Preparation of sensor foil pH solid‐state sensor
A “cocktail” containing the indicator dye (0.25 mg) and hydrogel D4 (50 mg) in THF (500 μL) was knife‐coated on a dust‐free, transparent PET support to obtain approximately 7.5 μm (for absorption measurements) and 2.5 μm (for emission measurements) thick sensing layers after solvent evaporation.
Carbon dioxide solid‐state sensor
To prepare the CO2 solid‐state sensors, the indicator dye (1 mg), ethyl cellulose (49 %, 100 mg), and optionally tributyl phosphate (TBP, 200 or 50 μL) were dissolved in an ethanol/toluene mixture (2:3, v/v, 1.9 g). The “cocktail” was purged with carbon dioxide and tetraoctylammonium hydroxide solution (20 % w/w TOAOH in MeOH, 100 μL) or tetrabutylammonium hydroxide solution (20 % w/w TBAOH in MeOH, 100 μL) was added. The “cocktails” were knife‐coated on a PET support to obtain an approximately 2.5 μm thick sensing layer after solvent evaporation. A PTFE hydrophobic membrane was placed on each sensor layer before solvent evaporation. The exact composition of the sensing materials is shown in Table 3.
Methods
Mass spectrometry was performed with a Micromass TofSpec 2E Time‐of‐Flight Mass Spectrometer at the Institute for Chemistry and Technology of Materials, Graz University of Technology. 1H NMR spectra were recorded with a 300 MHz Bruker Instrument (http://www.bruker.com) in CDCl3 as the solvent and tetramethylsilane as the standard. 13C NMR spectra were recorded with a 500 MHz Varian Inova 500 Instrument in CDCl3 as the solvent and tetramethylsilane as the standard.
Absorption spectra were recorded with a Cary 50 UV/Vis spectrophotometer from Varian (http://www.agilent.com) by using optical glass cuvettes from Hellma Analytics (http://www.hellma-analytics.com). Emission and excitation spectra were recorded with a FluoroLog 3 Spectrofluorometer from Horiba Scientific Jobin Yvon (http://www.horiba.com) equipped with a R2658 photomultiplier from Hamamatsu and corrected for detector response. Determination of absolute fluorescence quantum yields (Φ) was performed with the same spectrofluorometer from Horiba equipped with an integrating sphere Quanta‐phi.
Fluorescence decay times were measured by time‐correlated single photon counting (TCSPC) with a FluoroLog 3 Spectrofluorometer equipped with a DeltaHub module and NanoLEDs (λ=435 and 635 nm, Horiba) as excitation sources. Data analysis was performed with DAS6 software (http://www.horiba.com) by using a monoexponential fit.
The photostability of the dyes was accessed by irradiating water‐free toluene solutions with an array of 12 green high‐power LEDs (λ=528 nm, OSRAM Oslon SSL 80, http://www.led-tech.de) equipped with a cooling block and a focusing lens from Edmund optics (http://www.edmundoptics.de). The photostability of the deprotonated form of 2 a was accessed by irradiating water‐free toluene solutions with an array of 12 red high‐power LEDs (λ=635 nm, OSRAM Oslon SSL 80, http://www.led-tech.de) equipped with a cooling block and a focusing lens from Edmund optics (http://www.edmundoptics.de). The absorption spectra were acquired each for 15–30 min. The photon flux was 15 600 μmol s−1 m−2 for the green LEDs and 19 999 μmol s−1 m−2 for the red LEDs, as measured by a Li‐250A light meter from Li‐COR (http://www.licor.com).
The pH of buffer solutions (CAPS, MOPS, TRIS, and disodium phosphate) was adjusted with a pH meter (Education Line, Mettler Toledo, http://www.mt.com) by using a glass electrode (InLab Routine Pro, Mettler Toledo, http://www.mt.com). The pH meter was calibrated at 25 °C with standard buffer solutions of pH 4.01, 7.01, and 10.01 (Hanna Instruments, http://www.hannainst.com). The ionic strength (0.15 m) of the buffers was adjusted with sodium chloride as a background electrolyte.
For the leaching experiments, the sensor foil (hydrogel D4) was fixed in a home‐made flow‐through cell. Buffer solutions (pH 6.6 and 12.5, ionic strength 0.15 m) were pumped through the cell at a constant flow, and absorption spectra were recorded over 24 h. A peristaltic pump equipped with a flexible tube from Ismatec (http://www.ismatec.com; purple/black, ID: 2.29 mm) was used.
For carbon dioxide measurements, the sensor foil (CO2_1–9) was fixed in a home‐made flow‐through cell connected to a gas‐mixing device. Gas calibration mixtures were produced with a gas‐mixing device from Voegtlin (http://www.voegtlin.com, red‐y for gas flow), and a constant flow (200 mL min−1) was controlled by LabView software. The flow‐through cell was filled with water that was purged with gas mixtures. For measurements in the gas phase, a relative humidity of 85 % was adjusted by bubbling of the gas through a saturated potassium chloride solution. The temperature was controlled with a cryostat Thermostat Thermo Haake K10.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support by the Austrian Academy of Science (ÖAW) at the Institute of Analytical Chemistry and Food Chemistry, Graz University of Technology (DOC‐Fellowship of David Pfeifer), is gratefully acknowledged. The authors also thank Prof. Hansjoerg Weber, Institute of Organic Chemistry, Graz University of Technology, for help with the acquisition of the NMR spectra.
D. Pfeifer, I. Klimant, S. M. Borisov, Chem. Eur. J. 2018, 24, 10711.
References
- 1. Wang X., Meier R. J., Schmittlein C., Schreml S., Schäferling M., Wolfbeis O. S., Sens. Actuators B 2015, 221, 37–44. [Google Scholar]
- 2. Jeevarajan A. S., Vani S., Taylor T. D., Anderson M. M., Biotechnol. Bioeng. 2002, 78, 467–472. [DOI] [PubMed] [Google Scholar]
- 3. John G. T., Goelling D., Klimant I., Schneider H., Heinzle E., J. Dairy Res. 2003, 70, 327–333. [DOI] [PubMed] [Google Scholar]
- 4. Kühl M. in Methods in Enzymology, Elsevier, Amsterdam, 2005, pp. 166–199. [DOI] [PubMed] [Google Scholar]
- 5. Larsen M., Borisov S. M., Grunwald B., Klimant I., Glud R. N., Limnol. Oceanogr.: Methods 2011, 9, 348–360. [Google Scholar]
- 6. Leiner M. J. P., Anal. Chim. Acta 1991, 255, 209–222. [Google Scholar]
- 7. Narayanaswamy R., Wolfbeis O. S., Optical Sensors: Industrial, Environmental and Diagnostic Applications, Springer, Amsterdam, 2004. [Google Scholar]
- 8. Schäferling M., Angew. Chem. Int. Ed. 2012, 51, 3532–3554; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 3590–3614. [Google Scholar]
- 9. Meier R. J., Schreml S., Wang X., Landthaler M., Babilas P., Wolfbeis O. S., Angew. Chem. Int. Ed. 2011, 50, 10893–10896; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 11085–11088. [Google Scholar]
- 10. Aigner D., Dmitriev R. I., Borisov S. M., Papkovsky D. B., Klimant I., J. Mater. Chem. B 2014, 2, 6792–6801. [DOI] [PubMed] [Google Scholar]
- 11. Wolfbeis O. S., J. Mater. Chem. 2005, 15, 2657. [Google Scholar]
- 12. Wencel D., Abel T., McDonagh C., Anal. Chem. 2014, 86, 15–29. [DOI] [PubMed] [Google Scholar]
- 13. Lin J., TrAC Trends Anal. Chem. 2000, 19, 541–552. [Google Scholar]
- 14. Umezawa K., Citterio D., Suzuki K., Anal. Sci. 2014, 30, 327–349. [DOI] [PubMed] [Google Scholar]
- 15. Orellana G., Haigh D., Curr. Anal. Chem. 2008, 4, 273–295. [Google Scholar]
- 16. McDonagh C., Burke C. S., MacCraith B. D., Chem. Rev. 2008, 108, 400–422. [DOI] [PubMed] [Google Scholar]
- 17. Willoughby D., Thomas R. C., Schwiening C. J., Pflügers Arch. 1998, 436, 615–622. [DOI] [PubMed] [Google Scholar]
- 18. Kermis H. R., Kostov Y., Rao G., Analyst 2003, 128, 1181. [DOI] [PubMed] [Google Scholar]
- 19. Schröder C. R., Weidgans B. M., Klimant I., Analyst 2005, 130, 907. [DOI] [PubMed] [Google Scholar]
- 20. Murtagh J., Frimannsson D. O., O'Shea D. F., Org. Lett. 2009, 11, 5386–5389. [DOI] [PubMed] [Google Scholar]
- 21. Han J., Burgess K., Chem. Rev. 2010, 110, 2709–2728. [DOI] [PubMed] [Google Scholar]
- 22. Jokic T., Borisov S. M., Saf R., Nielsen D. A., Kühl M., Klimant I., Anal. Chem. 2012, 84, 6723–6730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ni Y., Wu J., Org. Biomol. Chem. 2014, 12, 3774. [DOI] [PubMed] [Google Scholar]
- 24. Strobl M., Rappitsch T., Borisov S. M., Mayr T., Klimant I., Analyst 2015, 140, 7150–7153. [DOI] [PubMed] [Google Scholar]
- 25. Aigner D., Freunberger S. A., Wilkening M., Saf R., Borisov S. M., Klimant I., Anal. Chem. 2014, 86, 9293–9300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Martin M. M., Lindqvist L., J. Lumin. 1975, 10, 381–390. [Google Scholar]
- 27. Cajlakovic M., Lobnik A., Werner T., Anal. Chim. Acta 2002, 455, 207–213. [Google Scholar]
- 28. Wolfbeis O. S., Marhold H., Chem. Ber. 1985, 118, 3664–3672. [Google Scholar]
- 29. Wolfbeis O. S., Koller E., Hoghmuth P., Bull. Chem. Soc. Jpn. 1985, 58, 731–734. [Google Scholar]
- 30. Offenbacher H., Wolfbeis O. S., Fürlinger E., Sens. Actuators 1986, 9, 73–84. [Google Scholar]
- 31. Hulth S., Aller R. C., Engström P., Selander E., Limnol. Oceanogr. 2002, 47, 212–220. [Google Scholar]
- 32. Whitaker J. E., Haugland R. P., Prendergast F. G., Anal. Biochem. 1991, 194, 330–344. [DOI] [PubMed] [Google Scholar]
- 33. Li Y., Wang Y., Yang S., Zhao Y., Yuan L., Zheng J., Yang R., Anal. Chem. 2015, 87, 2495–2503. [DOI] [PubMed] [Google Scholar]
- 34. He L., Lin W., Xu Q., Wei H., ACS Appl. Mater. Interfaces 2014, 6, 22326–22333. [DOI] [PubMed] [Google Scholar]
- 35. Wan Q., Chen S., Shi W., Li L., Ma H., Angew. Chem. Int. Ed. 2014, 53, 10916–10920; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 11096–11100. [Google Scholar]
- 36. Charier S., Ruel O., Baudin J.-B., Alcor D., Allemand J.-F., Meglio A., Jullien L., Angew. Chem. Int. Ed. 2004, 43, 4785–4788; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 4889–4892. [Google Scholar]
- 37. Niu W., Fan L., Nan M., Li Z., Lu D., Wong M. S., Shuang S., Dong C., Anal. Chem. 2015, 87, 2788–2793. [DOI] [PubMed] [Google Scholar]
- 38. Liu X.-D., Xu Y., Sun R., Xu Y.-J., Lu J.-M., Ge J.-F., Analyst 2013, 138, 6542. [DOI] [PubMed] [Google Scholar]
- 39. Staudinger C., Borisov S. M., Methods Appl. Fluoresc. 2015, 3, 042005. [DOI] [PubMed] [Google Scholar]
- 40. Kohl C., Weil T., Qu J., Müllen K., Chem. Eur. J. 2004, 10, 5297–5310. [DOI] [PubMed] [Google Scholar]
- 41. Langhals H., Ismael R., Yürük O., Tetrahedron 2000, 56, 5435–5441. [Google Scholar]
- 42. Seybold G., Dyes Pigm. 1989, 11, 303–317. [Google Scholar]
- 43. Rademacher A., Märkle S., Langhals H., Chem. Ber. 1982, 115, 2927–2934. [Google Scholar]
- 44. Langhals H., Karolin J., Johansson L. B.-Å., J. Chem. Soc. Faraday Trans. 1998, 94, 2919–2922. [Google Scholar]
- 45. Schnurpfeil G., Dyes Pigm. 1995, 27, 339–350. [Google Scholar]
- 46. Qu J., Kohl C., Pottek M., Müllen K., Angew. Chem. 2004, 116, 1554–1557. [DOI] [PubMed] [Google Scholar]
- 47. Baumstark D., Wagenknecht H.-A., Angew. Chem. Int. Ed. 2008, 47, 2612–2614; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 2652–2654. [Google Scholar]
- 48. Avlasevich Y., Li C., Müllen K., J. Mater. Chem. 2010, 20, 3814. [Google Scholar]
- 49. Aigner D., Borisov S. M., Klimant I., Anal. Bioanal. Chem. 2011, 400, 2475–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang Y., Zhao Z., Huang X., Xie Y., Liu C., Li J., Guan X., Zhang K., Cheng C., Xiao Y., RSC Adv. 2012, 2, 12644. [Google Scholar]
- 51. Dimitrakopoulos C. D., Malenfant P. R. L., Adv. Mater. 2002, 14, 99–117. [Google Scholar]
- 52. Aigner D., Borisov S. M., Petritsch P., Klimant I., Chem. Commun. 2013, 49, 2139. [DOI] [PubMed] [Google Scholar]
- 53. Holtrup F. O., Müller G. R. J., Quante H., De Feyter S., De Schryver F. C., Müllen K., Chem. Eur. J. 1997, 3, 219–225. [DOI] [PubMed] [Google Scholar]
- 54. Avlasevich Y., Müllen K., Chem. Commun. 2006, 4440–4442. [DOI] [PubMed] [Google Scholar]
- 55. Avlasevich Y., Müller S., Erk P., Müllen K., Chem. Eur. J. 2007, 13, 6555–6561. [DOI] [PubMed] [Google Scholar]
- 56. Langhals H., Esterbauer A. J., Kinzel S., New J. Chem. 2009, 33, 1829. [Google Scholar]
- 57. Würthner F., Chem. Commun. 2004, 1564–1579. [DOI] [PubMed] [Google Scholar]
- 58. Würthner F., Stolte M., Chem. Commun. 2011, 47, 5109. [DOI] [PubMed] [Google Scholar]
- 59. Sanguineti A., Sassi M., Turrisi R., Ruffo R., Vaccaro G., Meinardi F., Beverina L., Chem. Commun. 2013, 49, 1618. [DOI] [PubMed] [Google Scholar]
- 60. Johansson L. B.-Å., Langhals H., Spectrochim. Acta Part A 1991, 47, 857–861. [Google Scholar]
- 61. http://www.advbiomaterials.com/products/hydrophilic/HydroMed.pdf, 2015.
- 62.J. P. Broomfield, Corrosion of Steel in Concrete Understanding, Investigation and Repair, E & FN Spon [Etc.]: [Online:] Taylor & Francis, London [Etc.], 1997.
- 63. Zhou Q., Hill J., Byars E. A., Cripps J. C., Lynsdale C. J., Sharp J. H., Cem. Concr. Res. 2006, 36, 160–170. [Google Scholar]
- 64. Bowers K. J., Wiegel J., Extremophiles 2011, 15, 119–128. [DOI] [PubMed] [Google Scholar]
- 65. Hoondal G. S., Tiwari R. P., Tewari R., Dahiya N., Beg Q. K., Appl. Microbiol. Biotechnol. 2002, 59, 409–418. [DOI] [PubMed] [Google Scholar]
- 66. Jayakumar R., Jayashree S., Annapurna B., Seshadri S., Appl. Biochem. Biotechnol. 2012, 168, 1849–1866. [DOI] [PubMed] [Google Scholar]
- 67. Mills A., Chang Q., McMurray N., Anal. Chem. 1992, 64, 1383–1389. [Google Scholar]
- 68. Mills A., Chang Q., Analyst 1993, 118, 839. [Google Scholar]
- 69. Mills A., Chang Q., Sens. Actuators B 1994, 21, 83–89. [Google Scholar]
- 70. Mills A., Lepre A., Wild L., Sens. Actuators B 1997, 39, 419–425. [Google Scholar]
- 71. Mills A., Chang Q., Anal. Chim. Acta 1994, 285, 113–123. [Google Scholar]
- 72. Mills A., Monaf L., Analyst 1996, 121, 535. [Google Scholar]
- 73. von Bültzingslöwen C., McEvoy A. K., McDonagh C., MacCraith B. D., Klimant I., Krause C., Wolfbeis O. S., Analyst 2002, 127, 1478–1483. [DOI] [PubMed] [Google Scholar]
- 74. Aresta M., Carbon Dioxide Recovery and Utilization, Springer, Amsterdam, 2013. [Google Scholar]
- 75. Cook J. M., Karelitz R. L., Dalsis D. E., J. Chromatogr. Sci. 1985, 23, 57–63. [Google Scholar]
- 76. Baur J. E., Baur M. B., Franz D. A., J. Chem. Educ. 2006, 83, 577. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary