

Abstract
The use of readily available chiral trans-cyclohexanediamine-benzimidazole derivatives as bifunctional organocatalysts in the asymmetric electrophilic amination of unprotected 3-substituted oxindoles is presented. Different organocatalysts were evaluated; the most successful one contained a dimethylamino moiety (5). With this catalyst under optimized conditions, different oxindoles containing a wide variety of substituents at the 3-position were aminated in good yields and with good to excellent enantioselectivities using di-tert-butylazodicarboxylate as the aminating agent. The procedure proved to be also efficient for the amination of 3-substituted benzofuranones, although with moderate results. A bifunctional role of the catalyst, acting as Brønsted base and hydrogen bond donor, is proposed according to the experimental results observed.
Keywords: organocatalysis, electrophilic amination, benzimidazoles, asymmetric catalysis, oxindoles
1. Introduction
Substituted oxindoles containing a chiral center at the C3 position are attractive targets in organic synthesis since they are recognized as drug candidates and also for their use as intermediates in alkaloid synthesis. More particularly, the development of efficient methods to prepare 3-aminooxindoles is of current interest owing to the wide presence of these frameworks in bioactive natural products and pharmaceutically active compounds [1,2,3,4,5]. Among the different strategies developed for the construction of these oxindoles, asymmetric organocatalyzed electrophilic amination has emerged as a powerful tool because it is considered a simple and straightforward methodology [6,7,8,9]. Additional to this, the well-known advantages in the use of organocatalysts must be also taken into account: they are readily available or simple-to-prepare catalysts, easy to handle, and there is no risk of metal contamination in the final product [10,11]. For these reasons, it is not surprising that in the last decade, different studies have successfully described the organocatalytic asymmetric electrophilic 3-amination of oxindoles, giving rise to the desired compounds in high yields and enantioselectivities and with a broad substrate scope [12,13,14]. However, most of the works reported so far require the use of N-protected oxindoles, which implies a tedious protection-deprotection sequence.
In the last years, our research group has been involved in the synthesis and application of benzimidazole derivatives as bifunctional organocatalysts which activate different active methylene compounds through hydrogen bond interactions in different asymmetric organocatalyzed transformations [15,16,17,18,19]. More concretely, a few years ago we reported the successful employment of such organocatalysts in the enantioselective electrophilic amination of 1,3-dicarbonyl compounds [20,21]. Continuing with this research line, we disclose the application of 2-aminobenzimidazoles as catalysts in the asymmetric electrophilic amination of 3-substituted unprotected oxindoles (Figure 1).
Figure 1.

Benzimidazole-based organocatalyst in the asymmetric electrophilic amination of active methylene compounds.
2. Results and Discussion
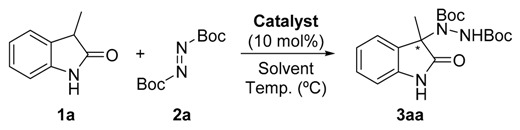
Firstly, the search for the optimal conditions was performed by choosing the model reaction between commercial available unprotected 3-methyl-2-oxindole (1a) and di-t-butylazodicarboxylate (2a), using 10 mol % of catalyst loading. Different parameters such as benzimidazole derivative (Figure 2), solvent, and temperature were examined (Table 1). The model reaction was not serendipitously selected because, to the best of our knowledge, only a few articles describe the successful enantioselective amination of such an unprotected oxindole [14,22,23,24]. As observed in the table, among the different benzimidazole derivatives tested (4–9) at room temperature and using toluene as solvent (Table 1, entries 1–6), catalyst 5 bearing a dimethylamino moiety—which has previously shown its efficiency for the amination of 1,3-dicarbonyl compounds [20]—provided the best results in terms of enantioselectivity (Table 1, entry 2). With the best catalyst at hand, other solvents were evaluated (Table 1, entries 7–11) without observing an improvement in the ee of the amination product 3aa. However, in the case of using hexane as solvent, similar results to those obtained in toluene were achieved (Table 1, entry 10). Therefore, both solvents were tested in a reaction performed at 0 °C for 72 h (Table 1, entries 12 and 13). Whereas the enantioselectivity in toluene increased up to 81% without detriment to conversion, a significant drop in the optical purity of 3aa was observed in hexane, probably due to the lower solubility of the reagents and catalyst in this solvent at lower temperatures. Attempts to further increase the ee of 3aa by lowering the reaction temperature were unsuccessful, probably for the same solubility reasons (Table 1, entries 14 and 15). It is also important to remark that the concentration barely affects the results observed for the best conditions. However, reducing the catalyst amount (5 mol %) resulted in a dramatic drop of enantioselectivity.
Figure 2.

Benzimidazole-derived organocatalysts tested.
Table 1.
Optimal condition screening a.
 | |||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Temp. (°C) | Conv. (%) b | ee (%) c |
| 1 | 4 | PhMe | 25 | 90 | 13 |
| 2 | 5 | PhMe | 25 | 80 | 73 |
| 3 | 6 | PhMe | 25 | 80 | 57 |
| 4 | 7 | PhMe | 25 | 92 | 5 |
| 5 | 8 | PhMe | 25 | 40 | rac. |
| 6 | 9 | PhMe | 25 | 87 | 3 |
| 7 | 5 | CH2Cl2 | 25 | 80 | 56 |
| 8 | 5 | Et2O | 25 | 85 | 66 |
| 9 | 5 | THF | 25 | 75 | 50 |
| 10 | 5 | C6H14 | 25 | 81 | 73 |
| 11 | 5 | MeOH | 25 | 79 | 5 |
| 12 | 5 | PhMe | 0 d | 89 | 81 |
| 13 | 5 | C6H14 | 0 d | 82 | 51 |
| 14 | 5 | PhMe | −20 d | 56 | 71 |
| 15 | 5 | PhMe | −78 e | 59 | 70 |
a Unless otherwise stated, general conditions were 1a (0.15 mmol), 2a (1.1 equiv.), organocatalyst (10 mol %) in toluene (1 mL) at the corresponding temperature for 24 h. b Conversions determined by 1H NMR from the crude reaction mixture. c Determined by chiral HPLC analysis (Daicel Chiralcel OD-H, see Materials and Methods section for details). d The reaction was run for 72 h. e The reaction was run for 96 h.
Next, we decided to evaluate the influence of the azodicarboxylate reagent. Thus, diethyl (2b) and diisopropyl (2c) derivatives were allowed to react with oxindole 1a under the best conditions (Scheme 1). As depicted, none of them improved upon the results obtained with 2a.
Scheme 1.

Influence of azodicarboxylate reagent.
Finally, the last parameter evaluated was the presence of a protecting group on the oxindole. We selected the Boc group due to its ease of removal. Surprisingly, the reaction produced the expected amination product in good yields but with low enantioselectivity (Scheme 2).
Scheme 2.

Influence of the N-Boc protecting group.
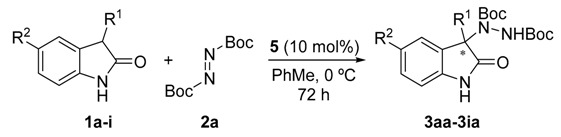
Once all the parameters which can have an effect in the reaction outcome were studied, the scope of the asymmetric benzimidazole 5 organocatalyzed electrophilic amination was evaluated. As previously mentioned, good yields and enantioselectivities were achieved with 3-methyloxindole (1a) (Table 2, entry 1). Similar results were obtained with the 5-bromo analogue 1b (Table 2, entry 2). Surprisingly, when 3-phenyloxindole (1c) was employed, a slight drop in both yield and enantioselectivity was observed (Table 2, entry 3). Better results, especially in term of ee values, were obtained with 3-allyloxindole (3d) and ester derivative 3e, reaching 84% and 85% ee, respectively (Table 2, entries 4 and 5). Next, a series of 3-benzyl-substituted oxindoles were evaluated. Thus, 3-benzyloxindole (3e) was firstly tested giving rise to good yield and enantioselectivity in compound 3fa (Table 2, entry 6). Surprisingly, excellent enantioselectivity was achieved when a 3-PMB-oxindole (3g) was employed (Table 2, entry 7). Encouraged by this result, a couple of para-substituted benzyl oxindoles were synthesized and tested (Table 2, entries 8 and 9). As can be observed in the table, the introduction of a NO2 group produced the amination product 3ha with good yields and enantioselectivity. In contrast, we were pleased to discover that the presence of CF3 gave rise to the product 3ia with perfect enantioselectivity.
Table 2.
Reaction scope a.
 | ||||
|---|---|---|---|---|
| Entry | 1 | 3 | Yield (%) b | ee (%) c |
| 1 | 1a (R1 = Me, R2 = H) | 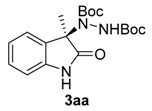 |
78 | 81 |
| 2 | 1b (R1 = Me, R2 = Br) | 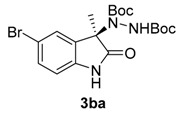 |
84 | 80 |
| 3 | 1c (R1 = Ph, R2 = H) | 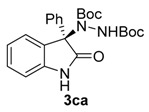 |
61 | 70 |
| 4 | 1d (R1 = Allyl, R2 = H) | 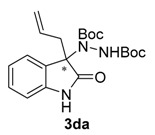 |
87 | 84 |
| 5 | 1e (R1 = CH2CO2Et, R2 = H) | 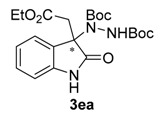 |
71 | 85 |
| 6 | 1f (R1 = Bn, R2 = H) | 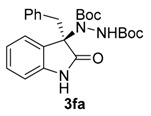 |
83 | 70 |
| 7 | 1g (R1 = p-MeOBn, R2 = H) | 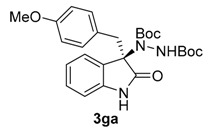 |
75 | 93 |
| 8 | 1h (R1 = p-NO2Bn, R2 = H) | 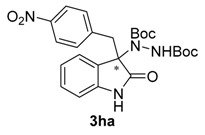 |
79 | 80 |
| 9 | 1i (R1 = p-CF3Bn, R2 = H) | 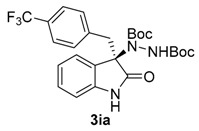 |
76 | 99 |
a Unless otherwise stated, general conditions were 1 (0.15 mmol), 2a (1.1 equiv.), 5 (10 mol %) in the corresponding toluene (1 mL) at 0 ºC for 72 h. b Isolated yield after column chromatography. c Determined by chiral HPLC analysis (see Materials and Methods section for details).
After having evaluated the scope of the methodology with different oxindoles and for the sake of comparison, we decided to test 5-methyl-3-phenylbenzofuranone under the optimal reaction conditions (Scheme 3). As depicted, lower enantioselectivity was achieved with this substrate when compared with the oxindole analogue (see Table 2, entry 3).
Scheme 3.

Enantioselective organocatalyzed electrophilic amination of benzofuranone 10.
Concerning a possible reaction mechanism, there is some experimental evidence that needs to be considered before hypothesizing a possible catalytic cycle. Firstly, when comparing some optical rotation values obtained from the amination products 3 with those described previously in the literature, we could conclude that catalyst 5 possessing an (R,R)-absolute configuration produced the corresponding (R)-products. Secondly, from the results obtained in the catalyst screening, it can be deduced that the dimethylamino moiety is essential for achieving good enantioselectivities, since all the catalysts lacking such a group produced low ee values. Finally, the fact that N-Boc-protected oxindole gave rise to the amination product in high yields but as racemic mixtures (Scheme 2) led us to think there might be another type of influence besides the possible steric hindrance. With all these data in hand, we hypothesize the following catalytic cycle (Figure 3), assuming the bifunctional role of catalyst 5: Organocatalyst 5, acting firstly as a Brønsted base, through the guanidine or dimethylamino moiety, could deprotonate the 3-substituted oxindole. Once the enolate is formed, the guanidine moiety can stablish hydrogen bond interactions not only with the enolate but also with the N–H bond from the amide group of oxindole, as depicted in intermediate A. Next, the protonated dimethylamino group can activate the diazocarboxylate by another hydrogen bond interaction (intermediate B), forming a close transition state in which the enolate reacts through the Re-face, affording the corresponding (R)-amination product and releasing the catalyst.
Figure 3.

Proposed reaction mechanism.
3. Materials and Methods
All reagents were purchased from commercial sources and used without further purification. Substrates which were not commercially available were synthesized according to known literature procedures (see Supporting Information for details). NMR spectra were performed on a Bruker AC-300 or Bruker Avance-400 (Bruker Corporation, Karlsruhe, Germany) using CDCl3 as solvent and TMS as internal standard unless otherwise stated. Optical rotations were measured on a Jasco P-1030 Polarimeter with a 5 cm cell (c given in g/100 mL). Enantioselectivities were determined by HPLC analysis (Agilent 1100 Series HPLC) equipped with a G1315B diode array detector and a Quat Pump G1311A (Agilent Technologies, Palo Alto, CA, United States) equipped with the corresponding Daicel chiral column; the retention time of the major enantiomer is highlighted in bold. HRMS were measured using HPLC–HRMS (ESI) equipment (Agilent 1100 Series LC/MSD Trap SL, Agilent Technologies). Analytical TLC was performed on Merck silica gel plates and the spots were visualized with UV light at 254 nm (Merck Millipore, Billerica, MA, USA). Flash chromatography employed Merck silica gel 60 (0.040–0.063 mm) (Merck Millipore, Billerica, MA, USA).
General Procedure for the Asymmetric Amination of 3-Substituted Oxindoles
In an open-air tube at room temperature (25 °C), the corresponding 3-substituted oxindole (0.10 mmol) was added to a solution of organocatalyst 5 (10 mol %) in toluene (1 mL). After 5 min, the mixture was introduced into a cooling bath at 0 °C and after 2 min, di-tert-butylazodicarboxilate (0.11 mmol, 1.1 eq.) was added in one portion. The reaction was then allowed to react for 3 days. After this time, water (5 mL) and ethyl acetate (5 mL) were added, and the aqueous layer was then re-extracted twice (2 × 10 mL). The combined organic phases were dried (MgSO4) and evaporated under reduced pressure. Finally, the reaction crude was purified by column chromatography on silica gel or TLC preparative using hexane/ethylacetate mixtures as eluent.
Di-tert-butyl (R)-1-(3-methyl-2-oxoindolin-3-yl)-hydrazine-1,2-dicarboxylate (3aa) [23]. Colorless oil; [α]D28 = −7.66 (c = 1.0, CHCl3, 81% ee). 1H-NMR (300 MHz, CDCl3) δH = 8.31 (s, 1H), 7.79 (d, J = 7.3 Hz, 1H), 7.20 (dd, J = 10.9, 4.3 Hz, 1H), 7.06 (t, J = 7.5 Hz, 1H), 6.82 (s, 1H), 1.53 (d, J = 5.7 Hz, 12H), 1.20 (s, 9H); 13C-NMR (75 MHz, CDCl3) δC = 180.35, 156.21, 153.14, 139.34, 133.55, 128.16, 124.05, 123.02, 109.89, 82.54, 81.24, 66.11, 28.25, 27.78, 23.55; chiral HPLC analysis: Chiralcel OD-H column, Hexane/iPrOH 95 : 05, flow rate = 1 mL/min, λ = 240 nm, retention times: 7.2, 8.3 min.
Di-tert-butyl 1-(1-(tert-butoxycarbonyl)-3-methyl-2-oxoindolin-3-yl)-hydrazine-1,2-dicarboxylate (3aa’) [25]. Colorless oil; 1H NMR (300 MHz, CDCl3) ) δH = 7.90 (d, J = 7.1 Hz, 1H), 7.80 (d, J = 8.0 Hz, 1H), 7.32 (d, J = 7.8 Hz, 1H), 7.20 (t, J = 7.4 Hz, 1H), 6.65 (s, 1H), 1.65 (s, 9H), 1.53 (d, J = 3.4 Hz, 12H), 1.14 (s, 9H); 13C-NMR (101 MHz, CDCl3) δC = 176.27, 156.24, 152.87, 149.24, 137.97, 128.44, 124.98, 123.69, 114.65, 84.33, 81.52, 65.81, 28.22, 28.11, 27.66, 24.38; chiral HPLC analysis: Chiralcel IA column, Hexane/iPrOH 90 : 10, flow rate = 1 mL/min, λ = 240 nm, retention times: 12.7, 18.1 min.
Diethyl 1-(3-methyl-2-oxoindolin-3-yl)-hydrazine-1,2-dicarboxylate (3ab) [23]. Colorless oil; 1H-NMR (300 MHz, CDCl3) δH = 8.45 (s, 1H), 7.77 (d, J = 7.4 Hz, 1H), 7.35 (s, 1H), 7.20 (t, J = 7.3 Hz, 1H), 7.05 (t, J = 7.5 Hz, 1H), 6.86 (d, J = 7.6 Hz, 1H), 4.30 (dd, J = 13.5, 6.9 Hz, 2H), 4.00 (d, J = 6.8 Hz, 2H), 1.55 (s, 3H), 1.34 (t, J = 7.1 Hz, 3H), 1.04 (s, 3H); 13C-NMR (101 MHz, CDCl3) δC = 179.59, 157.09, 154.20, 139.39, 132.56, 128.51, 124.18, 123.14, 109.84, 66.27, 62.83, 62.28, 22.95, 14.52, 13.98; chiral HPLC analysis: Chiralcel AD-H column, Hexane/iPrOH 90 : 10, flow rate = 1 mL/min, λ = 240 nm, retention times: 19.3, 33.2 min.
Diisopropyl (R)-1-(3-methyl-2-oxoindolin-3-yl)-hydrazine-1,2-dicarboxylate (3ac) [23]. Colorless oil; [α]D28 = −3.64 (c = 1.0, CHCl3, 60% ee); 1H-NMR (300 MHz, CDCl3) δH = 8.44 (s, 1H), 7.78 (d, J = 7.4 Hz, 1H), 7.20 (t, J = 7.3 Hz, 1H), 7.06 (dd, J = 13.4, 5.8 Hz, 2H), 6.86 (d, J = 7.6 Hz, 1H), 5.05 (dt, J = 12.4, 6.2 Hz, 1H), 4.73 (dt, J = 12.1, 6.0 Hz, 1H), 1.54 (s, 3H), 1.33 (dd, J = 6.2, 1.6 Hz, 6H), 1.08 (d, J = 6.1 Hz, 3H), 0.89 (bs, 3H); 13C-NMR (101 MHz, CDCl3) δC = 179.69, 156.88, 153.76, 139.38, 132.93, 128.42, 124.19, 123.09, 109.73, 70.81, 70.08, 66.12, 23.09, 22.04, 21.91, 21.65, 21.48; chiral HPLC analysis: Chiralcel AD-H column, Hexane/iPrOH 90 : 10, flow rate = 1 mL/min, λ = 240 nm, retention times: 14.2, 18.7 min.
Di-tert-butyl (R)-1-(5-bromo-3-methyl-2-oxoindolin-3-yl)-hydrazine-1,2-dicarboxylate (3ba) [24]. Yellow solid; [α]D28 = +18.64 (c = 1.0, CHCl3, 80% ee); 1H-NMR (300 MHz, CDCl3) δH = 8.80 (s, 1H), 7.93 (s, 1H), 7.32 (dd, J = 8.1, 1.8 Hz, 2H), 6.96–6.68 (m, 2H), 1.54 (m, 12H), 1.25 (s, 9H); 13C-NMR (101 MHz, CDCl3) δC = 178.15, 155.91, 152.78, 138.07, 131.16, 127.33, 124.07, 111.19, 82.77, 81.46, 66.89, 28.24, 27.87, 14.12; chiral HPLC analysis: Chiralcel OD-H column, Hexane/iPrOH 95 : 05, flow rate = 0.8 mL/min, λ = 240 nm, retention times: 10.6, 12.4 min.
Di-tert-butyl (R)-1-(2-oxo-3-phenylindolin-3-yl)-hydrazine-1,2-dicarboxylate (3ca) [24]. White solid; [α]D28 = −60.38 (c = 1.0, CHCl3, 70% ee); 1H NMR (300 MHz, CDCl3): δH = 8.25 (s, 1H), 8.09 (d, J = 7.3 Hz, 1H), 7.65–7.57 (m, 2H), 7.30–7.27 (m, 3H), 7.25 (s, 1H), 7.15 (t, J = 7.4 Hz, 1H), 6.82 (d, J = 7.6 Hz, 1H), 6.26 (bs, 1H), 1.32 (s, 9H), 1.21 (s, 9H); 13C-NMR (101 MHz, CDCl3) δC = 177.89, 154.81, 153.48, 140.03, 133.71, 130.59, 129.43, 128.65, 128.59, 128.15, 126.94, 122.64, 109.90, 82.76, 80.83, 72.64, 28.04, 27.81; chiral HPLC analysis: Chiralcel OD-H column, Hexane/iPrOH 90 : 10, flow rate = 1 mL/min, λ = 240 nm, retention times: 5.9, 9.0 min.
Di-tert-butyl 1-(3-allyl-2-oxoindolin-3-yl)-hydrazine-1,2-dicarboxylate (3da). Yellow oil; [α]D28 = −11.47 (c = 1.0, CHCl3, 84% ee); 1H-NMR (300 MHz, CDCl3) δH = 7.93 (s, 1H), 7.76 (d, J = 7.4 Hz, 1H), 7.20 (t, J = 7.6 Hz, 1H), 7.07 (t, J = 7.5 Hz, 1H), 6.71 (m, 2H), 5.42–5.24 (m, 1H), 4.97 (dd, J = 19.9, 13.6 Hz, 2H), 2.70 (d, J = 7.5 Hz, 2H), 1.54 (s, 9H), 1.22 (s, 9H); 13C-NMR (101 MHz, CDCl3) δC = 177.84, 156.13, 153.12, 139.88, 130.16, 128.36, 124.69, 123.00, 120.13, 109.35, 82.67, 81.39, 68.86 40.71, 28.26, 27.82; HRMS (ESI-MS) calculated for C32H44N4NaO4: 571.3260, found: 571.3310; chiral HPLC analysis: Chiralcel AD-H column, Hexane/iPrOH 90 : 10, flow rate = 1 mL/min, λ = 240 nm, retention times: 10.1, 16.1 min.
Di-tert-butyl-1-[3-(2-ethoxy-2-oxoethyl)-2-oxoindolin-3-yl]-hydrazine-1,2-dicarboxylate (3ea). Yellow oil; [α]D28 = −1.53 (c = 1.0, CHCl3, 85% ee); 1H-NMR (300 MHz, CDCl3) δH = 8.06 (s, 1H), 7.74 (d, J = 7.4 Hz, 1H), 7.21 (t, J = 7.3 Hz, 1H), 7.05 (t, J = 7.6 Hz, 1H), 6.86–6.66 (m, 2H), 3.95 (q, J = 7.1 Hz, 2H), 3.08 (d, J = 14.2 Hz, 1H), 2.90 (d, J = 14.1 Hz, 1H), 1.54 (s, 9H), 1.24 (s, 9H), 1.04 (t, J = 7.1 Hz, 3H); 13C-NMR (101 MHz, CDCl3) δC = 168.30, 155.89, 152.80, 140.29, 129.70, 128.96, 125.33, 122.91, 109.67, 82.91, 81.56, 66.66, 60.95, 41.32, 28.27, 27.85, 13.83; HRMS (ESI-MS) calculated for C34H48N4NaO8: 663.3370, found: 663.3375; chiral HPLC analysis: Chiralcel AD-H column, Hexane/iPrOH 70 : 30, flow rate = 1 mL/min, λ = 240 nm, retention times: 8.4, 11.9 min.
Di-tert-butyl (R)-1-(3-benzyl-2-oxoindolin-3-yl)-hydrazine-1,2-dicarboxylate (3fa) [24]. White solid; [α]D28 = −7.83 (c = 1.0, CHCl3, 70% ee); 1H-NMR (300 MHz, CDCl3) δH = 7.95 (dd, J = 5.3, 3.2 Hz, 1H), 7.26 (s, 1H), 7.11 (dd, J = 5.6, 3.2 Hz, 2H), 7.06–6.88 (m, 4H), 6.71 (d, J = 7.0 Hz, 2H), 6.44 (dd, J = 5.5, 3.1 Hz, 1H), 3.32 (d, J = 12.0 Hz, 1H), 3.15 (d, J = 12.1 Hz, 1H), 1.58 (s, 9H), 1.23 (d, J = 2.6 Hz, 9H); 13C-NMR (101 MHz, CDCl3) δC = 177.66, 156.33, 152.80, 139.96, 132.90, 130.45, 128.41, 127.39, 126.75, 124.88, 122.81, 109.06, 82.69, 81.47, 69.96, 42.19, 28.29, 27.82; chiral HPLC analysis: Chiralcel AD-H column, Hexane/iPrOH 90 : 10, flow rate = 1 mL/min, λ = 240 nm, retention times: 8.9, 10.6 min.
Di-tert-butyl (R)-1-(3-(4-methoxybenzyl)-2-oxoindolin-3-yl)-hydrazine-1,2-dicarboxylate (3ga) [24]. Yellow oil; [α]D28 = −102.12 (c = 1.0, CHCl3, 93% ee); 1H-NMR (300 MHz, CDCl3) δH = 7.97–7.88 (m, 1H), 7.11 (d, J = 4.1 Hz, 3H), 6.80 (s, 1H), 6.63 (d, J = 8.0 Hz, 2H), 6.53 (d, J = 8.1 Hz, 2H), 6.46 (s, 1H), 3.69 (s, 3H), 3.29 (d, J = 12.2 Hz, 1H), 3.10 (d, J = 12.1 Hz, 1H), 1.57 (s, 9H), 1.22 (s, 9H); 13C-NMR (101 MHz, CDCl3) δC = 177.67, 158.40, 156.29, 153.12, 131.42, 128.37, 124.81, 122.79, 112.84, 109.11, 82.66, 81.47, 69.93, 55.02, 41.36, 28.28, 27.83; chiral HPLC analysis: Chiralcel AD-H column, Hexane/iPrOH 90 : 10, flow rate = 1 mL/min, λ = 240 nm, retention times: 13.1, 15.9 min.
Di-tert-butyl-1-[3-(4-nitrobenzyl)-2-oxoindolin-3-yl]-hydrazine-1,2-dicarboxylate (3ha). Red oil; [α]D28 = −32.43 (c = 1.0, CHCl3, 80% ee); 1H-NMR (300 MHz, CDCl3) δH = 7.97 (dd, J = 5.2, 3.3 Hz, 1H), 7.84 (d, J = 8.8 Hz, 2H), 7.14 (dd, J = 5.6, 3.2 Hz, 2H), 6.94 (s, 1H), 6.87 (d, J = 8.8 Hz, 2H), 6.46 (dd, J = 5.5, 3.1 Hz, 1H), 3.40 (d, J = 11.9 Hz, 1H), 3.22 (d, J = 11.9 Hz, 1H), 1.57 (s, 9H), 1.22 (s, 9H); 13C-NMR (101 MHz, CDCl3) δC = 177.18, 156.42, 152.93, 147.01, 140.96, 139.71, 131.34, 129.02, 124.92, 123.21, 122.52, 109.46, 83.06, 81.81, 69.57, 41.88, 28.28, 27.81; HRMS (ESI-MS) calculated for C25H30N4O6: 482.2165, found: 480.2332; chiral HPLC analysis: Chiralcel AD-H column, Hexane/iPrOH 90 : 10, flow rate = 1 mL/min, λ = 240 nm, retention times: 18.9, 28.6 min.
Di-tert-butyl (R)-1-[02-oxo-3-(4-(trifluoromethyl)benzyl)-indolin-3-yl]hydrazine-1,2-dicarboxylate (3ia) [24]. Yellow oil; [α]D28 = −98.62 (c = 1.0, CHCl3, 99% ee); 1H-NMR (300 MHz, CDCl3) δH = 8.01–7.86 (bs, 1H), 7.40 (s, 1H), 7.22 (d, J = 8.1 Hz, 2H), 7.15–7.09 (m, 2H), 6.98 (s, 1H), 6.80 (d, J = 8.1 Hz, 2H), 6.43 (bs, 1H), 3.34 (d, J = 12.0 Hz, 1H), 3.18 (d, J = 12.0 Hz, 1H), 1.57 (s, 9H), 1.22 (s, 9H); 13C-NMR (101 MHz, CDCl3) δC = 177.32, 156.36, 152.98, 139.85, 137.24, 130.76, 130.24, 128.75, 124.84, 124.22, 122.98, 109.33, 82.88, 81.67, 69.67, 41.87, 28.27, 27.80; chiral HPLC analysis: Chiralcel AD-H column, Hexane/iPrOH 95 : 05, flow rate = 1 mL/min, λ = 240 nm, retention times: rt = 16.2, 20.4 min.
Di-tert-butyl (R)-1-(5-methyl-2-oxo-3-phenyl-2,3-dihydrobenzofuran-3-yl)-hydrazine-1,2-dicarboxylate (11a). Colorless oil; [α]D28 = −23.86 (c = 1.0, CHCl3, 55% ee); 1H-NMR (300 MHz, CDCl3) δH = 7.88 (s, 1H), 7.57 (dd, J = 6.7, 2.9 Hz, 2H), 7.36–7.32 (m, 3H), 7.23–7.13 (m, 1H), 6.97 (d, J = 8.2 Hz, 1H), 6.28 (s, 1H), 2.47 (s, 3H), 1.34 (s, 9H), 1.29 (s, 9H); 13C-NMR (101 MHz, CDCl3) δC = 174.61, 154.62, 153.21, 150.91, 133.92, 132.85, 130.84, 130.13, 129.27, 129.02, 128.46, 128.31, 127.55, 110.73, 110.36, 83.85, 81.18, 71.45, 28.01, 27.78, 21.46; HRMS (ESI-MS) calculated for C40H47N2NaO6: 674.3332, found: 674.2301; chiral HPLC analysis: Chiralcel AD-H column, Hexane/iPrOH 70 : 30, flow rate = 1 mL/min, λ = 240 nm, retention times: 7.4, 16.9 min.
4. Conclusions
In summary, in this work we have described the use of readily available chiral trans-cyclohexanediamine-benzimidazole derivative (5) as an organocatalyst for the asymmetric electrophilic amination of 3-substituted oxindoles employing di-t-butylazodicarboxylate as the aminating agent. In general, for this challenging transformation, which avoids the need for protection of the oxindole substrate, the amination products were obtained in good yields and with enantioselectivities varying from good to high when 10 mol % of catalyst was employed. On the contrary, moderate enantioselectivities were achieved with benzofuranone derivatives. Finally, according to some experimental finds and assuming the bifunctional role of the catalyst, a possible catalytic cycle has been postulated. However, a more comprehensive study of the reaction mechanism is necessary.
Acknowledgments
Financial support from the Spanish Ministerio de Economía, Industria y Competitividad (CTQ2015-66624-P) and the University of Alicante (VIGROB-173, VIGROB-285, UAUSTI17-06, UADIF17-27) is acknowledged. M.F. thanks the University of Edinburgh for an ERASMUS fellowship.
Supplementary Materials
The following are available online. General remarks and as well as NMR spectra and HPLC charts of amination compounds are provided.
Author Contributions
A.B. conceived and designed the experiments; L.B., A.B. and M.F. performed the experiments and analyzed the data; A.B., L.B. and M.F. wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds 3aa–3ia are available from the authors.
References
- 1.Barluenga J., Valdés C. Five-membered heterocycles: Indole and related systems. In: Alvarez-Builla J., Vaquero J.J., Barluenga J., editors. Modern Heterocyclic Chemistry. Volume 1. Wiley-VCH Verlag & Co.; Weinheim, Germany: 2011. pp. 495–498. [Google Scholar]
- 2.Dalpozzo R. Catalytic asymmetric synthesis of hetero-substituted oxindoles. Org. Chem. Front. 2017;4:2063–2078. doi: 10.1039/C7QO00446J. [DOI] [Google Scholar]
- 3.Dalpozzo R., Bartoli G., Bencivenni G. Recent advances in organocatalytic methods for the synthesis of disubstituted 2- and 3-indolinones. Chem. Soc. Rev. 2012;41:7247–7290. doi: 10.1039/c2cs35100e. [DOI] [PubMed] [Google Scholar]
- 4.Kaur J., Chimni S.S., Mahajana S., Kumarb A. Stereoselective synthesis of 3-amino-2-oxindoles from isatin imines: New scaffolds for bioactivity evaluation. RSC Adv. 2015;5:52481–52496. doi: 10.1039/C5RA06969F. [DOI] [Google Scholar]
- 5.Mohammadi S., Heiran R., Herrera R.P., Marqués-López E. Isatin as a strategic motif for asymmetric catalysis. ChemCatChem. 2013;5:2131–2148. doi: 10.1002/cctc.201300050. [DOI] [Google Scholar]
- 6.Vilaivan T., Bhanthumnavin W. Organocatalyzed asymmetric α-oxidation, α-aminoxylation and α-amination of carbonyl compounds. Molecules. 2010;15:917–958. doi: 10.3390/molecules15020917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vallribera A., Sebastian R.M., Shafir A. Azodicarboxylates as electrophilic aminating reagents. Curr. Org. Chem. 2011;15:1539–1577. doi: 10.2174/138527211795378083. [DOI] [Google Scholar]
- 8.Russo A., De Fusco C., Lattanzi A. Enantioselective organocatalytic α-heterofunctionalization of active methines. RSC Adv. 2012;2:385–397. doi: 10.1039/C1RA00612F. [DOI] [Google Scholar]
- 9.Govender T., Arvidsson P.I., Maguire G.E.M., Kruger H.G., Naicker T. Enantioselective organocatalyzed Transformations of β-ketoesters. Chem. Rev. 2016;116:9375–9437. doi: 10.1021/acs.chemrev.6b00156. [DOI] [PubMed] [Google Scholar]
- 10.Dalko P.I. Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions, and Applications. Wiley-VCH; Weinheim, Germany: 2013. [Google Scholar]
- 11.Alemán J., Cabrera S. Applications of asymmetric organocatalysis in medicinal chemistry. Chem. Soc. Rev. 2013;42:774–793. doi: 10.1039/C2CS35380F. [DOI] [PubMed] [Google Scholar]
- 12.Chauhan P., Chimni S.S. Organocatalytic asymmetric synthesis of 3-amino-2-oxindole derivatives bearing a tetra-substituted stereocenter. Tetrahedron Asymmetry. 2013;24:343–356. doi: 10.1016/j.tetasy.2013.03.002. [DOI] [Google Scholar]
- 13.Yu J.S., Zhou F., Liu Y.L., Zhou J.A. Journey in the catalytic synthesis of 3-substituted 3-aminooxindoles. Synlett. 2015;26:2491–2504. [Google Scholar]
- 14.Freckleton M., Baeza A., Benavent L., Chinchilla R. Asymmetric organocatalytic electrophilic heterofunctionalization of oxindoles. Asian J. Org. Chem. 2018 doi: 10.1002/ajoc.201800146. [DOI] [Google Scholar]
- 15.Almaşi D., Alonso D.A., Gómez-Bengoa E., Nájera C. Chiral 2-aminobenzimidazoles as recoverable organocatalysts for the addition of 1,3-dicarbonyl compounds to nitroalkenes. J. Org. Chem. 2009;74:6163–6168. doi: 10.1021/jo9010552. [DOI] [PubMed] [Google Scholar]
- 16.Gómez-Torres E., Alonso D.A., Gómez-Bengoa E., Nájera C. Conjugate addition of 1,3-dicarbonyl compounds to maleimides using a chiral C2-symmetric bis(2-aminobenzimidazole) as recyclable organocatalyst. Org. Lett. 2011;13:6106–6109. doi: 10.1021/ol202599h. [DOI] [PubMed] [Google Scholar]
- 17.Gómez-Torres E., Alonso D.A., Gómez-Bengoa E., Nájera C. Enantioselective synthesis of succinimides by michael addition of 1,3-dicarbonyl compounds to maleimides catalyzed by a chiral bis(2-aminobenzimidazole) organocatalyst. Eur. J. Org. Chem. 2013:1434–1440. doi: 10.1002/ejoc.201201046. [DOI] [Google Scholar]
- 18.Trillo P., Baeza A., Nájera C. Bis(2-aminobenzimidazole)-organocatalyzed asymmetric alkylation of activated methylene compounds with benzylic and allylic alcohols. Synthesis. 2014;46:3399–3407. [Google Scholar]
- 19.Serrano-Sánchez D., Baeza A., Alonso D.A. Organocatalytic asymmetric α-chlorination of 1,3-dicarbonyl compounds catalyzed by 2-aminobenzimidazole derivatives. Symmetry. 2016;8:3. doi: 10.3390/sym8010003. [DOI] [Google Scholar]
- 20.Trillo P., Gómez-Martínez M., Alonso D.A., Baeza A. 2-aminobenzimidazole organocatalyzed asymmetric amination of cyclic 1,3-dicarbonyl compounds. Synlett. 2015;26:95–100. [Google Scholar]
- 21.Benavent L., Puccetti F., Baeza A., Gómez-Martínez M. Readily available chiral benzimidazoles-derived guanidines as organocatalysts in the asymmetric α-amination of 1,3-dicarbonyl compounds. Molecules. 2017;22:1333–1344. doi: 10.3390/molecules22081333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qian ZQ., Zhou F., Du T.P., Wang B.L., Ding M., Zhao X.L., Zhou J. Asymmetric construction of quaternary stereocenters by direct organocatalytic amination of 3-substituted oxindoles. Chem. Commun. 2009:6753–6755. doi: 10.1039/b915257a. [DOI] [PubMed] [Google Scholar]
- 23.Cheng L., Liu L., Wang D., Chen Y.J. Highly enantioselective and organocatalytic α-amination of 2-oxindoles. Org. Lett. 2009;11:3874–3877. doi: 10.1021/ol901405r. [DOI] [PubMed] [Google Scholar]
- 24.Zhou F., Ding M., Liu Y.L., Wang C.H., Ji C.B., Zhang Y.Y., Zhou J. Organocatalytic asymmetric α-amination of unprotected 3-aryl and 3-aliphatic substituted oxindoles using di-tert-butyl azodicarboxylate. Adv. Synth. Catal. 2011;353:2945–2952. doi: 10.1002/adsc.201100379. [DOI] [Google Scholar]
- 25.Mouri S., Chen Z., Mitsunuma H., Furutachi M., Matsunaga S., Shibasaki M. Catalytic asymmetric synthesis of 3-aminooxindoles: Enantiofacial selectivity switch in bimetallic vs monometallic schiff base catalysis J. Am. Chem. Soc. 2010;132:1255–1257. doi: 10.1021/ja908906n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.