

Abstract
Leishmania major (L. major) is a protozoan parasite that causes cutaneous leishmaniasis. About 12 million people are currently infected with an annual incidence of 1.3 million cases. The purpose of this study was to synthesize a small library of novel thiophene derivatives, and evaluate its parasitic activity, and potential mechanism of action (MOA). We developed a structure–activity relationship (SAR) study of the thiophene molecule 5A. Overall, eight thiophene derivatives of 5A were synthesized and purified by silica gel column chromatography. Of these eight analogs, the molecule 5D showed the highest in vitro activity against Leishmania major promastigotes (EC50 0.09 ± 0.02 µM), with an inhibition of the proliferation of intracellular amastigotes higher than 75% at only 0.63 µM and an excellent selective index. Moreover, the effect of 5D on L. major promastigotes was associated with generation of reactive oxygen species (ROS), and in silico docking studies suggested that 5D may play a role in inhibiting trypanothione reductase. In summary, the combined SAR study and the in vitro evaluation of 5A derivatives allowed the identification of the novel molecule 5D, which exhibited potent in vitro anti-leishmanial activity resulting in ROS production leading to cell death with no significant cytotoxicity towards mammalian cells.
Keywords: thiophene compounds, Leishmania major, cutaneous leishmaniasis, drug screening, chemotherapy, structure–activity relationship (SAR), in silico docking, reactive oxygen species
1. Introduction
Leishmaniasis is a devastating neglected tropical disease (NTD) [1] caused by the protozoan parasite of the genus Leishmania. The parasite is transmitted from animals to humans through the bite of infected females Lutzomyia or Phlebotomus sand flies [2]. Over 20 species and subspecies of Leishmania infect humans, causing three major clinical forms of the disease: cutaneous (CL), visceral, and mucocutaneous leishmaniasis [3]. The prevalence of CL, the most common form of leishmaniasis, is estimated between 0.7 and 1.3 million new annually cases worldwide [4], and it is commonly caused by Leishmania major (L. major) or L. mexicana. CL presents as singular ulcerative or nodular lesions at the bite site that may resolve into scar tissue, often leading to scarring and social stigma [5]. The disease is present in both the Old World in regions of the Middle East, Africa, Central Western and Easter Europe; and the New World in regions of Central and South America, and more recently in North America [5].
Currently, there is no preventative or therapeutic human vaccine available against any clinical manifestation of the disease, and available treatments such as pentavalent antimonials (Glucantime and Pentostam), liposomal amphotericin B (AmBisome®), and miltefosine (IMPAVIDO®) present several disadvantages [6]. Pentavalent antimonial treatments are the first line of action, however, systemic therapy its required for more than 20 days, with toxic side effects including cardiotoxicity and hepatotoxicity [7,8]. Amphotericin B is highly active, but has extensive toxicity complications (nausea, vomiting, rigors, fever, hypertension or hypotension, and hypoxia) that usually lead to treatment interruption; besides, its administration requires hospitalization and its high cost limits its use in developed countries [5,9]. Miltefosine is the only oral agent against leishmaniasis, however, it presents several limitations such as embryo-fetal toxicity, fetal death, and its long half-life (150 h) may facilitate the emergence of drug resistance [10,11]. Thus, these facts clearly emphasize the urgent priority for the development of novel chemotherapies against leishmaniasis.
Considering the current interest in the search of antileishmanial agents, we previously reported for the first time that arylalkylamine type-compounds exhibit anti-Leishmania activity with no toxicity to mammalian cells [12,13]. Toward our medicinal chemistry effort in developing novel compounds with anti-parasitic activity and drug like properties (e.g., improved solubility, potency, stability and less or low toxicity), we assessed the antileishmanial activity of a series of novel compounds based on the thiophene scaffold with pharmaceutical properties: low toxicity, improved potency and solubility [13]. In this context, in the present study, we evaluated nine synthetic thiophene molecules derivatives against L. major.
We set a goal to design and synthesize a scaffold with antileishmanial activity in a one- or two-step synthesis using simple and efficient chemical transformations with high yield, high atom economy and inexpensive starting material. In this study, we focused on the creation of substituted thiophenes which are considered among the privileged structures in drug discovery [14]. Substituted thiophenes are known with their various biological activities such as anti-microbial, anti-cancer, and anti-inflammatory properties [14]. Therefore, the development of novel thiophene compounds with activity against Leishmania is crucial and urgent, as they may also complement current drugs and overcome drug resistance.
2. Results
2.1. Synthetic Chemistry
Toward synthesizing the substituted thiophene 5A efficiently, we used the well establish three components coupling reaction between a ketone, cyanoacetate and elemental sulfur [15] followed by a simple acylation reaction. The synthesis of 5A is depicted in Figure 1A. After synthesizing 5A, we developed the Structure–Activity Relationship (SAR) of 5A and created eight analogs, as shown in Figure 1B. The purity of each analog was confirmed by 1H-NMR, 13C-NMR and MS, and then novel thiophene-like library compounds (Figure 2) were assessed for their potential in vitro antileishmanial activity.
Figure 1.

(A) Synthesis of thiophene 5A (Ethyl 5,5,7,7-tetramethyl-2-(4-(trifluoromethyl)benzamido)-4,5,6,7-tetrahydrothieno [2,3-c]pyridine-3-carboxylate). (B) Synthetic route for the creation of thiophene compounds.
Figure 2.

Chemical structures of the nine thiophene compounds.
2.2. Efficacy and Cytotoxicity of Parent Compound 5A
First, the efficacy of thiophene derivative 5A was evaluated against transgenic L. major promastigotes expressing firefly luciferase (L. major-luc) as previously described [16,17]. Compound 5A at a range of 0.31–10 µM was incubated with 2 × 106 L. major-luc promastigotes per mL for 72 h at 28 °C, and in vitro parasite viability (% survival) was measured by luciferase activity. Compound 5A displayed an approximate 50% effective concentration (EC50) of 0.34 µM against L. major promastigotes (Figure 3 and Table 1). Furthermore, we investigated the potential cytotoxicity of 5A by alamarBlue™ Cell Viability Assay (Thermo Fisher Scientific, Waltham, MA, USA) [18] and 5A displayed a selective index (S.I.) of 30.58 against BALB/c intraperitoneal mouse macrophages (IPФ), and 51.91 in monkey kidney cells (LLC-MK2) (Table 1).
Figure 3.

Antiparasitic effect of thiophene derivative 5A. Viability of L. major-luc promastigotes incubated with 5A at 0.31 to 10 µM for 72 h. Evaluation of intraperitoneal mouse macrophages (IPΦ) cytotoxicity for 48 h, or monkey kidney cells (LLC-MK2) treated with 5A at concentrations of 0.31 to 40 µM for 72 h. Controls treated with 1% DMSO, or amphotericin B (amp B) at 5 µM.
Table 1.
Antiparasitic activity in L. major-luc promastigotes, and cytotoxicity to intraperitoneal mouse macrophages (IPΦ) or monkey kidney cells (LLC-MK2) of 5A and analogs. EC50 Median effective concentration. ± values are the estimated EC50 interval. CC50 Median cytotoxic concentration. ± values are the estimated CC50 interval S.I. Selective Index (CC50 mammalian cells)/(EC50 in L. major-luc promastigotes).
| Compound | Leishmania major-luc | Mammalian Cells | Mammalian Cells |
|---|---|---|---|
| Promastigotes | IPΦ | LLC-MK2 | |
| EC50 (μM) | CC50 (μM) [S.I.] | CC50 (μM) [S.I.] | |
| 5A | ~0.3410 | ~ 10.40 [30.50] | 17.69 ± 1.12 [52.85] |
| 5B | 5.98 ± 1.72 | N/A | N/A |
| 5C | 4.73 ± 0.69 | N/A | N/A |
| 5D | 0.09 ± 0.02 | 27.89 ± 3.19 [310] | >80 |
| 5E | 0.78 ± 0.11 | 16.59 ± 1.52 [21.27] | 80 ± 4.45 [102.56] |
| 5F | >12.50 | N/A | N/A |
| 5G | >12.50 | N/A | N/A |
| 5H | 3.05 ± 0.47 | N/A | N/A |
| 5I | 5.5 ± 1.80 | N/A | N/A |
2.3. In Vitro Anti-Leishmanial Activity of Thiophene Derivatives and Their Cytotoxicity
Consequently, to lower the toxicity and increase the parasitic activity, eight new thiophene molecules (Figure 2) were evaluated. First, the thiophene compounds were tested in the presence of increasing drug concentrations (1.56–12.5 µM), followed by incubation with L. major-luc promastigotes (2 × 106/mL) for 72 h at 28 °C. The experiment was performed using the same conditions as described for the parent drug 5A. As summarized in Table 1, all eight thiophene molecules showed promising antileishmanial activity against L. major promastigotes with an EC50 ranging from 0.09 to 6.25 µM (Figure 4A). However, the best thiophene compounds were 5D (EC50 0.09 ± 0.02 µM) and 5E (EC50 0.78 ± 0.11 µM) (Figure 4B and Table 1).
Figure 4.

(A) Antiparasitic effect of the eight thiophene derivatives. Viability of L. major-luc promastigotes treated with the eight thiophene derivatives at a concentration of 1.56 to 12.5 µM for 72 h. (B) Evaluation of L. major-luc promastigotes treated with 5D or 5E thiophene derivatives at lower concentrations (0.005 to 1.56 µM) for 72 h. Controls treated with 1% DMSO, or amphotericin B (amp B) at 5 µM. (C) Cytotoxicity evaluation of intraperitoneal mouse macrophages (IPΦ) treated with 5D or 5E thiophene derivatives at concentrations from 0.63 to 20 µM for 48 h. (D) Cytotoxicity evaluation of monkey kidney cells (LLC-MK2) treated with 5D or 5E thiophene derivatives at a concentrations from 0.63 to 80 µM for 72 h. Controls treated with 1% DMSO, amp B at 5 µM, or 5% of hydrogen peroxide (H2O2). Cells were stained with Hoechst 33342 (healthy cell) and Propidium Iodide (compromised cell).
Consequently, cytotoxicity assays were performed by incubating LLC-MK2 or IPФ with compounds. First, 5D or 5E were incubated with 1 × 105 LLC-MK2/mL or 1 × 105 IPФ/mL for 72 and 48 h, respectively. Interestingly, compound 5D did not display perceptible toxicity against LLC-MK2 at concentrations up to 80 µM (Figure 4C,D). In the case of IPФ, 5D exhibited a CC50 value of 27.89 ± 3.19 µM and an excellent S.I. of 310. As 5E compound CC50 values of 80 ± 4.45 µM in LLC-MK2 cells and 16.59 ± 1.52 µM in IPФ, with S.I. values of 102.56 and 21.27, respectively. More importantly, we determined that both 5D and 5E displayed lower cytotoxicity to mammalian cells and higher parasitic activity than parent compound 5A (Table 1).
2.4. In Vitro Efficacy of Thiophene 5D Against Intracellular Amastigotes
Additionally, efficacy of thiophene compounds 5D and 5E was tested against the infectious intracellular amastigote form of L. major, by High-Content Imaging Assay (HCIA) on infected intraperitoneal mouse macrophages. As observed in Figure 5A, in comparison with untreated control and 1% DMSO, 5D and 5E inhibited the proliferation of the intracellular amastigotes by more than 75% and 50%, respectively, at a 0.625 µM concentration. Furthermore, as observed in Figure 5B, a reduced number of infected cells were observed after 5D or 5E treatment (2.5 µM) when compared to control treated with 1% DMSO.
Figure 5.

(A) High content imaging assay (HCIA) analysis of intraperitoneal mouse macrophages (IPΦ) infected with L. major-luc amastigotes, followed by treatment with 5D or 5E from 0.63 to 10 µM for 48 h. Controls included untreated, 1% DMSO, amphotericin B (amp B) at 5 µM, or parent drug 5A. Data are represented as the percentage (%) of infected IPΦ with three or more amastigotes per cell. Note: Data for 5A at concentration 5 and 10 µM were not generated because 5A was cytotoxic for IPΦ at such concentration. (B) Representative monochromatic images of infected IPΦ with L. major after 48 h treatment with 5D or 5E at 2.5 µM, amp B (5 µM), or 1% DMSO.
2.5. Molecule 5D Induces ROS in L. major
Based on our previous study [12], it was hypothesized that 5D may induce parasite death through the production of ROS. Thus, 2 × 106 L. major promastigotes per mL were incubated with 5D (EC50 0.09 ± 0.02 µM). After 24 h, ROS levels were measured by the addition of 10 µM of the cell-permeable dye H2DCFDA (Thermo Fisher Scientific, Waltham, MA, USA), and fluorescence was monitor for an additional 7 h using a fluorometer. As expected, ROS levels in 5D treated parasites were 14.5-fold higher compared to vehicle control 1% DMSO (Figure 6).
Figure 6.

Generation of reactive oxygen species in L. major treated for 31 h with 5D at 0.90 µM (EC50). Values shown are the mean and standard error of five different replicates minus basal fluorescence. Control treated with 1% DMSO or hydrogen peroxide (H2O2) at 100 µM.
2.6. Docking of 5D on TryR from Leishmania
Next, to determine the possible molecular mechanism responsible for the antileishmanial activity of 5D, docking studies on TryR from L. infantum (PDB id: 2JK6) were performed. Using Glide Standard Precision [19] and Extra Precision (XP), we performed Rigid Receptor Docking analysis of control (Quinacrine Mustard) and 5D. The 3D ligand structures were docked against the best potential binding site of 2JK6. Glide SP and XP only accounts for the ligand being dynamic however the protein remains rigid. Docking box coordinates and dimensions remained all at default (20 × 20 × 20 Å). Glide XP gives an output of a docking score, which was analyzed by the lowest number, or whichever is more negative to be the highest scoring ligand. The docking results, summarized in Table 2, showed the control (Quinacrine Mustard) with higher binding affinity than 5D in both SP and XP. However, the XP docking score did not differ by much, indicating more rigorous docking analysis is needed. Thus, both ligands were taken to Schrodinger’s Flexible receptor docking.
Table 2.
In silico study of 5D and control Quinacrine Mustard.
| Receptor | Ligand | Structure | Glide SP (XP) | IFD XP (IFD Score) |
|---|---|---|---|---|
| TryR (2JK6) | 5D |
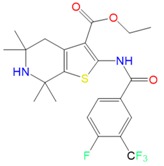
|
−4.6 (−5.5) | −10.0 |
| Quinacrine Mustard (control) |
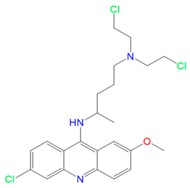
|
−6.9 (−6.7) | −9.2 |
Schrodinger IFD protocol for all IFD jobs was used [20]. The IFD program makes use of both Glide (for docking) and Prime (for protein structure modeling). The combination of the two software packages allows a more accurate ligand binding calculation. We performed the re-docking with Glide XP for the refined docking results [21]. The IFD data presented in Table 2 show that our lead molecule 5D had a better binding affinity. Docking scores from Rigid Receptor Docking and Flexible Receptor
Docking differed significantly. This is accounted for the protein dynamic movement during drug binding in IFD. Furthermore, Figure 7A,B presents the IFD binding pocket of the protein–ligand complex. Figure 7A shows our lead molecule 5D which displays hydrogen bond interactions with SER 1632, ARG 287, VAL 55, and also with CYS 57. Compound 5D also exhibits π-cation interaction with residue ARG 287. Quinacrine Mustard interacted with a new set of residues and showed only two hydrogen bonds between MET 333 and ALA 365 (Figure 7B). The control also formed salt bridges with ASP 327 as well as GLU 202. π–π and π–cation interaction was also shown between TYR 198 and LYS 60, respectively. These results provided evidence that the possible MOA of 5D may be through the inhibition of TryR, an essential enzyme to the thiol metabolism of the parasite [22,23], and promising chemotherapeutic target against leishmaniasis [24].
Figure 7.

Trypanothione Reductase from L. infantum (PDB: 2JK6) Protein–ligand complex of Induced Fit Docking (IFD): (A) ligand interaction diagram of 5D in complex with 2JK6 and 2D ligand interaction and; (B) ligand interaction diagram of Quinacrine Mustard in complex with 2JK6 and 2D ligand interaction.
3. Discussion
There is an urgent need for new therapeutics that are more effective and less toxic than conventional treatments used to treat infectious diseases, including leishmaniasis [25]. Thiophenes derivatives are known for their therapeutic applications and have shown promising results to treat different types of cancer, degenerative diseases, HIV, and malaria [26,27,28,29,30,31,32,33]. Thus, we evaluated the anti-leishmania activity and selectivity of nine thiophene derivatives against L. major, and potential MOA was elucidated for our best candidate, 5D.
Thiophene derivatives 5A, 5D and 5E exhibited potent parasitic activity against L. major promastigotes (Table 1). Experimental models involving macrophages are ideal to study leishmaniasis since they are the major host cell for Leishmania spp. [34]. Thus, our three best candidates were further evaluated against the most important form of the parasite, intracellular amastigotes, in an in vitro infection model of murine macrophages. In this case, 5D presented the best anti-leishmanial activity by decreasing the proliferation of the parasite by 80%.
The in vitro toxicity of 5D and 5E was evaluated towards IPФ and LCC-MK2 cells. Our best two compounds were safer for the two cytotoxic models than the reference drug, amphotericin B, which is already known for its cytotoxic effects [35]. Even though amphotericin B presented similar activity as 5D against promastigotes and amastigotes, this result further supports the application of 5D and 5E as anti-leishmanial agents. Furthermore, the selectivity presented by 5D was remarkably higher than the parent compound 5A (10-fold higher), demonstrating the success to increase the anti-leishmanial activity and reduced cytotoxicity effects when compared to our previously reported arylalkylamine type-compound [12].
Next, we studied the potential MOA of derivative 5D. ROS can be generated in response to some drugs, resulting in destruction of cellular macromolecular components inducing cell death by affecting parasite mitochondrial function [36,37]. Here, we observed that 5D induced ROS production in L. major promastigotes after 31 h of treatment. The redox homeostasis in Leishmania is achieved through the activity of several superoxide dismutases, heme peroxidases, as well as of a series of thiol-containing proteins that directly or indirectly depend on trypanothione reductase [23,38]. In this regard, the trypanothione metabolism is unique to trypanosomatids and its main detoxification pathway [39]. This pathway protects parasites from oxidative stress and participates in several cellular processes that are carried out by glutathione in other organisms. Moreover, there are several trypanothione-dependent pathways that include enzymes such as tryparedoxin peroxidase (detoxication of hydroperoxide), ascorbate peroxidase (homeostasis of ascorbate), ribonucleotide reductase (synthesis of DNA precursors), and others [40,41]. With this idea on mind, we decided to explore in silico docking analysis to assess the possibility of L. major TryR as the target of 5D. Moreover, our results suggested that TryR interacts with 5D, however we do not exclude the possibility that other redox metabolism enzymes could be also targeted by compound.
In conclusion, to discover new chemotherapy agents against leishmaniasis, we efficiently synthetized nine thiophene type-compounds including 5A, following a two-step synthesis from low-priced commercially available starting materials. We then showed that our novel thiophene type-compounds possess high in vitro antileishmanial activity. Based on our SAR study, 5D analog was selected as the most promising lead compound among this library with excellent antiparasitic and S.I. Furthermore, 5D may act against trypanothione metabolism, followed by the production of ROS in the parasite; nevertheless, biological studies with recombinant TryR enzyme needs to be performed to further support this assumption. Overall, 5D represents a potential chemotherapeutic agent for the treatment of leishmaniasis, and further evaluation in a pre-clinical mouse model of cutaneous leishmaniasis is currently in progress in our laboratory.
4. Materials and Methods
4.1. General
Unless otherwise noted, all commercial reagents were used as purchased from Aldrich, St. Louis, MO, USA. All the reactions were monitored by thin-layer chromatography (TLC) that was performed on silica gel plates GF254. Compounds were visualized under a UV lamp. Flash chromatography was performed using silica gel (200–300 mesh) with various ratios of Dichloromethane: Methanol solvents as indicated in the text. Spectroscopy: 1H and 13C-NMR spectra were obtained on a Bruker DPX 400 MHz. Chemical shifts (δ) are quoted in parts per million (ppm), to the nearest 0.01 ppm and internally referenced relative to the solvent nuclei. 1H-NMR spectral data are reported with their chemical shift in parts per million (ppm). The multiplicity in 1H-NMR is abbreviated as follows: brs: broad; s: singlet; d: doublet; t: triplet; q: quartet; quint: quintet; sext: sextet; m: multiplet; or as a combination (e.g., dd, dt, etc.). The coupling constant (J) in hertz, integration and proton count were determined.
Spectrometer: Liquid chromatography/mass spectra (LC-MS) [+ESI] were taken on double focusing sector type mass spectrometer HX-110A. Maker JEOL Ltd. Tokyo, Japan (resolution of 10,000 and 10 KV accel. Volt. Ionization method; FAB (Fast Atom Bombardment) used Xe 3 KV energy. Used Matrix, NBA (m-Nitro benzyl alcohol)). Melting points were measured using a Mel-Temp melting point apparatus.
4.2. Chemical Synthesis
4.2.1. General Synthetic Procedure 1
Synthesis of the Ethyl 5,5,7,7-tetramethyl-2-(4-(trifluoromethyl) benzamido)-4,5,6,7-tetrahydrothieno[2,3-c]pyridine-3-carboxylate (5A)
Briefly, the 2,2,6,6-tetramethylpiperidine ketone (1, 1 equivalent) was mixed with 2-cyanoacetate esters (2, 1 equivalent) and elemental sulfur (1 equivalent) in the presence of diethylamine (3 equivalents) in ethanol (ETOH) at 60 °C for 17 h. The crude was precipitated by adding water and filtered to provide the desired known 2-aminothiophene intermediate (3) [12]. The latter was reacted with 4-trifluomethy benzoyl chloride in the presence of diisopropylethylamine in dry dichloromethane at 0 °C for 1 h and then at room temperature for 4 h. The solvent was evaporated then the crude mixture was purified by flash-column chromatography on silica gel, using a mixture of solvent of dichloromethane: methanol (DCM: MeOH) at ratios from 100:1 to 50:1, to provide the desired ethyl 5,5,7,7-tetramethyl-2-(4-(trifluoromethyl)benzamido)-4,5,6,7-tetrahydrothieno [2,3-c]pyridine-3-carboxylate (5A) as a light brown solid. The purity of 5A was confirmed by 1H-NMR, 13C-NMR, LC/MS and melting point.
4.2.2. General Synthetic Procedure 2
Synthesis of 5,5,7,7-tetramethyl-2-(2-(trifluoromethyl)benzamido)-4,5,6,7-tetrahydrothieno [2,3-c]pyridine-3-carboxylic acid (5G)
To ethyl 5,5,7,7-tetramethyl-2-(2-(trifluoromethyl)benzamido)-4,5,6,7-tetrahydrothieno [2,3-c]pyridine-3-carboxylate compound (5B; 1 equiv.) in 1 mL of tetrahydrofuran (THF), was added 5 equiv. of sodium hydroxide (NaOH) in water (1 mL). The mixture was stirred at room temperature during 17 h. The THF solvent was evaporated then mixture was acidified with HCl 1M to pH = 4. The protonated acid compound was extracted with ethylacetate (EtOAc) (3×; 50 mL) then dried under magnesium sulfate (MgSO4), and the solvent was evaporated to provide the desired 5,5,7,7-tetramethyl-2-(2-(trifluoromethyl)benzamido)-4,5,6,7-tetrahydrothieno [2,3-c]pyridine-3-carboxylic acid (5G), (Figure 2). The purity of 5G analog was confirmed by 1H-NMR, 13C-NMR, and LC/MS.
Following the general synthetic procedure 1, and in the presence of various substituted benzoyl chlorides, we created the analogs 5B–5E, and 5H–5I.
Ethyl-5,5,7,7-tetramethyl-2-(2-(trifluoromethyl)benzamido)-4,5,6,7-tetrahydrothieno[2,3-c]pyridine-3-carboxylate (5B)
Brown solid, yield 75%, m.p. 181.5–182.5 °C. 1H-NMR (400 MHz, CDCl3) δ 11.68 (s, 1H, NHCO), 7.78–7.65 (m, 4H, ArH), 4.32 (q, J = 7.0 Hz, 3H, CH2), 2.70 (s, 2H, CH2), 1.92 (m, 1H, NH), 1.39–1.16 (m, 15H, 5 × CH3). 13C-NMR (101 MHz, CDCl3) δ 166.18, 163.70, 147.33, 135.09, 133.90, 132.18, 130.71, 128.50, 128.32, 126.68, 112.30, 60.61, 51.89, 49.84, 39.53, 34.13, 29.99, 14.00. LC-MS (ESI) for C22H25F3N2O3S, M + 1 = 455.22.
Ethyl-5,5,7,7-tetramethyl-2-(2-(trifluoromethyl)benzamido)-4,5,6,7-tetrahydrothieno[2,3-c]pyridine-3-carboxylate (5C)
Brown solid, yield 70%, m.p. 132.6–133.6 °C. 1H-NMR (400 MHz, CDCl3) δ 11.56 (s, 1H, NHCO), 7.52–7.40 (m, 3H, ArH), 4.10 (q, J = 7.1 Hz, 3H, CH2), 2.53 (s, 2H, CH2), 1.32–1.03 (s, 15H, 5 × CH3). 13C-NMR (101 MHz, CDCl3) δ 166.05, 162.14, 146.93, 138.40, 135.50, 135.04, 130.73, 128.58, 128.43, 128.25, 126.30, 112.53, 60.69, 52.17, 50.17, 39.27, 33.88, 29.71, 13.96. (ESI+, M + 1 = 489.02. LC-MS (ESI) for C22H24ClF3N2O3S, M + 1 = 489.02.
Ethyl-2-(2-fluoro-3-(trifluoromethyl)benzamido)-5,5,7,7-tetramethyl-4,5,6,7-tetrahydrothieno[2,3-c]-pyridine-3-carboxylate (5D)
Light brown solid, yield 76%, m.p. 145.4–146.4 °C. 1H-NMR (400 MHz, CDCl3) δ 12.40 (d, J = 10.3 Hz, 1H, NHCO), 8.49–8.30 (m, 1H, ArH), 7.83 (dd, J = 10.5, 3.9 Hz, 1H, ArH), 7.45 (t, J = 7.8 Hz, 1H, ArH), 4.42 (q, J = 7.1 Hz, 2H, CH2), 2.74 (s, 2H, CH2), 1.54 (s, 6H, 2 × CH3), 1.42 (t, J = 7.1 Hz, 3H, CH3), 1.23 (t, J = 7.1 Hz, 6H, 2 × CH3). 13C-NMR (101 MHz, CDCl3) δ 165.70, 158.50, 146.67, 136.26, 135.74, 131.23, 129.15, 124.87, 123.48, 121.53, 119.30, 113.11, 60.86, 52.03, 50.01, 39.71, 34.34, 30.23, 14.29. LC-MS (ESI) for C22H24F4N2O3S, M + 1 = 473.05.
Ethyl-2-(4-fluoro-3-(trifluoromethyl)benzamido)-5,5,7,7-tetramethyl-4,5,6,7-tetrahydrothieno[2,3-c]-pyridine-3-carboxylate (5E)
Light brown solid, yield 66%, m.p. 116.0–117.0 °C. 1H-NMR (400 MHz, CDCl3) δ 12.40 (s, 1H, NHCO), 8.25 (dd, J = 6.4, 1.4 Hz, 1H, ArH), 8.10 (ddd, J = 7.9, 4.1, 2.2 Hz, 1H, ArH), 7.29 (t, J = 9.1 Hz, 1H, ArH), 4.34 (q, J = 7.1 Hz, 2H, CH2), 2.65 (s, 2H, CH2), 1.46 (s, 6H, 2 × CH3), 1.36 (t, J = 7.1 Hz, 3H, CH3), 1.16 (d, J = 11.3 Hz, 6H, 2 × CH3). 13C-NMR (101 MHz, CDCl3) δ 166.89, 160.81, 148.04, 135.17, 132.73, 128.95, 128.58, 127.43, 119.35, 117.73, 117.51, 112.28, 60.92, 53.37, 52.12, 50.08, 39.57, 34.13, 30.03, 14.18. LC-MS (ESI) for C22H24F4N2O3S, M + 1 = 473.05.
Methyl-2-(4-fluoro-3-(trifluoromethyl)benzamido)-5,5,7,7-tetramethyl-4,5,6,7-tetrahydrothieno[2,3-c]-pyridine3-carboxylate (5H)
Light brown solid, yield 72%, m.p. 122.0–123.0 °C. 1H-NMR (400 MHz, CDCl3) δ 12.45 (s, 1H, NHCO), 8.32 (s, 1H, ArH), 8.18 (s, 1H, ArH), 7.37 (t, J = 8.0 Hz, 1H, ArH), 3.93 (s, 3H, CH3), 2.71 (s, 2H, CH2), 1.53 (s, 6H, 2 × CH3), 1.27–1.16 (m, 9H, 3 × CH3).13C-NMR (101 MHz, CDCl3) δ 167.47, 161.06, 160.86, 148.44, 132.84, 132.74, 129.03, 128.46, 127.53, 117.86, 117.64, 112.17, 52.45, 51.80, 50.45, 39.49, 34.13, 29.98 LC-MS (ESI) for C21H22F4N2O3S, M + 1 = 459.07.
Methyl-5,5,7,7-tetramethyl-2-(2-(trifluoromethyl)benzamido)-4,5,6,7-tetrahydrothieno[2,3-c]pyridine-3-carboxylate (5I)
Light brown solid, yield 60%, m.p. 184.2–185.2 °C. 1H-NMR (400 MHz, CDCl3) δ1H-NMR (400 MHz, CDCl3) δ 11.67 (s, 1H, NHCO), 7.78 (d, J = 7.4 Hz, 1H, ArH), 7.66 (dd, J = 10.9, 4.5 Hz, 3H, ArH), 3.85 (s, 3H, CH3), 2.68 (s, 2H, CH2), 1.52 (s, 6H, 2 × CH3), 1.23 (s, 6H, 2 × CH3). 13C-NMR (101 MHz, CDCl3) δ 166.90, 164.03, 147.80, 135.24, 134.04, 132.27, 130.83, 128.57, 127.92, 126.90, 126.85, 112.18, 52.18, 51.64, 50.17, 39.59, 34.26, 30.12 LC-MS (ESI) for C21H23F3N2O3S, M + 1 = 441.07.
Following the general synthetic procedure 2, we created the analogs 5F and 5G.
2-(5-chloro-2-(trifluoromethyl)benzamido)-5,5,7,7-tetramethyl-4,5,6,7-tetrahydrothieno[2,3-c]pyridine-3-carboxylic acid (5F)
Brown oilesh, yield 51% 1H-NMR (400 MHz, CDCl3) δ 11.69 (s, 1H, NHCO), 7.57–7.44 (m, 3H, ArH), 2.53 (s, 2H, CH2), 1.32–1.03 (s, 12H, 4 × CH3). 13C-NMR (101 MHz, CDCl3) δ 167.09, 164.14, 161.83, 139.04, 135.50, 135.04, 130.73, 128.58, 128.43, 128.25, 126.30, 112.53, 52.17, 50.17, 39.27, 33.88, 29.71. LC-MS (ESI) for C22H24ClF3N2O3S, M + 1 = 461.03.
5,5,7,7-tetramethyl-2-(2-(trifluoromethyl)benzamido)-4,5,6,7-tetrahydrothieno[2,3-c]pyridine-3-carboxylic acid (5G)
Brown oilesh, yield 55%. 1H-NMR (400 MHz, CDCl3) δ 11.70 (s, 1H, NHCO), 7.92–7.62 (m, 4H, ArH), 2.70 (s, 2H, CH2), 1.39–1.16 (m, 12H, 4 × CH3). 13C-NMR (101 MHz, CDCl3) δ 167.08, 163.70, 162.03, 136.09, 134.01, 132.18, 130.71, 128.50, 128.32, 126.68, 112.30, 51.89, 49.84, 39.53, 34.13, 29.99. LC-MS (ESI) for C22H25F3N2O3S, M + 1 = 427.02.
4.3. Cell Maintenance
Transgenic L. major promastigotes expressing firefly luciferase (L. major-luc) (Lmj-FV1-LUC-TK [L. major strain Friedlin {MHOM/JL/80/Friedlin}] were maintain in M199 medium supplemented with hemin, 10% inactivated fetal bovine serum (iFBS) (Gibco, Thermo Fisher Scientific, Waltham, MA, USA), and 1% of 10,000 units/mL penicillin and 10 mg/mL streptomycin (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) as described [42]. Intraperitoneal murine macrophages (IPФ) were obtained from BALB/c mice [43]. Monkey kidney epithelial cells (LLC-MK2) (ATCC # CCL-7) (American Type Culture Collection, Manassas, VA, USA), and IPФ were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM), supplemented with 10% iFBS, along with 1% of 10,000 units/mL penicillin and 10 mg/mL streptomycin. The procedures were performed minimizing the distress and pain for animals following the NIH guidance and animal protocol (A-201107-1) approved by UTEP’s Institutional Animal Care and Use Committee (IACUC).
4.4. Luciferase Assay—Viability of Leishmania Major promastigotes
The antiparasitic activity of the 9 thiophene compounds was determined by adding the analogs together with 2 × 106 L. major-luc promastigotes per mL in 96-well NUNC white microplates (Thermo Fisher Scientific, Waltham, MA, USA) followed by incubation for 72 h at 28 °C. Then, parasite survival was measured by luciferase activity with the addition of the substrate 5′-fluoroluciferin (ONE-Glo luciferase assay system; Promega, Madison, WI, USA), using a luminometer (Luminoskan; Thermo Fisher Scientific, Waltham, MA, USA). The luminescence intensity was a direct measure of the parasite survival, and 50% effective concentration (EC50) was determined for each drug and summarized in Table 1.
4.5. Assessment of Thiophene Compound Mammalian Cell Cytotoxicity
The potential cytotoxicity of 5A was tested by alamarBlueTM Cell Viability Assay (Thermo Fisher Scientific, Waltham, MA, USA) as previously described [18]. Briefly, 1 × 106/mL rhesus monkey kidney epithelial cells (LLC-MK2), and 1 × 106/mL BALB/c IPФ were seeded in a 96-well clear bottom black microplate (BD Biosciences, Franklin Lakes, NJ, USA). Cells were incubated in the presence of increasing drug concentrations for 72 h (LLC-MK2) or 48 h (IPФ) at 37 °C, 5% CO2, followed by addition of alamarBlueTM. Fluorescence was measured using a fluorometer (Fluoroskan; Thermo Fisher Scientific, Waltham, MA, USA). Compound 5D or 5E were incubated with 1 × 105 cells/mL (LLC-MK and IPФ) for 72 or 48 h, respectively, at 37 °C, 5% CO2. After the incubation period, a dilution of 20:1000 in PBS from a stock at 1 mg/mL of Propidium Iodide (PI) and Hoechst 33342 (Thermo Scientific, Waltham, MA, USA) were added for survival discrimination as previously described [44]. Analysis was performed by High-Content Imaging Assay (HCIA) using an IN Cell 2000 Analyzer Bioimaging System (GE Healthcare, Chicago, IL, USA) for LLC-MK2 cells, and BD Pathway 855 High-resolution Bioimager System (BD Biosciences, Franklin Lakes, NJ, USA) for IPФ. The 50% cytotoxic concentration (CC50) and selective index (S.I.) was determined and summarized in Table 1.
4.6. High-Content Imaging Assay—Proliferation Experiments
BALB/c IPФ were acquired and seeded at a density of 1 × 105 cells/mL for 2 h at 37 °C, 5% CO2. After adherence, IPФ were infected with 1 × 106/mL metacyclic promastigotes of L. major-luc, at a ratio of 10:1 parasites per macrophage. Subsequently, infected IPФ were incubated with derivatives 5A, 5D and 5E at increasing concentrations (0.625 to 10 µM) for 48 h treatment. Afterwards, cells were fixed with 4% paraformaldehyde, and stained with (1.25:100) Alexa Fluor™ 488 Phalloidin (Thermo Fisher Scientific, Waltham, MA, USA) and (1:1000) DAPI (Sigma Aldrich, St. Louis, MO, USA). Then, the numbers of infected cells and amastigotes were determined by HCIA using an IN Cell 2000 Analyzer Bioimaging System (GE Healthcare, Chicago, IL, USA). Parameters were set for the excitation and emission spectra of Alexa Fluor™ 488 Phalloidin and DAPI, and a constraint of 3 or more parasites per macrophage was set as previously reported [44,45].
4.7. Measurement of Reactive Oxygen Species Levels
L. major promastigotes (2 × 106 cells/mL) were incubated for 24 h with 5D (EC50 0.90 µM) in a 96-well clear bottom black microplate (BD Biosciences, Franklin Lakes, NJ, USA). Controls treated with 1% DMSO, 100 µM of hydrogen peroxide (H2O2) as positive control, or M199 medium. After incubation period, 10 µM of H2DCFDA (Thermo Fischer Scientific, Waltham, MA, USA) (H2DCFDA/DMSO, 1 mg/mL) was added per well followed by 20 min of incubation at 37 °C. Fluorescence was measured for an additional 7 h using a fluorometer (Fluoroskan; Thermo Fisher Scientific, Waltham, MA, USA) at 527 nm using an excitation wavelength of 485. For all measurements, basal fluorescence was subtracted.
All graphs, EC50 and CC50 values were produced using Graph Pad Prism 7 Software (GraphPad Software, Inc., La Jolla, CA, USA).
4.8. Docking Studies—Pre-Docking Preparation
The structure of trypanothione reductase bound to Flavin adenine dinucleotide (PDB ID: 2JK6) [46] were obtained from the Protein Data Bank. The Protein Preparation Wizard in Maestro was used to minimize the protein structure, add hydrogens and charges, and find any missing residues. The two-dimensional structures of 5D and Quinacrine Mustard, a recently experimentally approved drug as a control, were drawn using the molecular structure editor ChemDraw Software (PerkinElmer, Waltham, MA, USA) and processed by LigPrep Schrödinger (Schrödinger, LLC., New York, NY, USA) to generate the 3D structures.
4.9. Binding Site Analysis
Maestro’s SiteMap tool (Schrödinger, LLC., New York, NY, USA) was used to predict the likely binding sites of trypanothione reductase. The SiteMap tool uses a series of algorithm that generates a map of hydrophobic and hydrophilic surfaces on the protein surface [47]. Hydrophilic surface maps are divided into donor, acceptor, and metal-binding regions. Five potential binding sites were identified with at least 15 site points. However, the top SiteMap was chosen to be the receptor grid.
Author Contributions
Conceptualization, E.I., R.S. and R.A.M.; Methodology, E.I., R.S. and R.A.M.; Software, H.A.; Validation, F.R., E.I., G.P.C., H.A., R.S. and R.A.M.; Formal Analysis, F.R., E.I., G.P.C., H.A., T.E.M.M.C., R.S. and R.A.M.; Investigation, F.R., E.I., G.P.C., H.A. and T.E.M.M.C.; Resources, R.S. and R.A.M.; Data Curation, F.R., E.I., R.S. and R.A.M.; Writing—Original Draft Preparation, F.R., E.I., R.S. and R.A.M.; Writing—Review and Editing, F.R., E.I., R.S. and R.A.M.; Visualization, F.R., E.I., H.A., R.S. and R.A.M.; Supervision, R.S. and R.A.M.; Project Administration, R.S. and R.A.M.; and Funding Acquisition, R.S. and R.A.M.
Funding
This research was funded by: UTEP/BBRC Core Facilities: Biomolecule Analysis (BACF), Cytometry, Screening and Imaging (CSI), and Genomic Analysis (GACF) supported by NIGMS grant No. 5G12MD007592. The synthesis and docking sections were performed by Rachid Skouta research laboratory partially supported by the Lung Cancer Research Foundation and the Green Fund. E.I. was supported by the RISE Scholars Program at UTEP through grant No. R25GM069621-11 from the NIGMS. G.P.C. was supported by The UT System Louis Stokes Alliance for Minority Participation (LSAMP) funded by the National Science Foundation (grant number HRD-1202008). TEMMC was supported by the National Institutes of Science and Technology (INCT-IDN, Brazil) Science without Borders Program/CNPq.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds are not available from the authors.
References
- 1.Alvar J., Velez I.D., Bern C., Herrero M., Desjeux P., Cano J., Jannin J., den Boer M., WHO Leishmaniasis Control Team Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE. 2012;7:e35671. doi: 10.1371/journal.pone.0035671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aoun K., Bouratbine A. Cutaneous leishmaniasis in North Africa: A review. Parasite. 2014;21:14. doi: 10.1051/parasite/2014014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cappai R., Morris L., Aebischer T., Bacic A., Curtis J.M., Kelleher M., McLeod K.S., Moody S.F., Osborn A.H., Handman E. Ricin-resistant mutants of Leishmania major which express modified lipophosphoglycan remain infective for mice. Parasitology. 1994;108:397–405. doi: 10.1017/S0031182000075946. [DOI] [PubMed] [Google Scholar]
- 4.World Health Organization Leishmaniasis-Fact Sheet N’375. [(accessed on 9 May 2016)];2014 Available online: http://www.who.int/mediacentre/factsheets/fs375/en/
- 5.McGwire B.S., Satoskar A.R. Leishmaniasis: Clinical syndromes and treatment. QJM. 2014;107:7–14. doi: 10.1093/qjmed/hct116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rodrigues K.A., Dias C.N., Néris P.L., Rocha Jda C., Scotti M.T., Scotti L., Mascarenhas S.R., Veras R.C., de Medeiros I.A., Keesen Tde S., et al. 2-Amino-thiophene derivatives present antileishmanial activity mediated by apoptosis and immunomodulation in vitro. Eur. J. Med. Chem. 2015;106:1–14. doi: 10.1016/j.ejmech.2015.10.011. [DOI] [PubMed] [Google Scholar]
- 7.Croft S.L., Coombs G.H. Leishmaniasis--current chemotherapy and recent advances in the search for novel drugs. Trends Parasitol. 2003;19:502–508. doi: 10.1016/j.pt.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 8.Kato K.C., Morais-Teixeira E., Reis P.G., Silva-Barcellos N.M., Salaun P., Campos P.P., Dias Correa-Junior J., Rabello A., Demicheli C., Frezard F. Hepatotoxicity of pentavalent antimonial drug: Possible role of residual Sb(III) and protective effect of ascorbic acid. Antimicrob. Agents Chemother. 2014;58:481–488. doi: 10.1128/AAC.01499-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laniado-Laborin R., Cabrales-Vargas M.N. Amphotericin B: Side effects and toxicity. Rev. Iberoam. Micol. 2009;26:223–227. doi: 10.1016/j.riam.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 10.Nagle A.S., Khare S., Kumar A.B., Supek F., Buchynskyy A., Mathison C.J.N., Chennamaneni N.K., Pendem N., Buckner F.S., Gelb M.H., et al. Recent developments in drug discovery for leishmaniasis and human African trypanosomiasis. Chem. Rev. 2014;114:11305–11347. doi: 10.1021/cr500365f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turner K.G., Vacchina P., Robles-Murguia M., Wadsworth M., McDowell M.A., Morales M.A. Fitness and Phenotypic Characterization of Miltefosine-Resistant Leishmania major. PLoS Negl. Trop. Dis. 2015;9:e0003948. doi: 10.1371/journal.pntd.0003948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iniguez E.A., Perez A., Maldonado R.A., Skouta R. Novel arylalkylamine compounds exhibits potent selective antiparasitic activity against Leishmania major. Bioorg. Med. Chem. Lett. 2015;25:5315–5320. doi: 10.1016/j.bmcl.2015.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Skouta R.M.R.A. Preparation of Tetramethyltetrahydrothienopyridine Derivatives for Use as Parasiticides. Appl. Publ 20160362420 A1. U.S. Patent. 2016 Dec 15;
- 14.Keri R.S., Chand K., Budagumpi S., Balappa Somappa S., Patil S.A., Nagaraja B.M. An overview of benzo[b]thiophene-based medicinal chemistry. Eur. J. Med. Chem. 2017;138:1002–1033. doi: 10.1016/j.ejmech.2017.07.038. [DOI] [PubMed] [Google Scholar]
- 15.Sensfuss U., Habicher W.D. 2-aminothiophenes from triacetonamine: A convenient way to novel sterically hindered piperidine derivatives. Heteroat. Chem. 1998;9:529–536. doi: 10.1002/(SICI)1098-1071(1998)9:6<529::AID-HC1>3.0.CO;2-#. [DOI] [Google Scholar]
- 16.Thalhofer C.J., Graff J.W, Love-Homan L., Hickerson S.M., Craft N., Beverley S.M., Wilson M.E. In vivo imaging of transgenic Leishmania parasites in a live host. J. Vis. Exp. 2010:1980. doi: 10.3791/1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Capul A.A., Barron T., Dobson D.E., Turco S.J., Beverley S.M. Two functionally divergent UDP-Gal nucleotide sugar transporters participate in phosphoglycan synthesis in Leishmania major. J. Biol. Chem. 2007;282:14006–14017. doi: 10.1074/jbc.M610869200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lara D., Feng Y., Bader J., Savage P.B., Maldonado R.A. Anti-trypanosomatid activity of ceragenins. J. Parasitol. 2010;96:638–642. doi: 10.1645/GE-2329.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khare S., Nagle A.S., Biggart A., Lai Y.H., Liang F., Davis L.C., Barnes S.W., Mathison C.J.N., Myburgh E., Gao M.-Y., et al. Proteasome inhibition for treatment of leishmaniasis, Chagas disease and sleeping sickness. Nature. 2016;537:229–233. doi: 10.1038/nature19339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schrödinger Suite 2018-1 Induced Fit Docking Protocol. [(accessed on 10 February 2018)]; Available online: https://www.schrodinger.com/induced-fit.
- 21.Clark A.J., Tiwary P., Borrelli K., Feng S., Miller E.B., Abel R., Friesner R.A., Berne B.J. Prediction of Protein-Ligand Binding Poses via a Combination of Induced Fit Docking and Metadynamics Simulations. J. Chem. Theory Comput. 2016;12:2990–2998. doi: 10.1021/acs.jctc.6b00201. [DOI] [PubMed] [Google Scholar]
- 22.Taylor M.C., Kelly J.M., Chapman C.J., Fairlamb A.H., Miles M.A. The structure, organization, and expression of the Leishmania donovani gene encoding trypanothione reductase. Mol. Biochem. Parasitol. 1994;64:293–301. doi: 10.1016/0166-6851(94)00034-4. [DOI] [PubMed] [Google Scholar]
- 23.Tovar J., Wilkinson S., Mottram J.C., Fairlamb A.H. Evidence that trypanothione reductase is an essential enzyme in Leishmania by targeted replacement of the tryA gene locus. Mol. Microbiol. 1998;29:653–660. doi: 10.1046/j.1365-2958.1998.00968.x. [DOI] [PubMed] [Google Scholar]
- 24.Khan M.O. Trypanothione reductase: A viable chemotherapeutic target for antitrypanosomal and antileishmanial drug design. Drug Target Insights. 2007;2:129–146. doi: 10.1177/117739280700200007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feasey N., Wansbrough-Jones M., Mabey D.C., Solomon A.W. Neglected tropical diseases. Br. Med. Bull. 2010;93:179–200. doi: 10.1093/bmb/ldp046. [DOI] [PubMed] [Google Scholar]
- 26.Haidle A.M., Zabierek A.A., Childers K.K., Rosenstein C., Mathur A., Altman M.D., Chan G., Xu L., Bachman E., Mo J.R., et al. Thiophene carboxamide inhibitors of JAK2 as potential treatments for myleoproliferative neoplasms. Bioorg. Med. Chem. Lett. 2014;24:1968–1973. doi: 10.1016/j.bmcl.2014.02.064. [DOI] [PubMed] [Google Scholar]
- 27.Huang H., Li H., Yang S., Chreifi G., Martasek P., Roman L.J., Meyskens F.L., Poulos T.L., Silverman R.B. Potent and selective double-headed thiophene-2-carboximidamide inhibitors of neuronal nitric oxide synthase for the treatment of melanoma. J. Med. Chem. 2014;57:686–700. doi: 10.1021/jm401252e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De Oliveira J.F., da Silva A.L., Vendramini-Costa D.B., da Cruz Amorim C.A., Campos J.F., Ribeiro A.G., de Moura R.O., Neves J.L., Ruiz A.L.T.G., de Carvalho J.E., et al. Synthesis of thiophene-thiosemicarbazone derivatives and evaluation of their in vitro and in vivo antitumor activities. Eur. J. Med. Chem. 2015;104:148–156. doi: 10.1016/j.ejmech.2015.09.036. [DOI] [PubMed] [Google Scholar]
- 29.Marchais-Oberwinkler S., Xu K., Wetzel M., Perspicace E., Negri M., Meyer A., Odermatt A., Moller G., Adamski J., Hartmann R.W. Structural Optimization of 2,5-Thiophene Amides as Highly Potent and Selective 17 beta-Hydroxysteroid Dehydrogenase Type 2 Inhibitors for the Treatment of Osteoporosis. J. Med. Chem. 2013;56:167–181. doi: 10.1021/jm3014053. [DOI] [PubMed] [Google Scholar]
- 30.Opsenica I.M., Verbic T.Z., Tot M., Sciotti R.J., Pybus B.S., Djurkovic-Djakovic O., Slavic K., Solaja B.A. Investigation into novel thiophene- and furan-based 4-amino-7-chloroquinolines afforded antimalarials that cure mice. Bioorg. Med. Chem. 2015;23:2176–2186. doi: 10.1016/j.bmc.2015.02.061. [DOI] [PubMed] [Google Scholar]
- 31.Patil D., Dash R.P., Thakur S.K., Pandya A.N., Venkatesh P., Vasu K.K., Nivsarkar M. Implication of novel thiazolo-thiophene derivative (MCD-KV-10) for management of asthma. J. Enzym. Inhib. Med. Chem. 2015;30:229–239. doi: 10.3109/14756366.2014.913035. [DOI] [PubMed] [Google Scholar]
- 32.Ghorab M.M., Al-Dhfyan A., Al-Dosari M.S., El-Gazzar M.G., AlSaid M.S. Antiproliferative activity of novel thiophene and thienopyrimidine derivatives. Drug Res. 2014;64:313–320. doi: 10.1055/s-0033-1358679. [DOI] [PubMed] [Google Scholar]
- 33.Ashok P., Lu C.L., Chander S., Zheng Y.T., Murugesan S. Design, Synthesis, and Biological Evaluation of 1-(thiophen-2-yl)-9H-pyrido[3,4-b]indole Derivatives as Anti-HIV-1 Agents. Chem. Biol. Drug Des. 2015;85:722–728. doi: 10.1111/cbdd.12456. [DOI] [PubMed] [Google Scholar]
- 34.Arango Duque G., Descoteaux A. Leishmania survival in the macrophage: Where the ends justify the means. Curr. Opin. Microbiol. 2015;26:32–40. doi: 10.1016/j.mib.2015.04.007. [DOI] [PubMed] [Google Scholar]
- 35.Butler W.T. Pharmacology, toxicity, and therapeutic usefulness of amphotericin B. JAMA. 1966;195:371–375. doi: 10.1001/jama.1966.03100050079024. [DOI] [PubMed] [Google Scholar]
- 36.Amer A.O., Swanson M.S. A phagosome of one’s own: A microbial guide to life in the macrophage. Curr. Opin. Microbiol. 2002;5:56–61. doi: 10.1016/S1369-5274(02)00286-2. [DOI] [PubMed] [Google Scholar]
- 37.Fonseca-Silva F., Inacio J.D.F., Canto-Cavalheiro M.M., Almeida-Amaral E.E. Reactive oxygen species production and mitochondrial dysfunction contribute to quercetin induced death in Leishmania amazonensis. PLoS ONE. 2011;6:e14666. doi: 10.1371/journal.pone.0014666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu J., Holmgren A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014;66:75–87. doi: 10.1016/j.freeradbiomed.2013.07.036. [DOI] [PubMed] [Google Scholar]
- 39.Irigoin F., Cibils L., Comini M.A., Wilkinson S.R., Flohe L., Radi R. Insights into the redox biology of Trypanosoma cruzi: Trypanothione metabolism and oxidant detoxification. Free Radic. Biol. Med. 2008;45:733–742. doi: 10.1016/j.freeradbiomed.2008.05.028. [DOI] [PubMed] [Google Scholar]
- 40.Schmidt A., Krauth-Siegel R.L. Enzymes of the trypanothione metabolism as targets for antitrypanosomal drug development. Curr. Top. Med. Chem. 2002;2:1239–1259. doi: 10.2174/1568026023393048. [DOI] [PubMed] [Google Scholar]
- 41.Wilkinson S.R., Horn D., Prathalingam S.R., Kelly J.M. RNA interference identifies two hydroperoxide metabolizing enzymes that are essential to the bloodstream form of the african trypanosome. J. Biol. Chem. 2003;278:31640–31646. doi: 10.1074/jbc.M303035200. [DOI] [PubMed] [Google Scholar]
- 42.Martinez A., Carreon T., Iniguez E., Anzellotti A., Sanchez A., Tyan M., Sattler A., Herrera L., Maldonado R.A., Sanchez-Delgado R.A. Searching for new chemotherapies for tropical diseases: Ruthenium-clotrimazole complexes display high in vitro activity against Leishmania major and Trypanosoma cruzi and low toxicity toward normal mammalian cells. J. Med. Chem. 2012;55:3867–3877. doi: 10.1021/jm300070h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Capul A.A., Hickerson S., Barron T., Turco S.J., Beverley S.M. Comparisons of mutants lacking the Golgi UDP-galactose or GDP-mannose transporters establish that phosphoglycans are important for promastigote but not amastigote virulence in Leishmania major. Infect. Immun. 2007;75:4629–4637. doi: 10.1128/IAI.00735-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lema C., Varela-Ramirez A., Aguilera R.J. Differential nuclear staining assay for high-throughput screening to identify cytotoxic compounds. Curr. Cell. Biochem. 2011;1:1–14. [PMC free article] [PubMed] [Google Scholar]
- 45.Iniguez E., Sanchez A., Vasquez M.A., Martinez A., Olivas J., Sattler A., Sanchez-Delgado R.A., Maldonado R.A. Metal-drug synergy: New ruthenium(II) complexes of ketoconazole are highly active against Leishmania major and Trypanosoma cruzi and nontoxic to human or murine normal cells. J. Biol. Inorg. Chem. 2013;18:779–790. doi: 10.1007/s00775-013-1024-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baiocco P., Colotti G., Franceschini S., Ilari A. Molecular basis of antimony treatment in leishmaniasis. J. Med. Chem. 2009;52:2603–2612. doi: 10.1021/jm900185q. [DOI] [PubMed] [Google Scholar]
- 47.Halgren T. New method for fast and accurate binding-site identification and analysis. Chem. Biol. Drug Des. 2007;69:146–148. doi: 10.1111/j.1747-0285.2007.00483.x. [DOI] [PubMed] [Google Scholar]