

Abstract
To complement existing affinity purification (AP) approaches for the identification of protein-protein interactions (PPI), enzymes have been introduced that allow the proximity-dependent labeling of proteins in living cells. One such enzyme, BirA* (used in the BioID approach), mediates the biotinylation of proteins within a range of approximately 10 nm. Hence, when fused to a protein of interest and expressed in cells, it allows the labeling of proximal proteins in their native environment. As opposed to AP that relies on the purification of assembled protein complexes, BioID detects proteins that have been marked within cells no matter whether they are still interacting with the protein of interest when they are isolated. Since it biotinylates proximal proteins, one can moreover capitalize on the exceptional affinity of streptavidin for biotin to very efficiently isolate them. While BioID performs better than AP for identifying transient or weak interactions, both AP- and BioID-mass spectrometry approaches provide an overview of all possible interactions a given protein may have. However, they do not provide information on the context of each identified PPI. Indeed, most proteins are typically part of several complexes, corresponding to distinct maturation steps or different functional units. To address this common limitation of both methods, we have engineered a protein-fragments complementation assay based on the BirA* enzyme. In this assay, two inactive fragments of BirA* can reassemble into an active enzyme when brought in close proximity by two interacting proteins to which they are fused. The resulting split-BioID assay thus allows the labeling of proteins that assemble around a pair of interacting proteins. Provided these two only interact in a given context, split-BioID then allows the analysis of specific context-dependent functional units in their native cellular environment. Here, we provide a step-by-step protocol to test and apply split-BioID to a pair of interacting proteins.
Keywords: Biochemistry, Issue 134, BioID, proteomics, protein engineering, protein-fragments complementation assay, protein-protein interactions, proximity labeling
Introduction
As most cellular functions are performed by proteins that dynamically assemble macromolecular complexes, the identification of protein-protein interactions (PPI) is a major endeavor in biomedical research. Indeed, PPI are often deregulated in disease and represent potential targets for therapeutics1. The most widely used method for the identification of PPI is the affinity purification (AP) approach in which, following cell lysis, a protein of interest is specifically purified on a matrix and associated proteins are subsequently identified by mass spectrometry (MS). While AP-MS is a powerful approach, it typically does not perform well on poorly soluble protein complexes, very transient interactions or PPI that require an intact subcellular structure. Moreover, the interpretation of the data can be complicated by the dynamic nature of PPI networks, as a single protein is often part of several distinct protein complexes.
Proximity-labeling techniques such as BioID2 or APEX23,4 were recently developed to address some of the limitations of the AP-MS approaches. In BioID, the enzyme BirA* (corresponding to a G115R variant of the wild type E. coli enzyme) catalyzes the formation of labile biotinyl-AMP (bio-AMP) that can react with primary amines. As opposed to the wild type enzyme, that retains bio-AMP in its active center, BirA* releases bio-AMP allowing its diffusion to its neighboring environment. Hence, when fused to a protein of interest and expressed in cells, proximal proteins can be biotinylated within an estimated range of 10 nm5. These marked proximal proteins are then isolated by streptavidin pulldown and identified by MS. As opposed to AP-MS, BioID requires the expression of a fusion protein. It can thus only be applied to proteins whose function is not hampered by tagging. Moreover, the speed of labeling is slow, typically 6–24 h2,6, making the detection of short-lived proteins challenging. Yet, compared to AP-MS, BioID-MS offers several key advantages: first, its captures the interactions in their native cellular environment; second, labeled proteins rather than assembled complexes are isolated following cell lysis; third, streptavidin pulldowns allow using denaturing buffers and harsh washing conditions. Hence, the method is more sensitive to detect transient or weak interactions7 or interactions that occur on a specific and hard to isolate subcellular structure8.
However, most proteins are usually part of larger complexes that can remodel according to cellular cues or to the function that needs to be performed. Hence, a single protein is typically part of several complexes, corresponding to distinct functional units, involving distinct and/or overlapping PPI. Both approaches give an overview of all associations a given protein may have, but they fail to address the context of individual PPI. To increase the resolution of the latter, we have designed a protein-fragments complementation assay (PCA) in which two inactive fragments of BirA* (NBirA*, that contains the catalytic domain, and CBirA* that can be viewed as the re-activation domain) can reassemble into an active enzyme when brought in close proximity by two interacting proteins9. The resulting split-BioID assay focuses the proximity-dependent biotinylation on proteins that assemble around a pair of interacting proteins and thus allows the identification of context dependent protein assemblies. We recently demonstrated the outstanding resolution power of split-BioID by resolving two distinct protein complexes involved in the miRNA-mediated gene-silencing pathway9.
Altogether, in a single and simple assay, split-BioID allows discovering and specifically assigning PPI to defined functional units in which a given protein is involved, provided an additional interacting protein of the corresponding protein complex is known.
Protocol
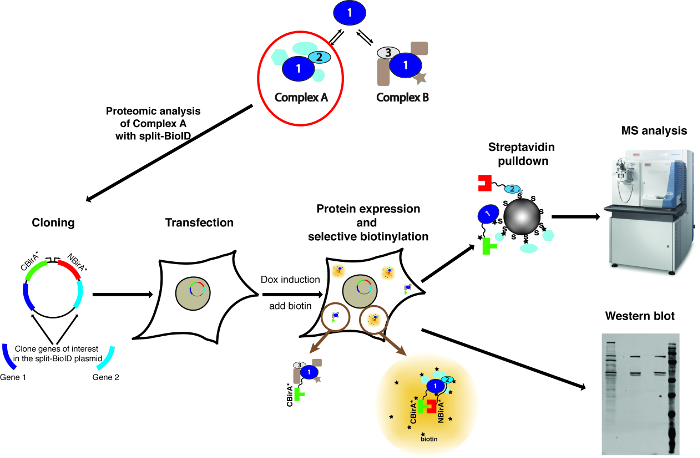
NOTE: An overview of the method is shown on Figure 1.
1. Planning of the Cloning Strategy
Select two putatively interacting proteins to be tested. NOTE: Each of the two proteins will be fused to one split-BioID fragment: either NBirA* or CBirA*. As a negative control, the CBirA* fusion proteins will be tested with NBirA* fused to the green fluorescent protein (NBirA*-GFP). As an additional control, the NBirA* fusions may be tested alone, without cognate CBirA* fusion or in combination with a CBirA* fused to an unrelated protein. It is advisable not to use CBirA*-GFP as a negative control as it was shown to consistently lead to major background when combined to any NBirA* fusion protein9. The cause of this observation may be the expression level of CBirA*-GFP, much higher than any other CBirA* fusion proteins we have used so far, that may lead to significant reassociation with the NBirA* fragment.
- Once two proteins are selected, check the literature to find whether both have already been successfully tagged in functional studies (for example as a GFP-tagged fusion protein).
- If such studies exist, note the position of the tag (at the N- or C-terminus) and use the same orientation to tag the proteins of interest with the split-BioID fragments.
- If no such study exists, plan constructs coding for both proteins tagged at the N- and C-terminus and plan an assay to test the functionality of the fusion proteins (for example, a rescue experiment in a cell line depleted for the WT protein). NOTE: When pairing two fusion proteins using the plasmids described in Figure 2 that encode long flexible linkers, the orientation of the fusion proteins (BirA* fragments fused upstream or downstream of the proteins of interest) generally does not matter. Indeed using FRB and FKBP as model proteins, it was shown that all four possible iterations (both fragments at the N-termini, both at the C-termini, one at the N-terminus and the other the C-terminus, and vice versa) yield comparable biotinylation9.
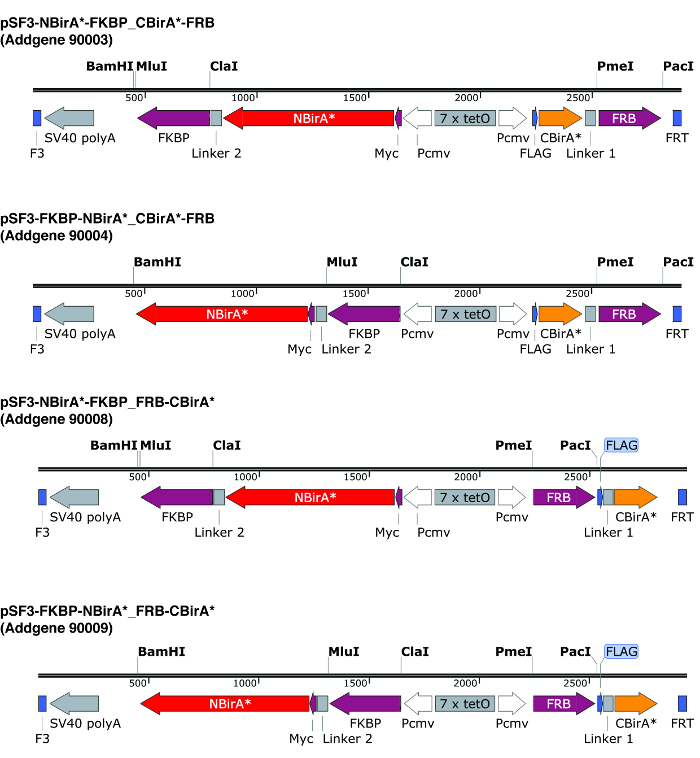
Design primers to amplify the ORFs of the two proteins of interest for cloning into the split-BioID plasmids. Pay attention that the translation frames are the same as the BirA* fragments. If using the plasmids described in Figure 2, use the restriction enzymes PmeI and PacI to construct the CBirA* fusion, and the enzymes ClaI and MluI for the NBirA* fusion. NOTE: The plasmids depicted in Figure 2 carry a bi-directional tet-responsive element flanked by F3/FRT recombination sites. This allows the regulated co-expression at roughly the same level of both fusion proteins from the same plasmid10, and the possibility to rapidly construct stable cell lines by recombination-mediated cassette exchange (RMCE). In these plasmids, NBirA* has a myc tag while CBirA* has a FLAG tag. The ORFs can of course also be cloned on individual plasmids with constitutive promoters. CRITICAL STEP: When testing two interacting proteins, it is recommended to compare NBirA* fused to protein 1 combined with CBirA* fused to protein 2, and NBirA* fused to protein 2 combined with CBirA* fused to protein 1. Indeed, it is frequently observed that one iteration works better than the other.
2. Cloning the ORFs of the Genes of Interest into the Split-BioID Plasmid
NOTE: In this example, two proteins that can be tagged at the N-terminus are considered. Four conditions will be tested and compared to non-transfected cells (Table 1).
Use polymerase chain reaction (PCR) to amplify the ORFs of the two proteins to be tested with the appropriate primers. Design primers that introduce the ClaI and MluI restriction sites for the NBirA* fusion proteins, and the PacI and PmeI sites for the CBirA* fusion proteins. NOTE: The PCR can be performed with any commercial, preferentially proofreading, DNA polymerase. Follow the standard protocol from the handbook and adapt it to the melting temperature of the primers and the length of the ORF to be amplified according to the manufacturer guidelines.
- Subclone the first ORF
- Digest both plasmid (about 2 μg) and PCR-amplified ORF1 (to be fused to NBirA*) with ClaI and MluI. Perform digestion reactions using 1 μL of each enzyme, mixed with the appropriate volume of DNA and reaction buffer in a total volume of 20 μL in a 1.5 mL tube for 1 h at 37 °C.
- Run both digestion samples on a 1% agarose-TAE DNA gel. Excise the bands corresponding to digested plasmid and ORF1 with a clean scalpel and transfer to 1.5 mL tubes.
- Purify both bands using a standard DNA extraction kit.
- Ligate digested plasmid and ORF1 using standard reagents.
- If using the ligation kit indicated in Table of Materials, ligate 100 ng of DNA containing three to five time molar excess insert over plasmid in a total volume of 4.5 μL. Add 5 μL of 2x T4 ligase buffer and 0.5 μL of T4 DNA ligase from the ligation kit. Perform the ligation reaction in a 1.5 mL tube for 10 min at room temperature (RT).
- Transform into standard DH-5α E. coli competent cells (prepared according to Inoue's method11).
- Mix 3 μL of the ligation reaction with 50 μL of competent cells in a 1.5 mL tube on ice and incubate for 30 min. Transfer the cells to a heat block set to 42 °C for 30-45 s and then incubate back on ice for 2 min. Add 250 μL of pre-warmed (37 °C) lysogeny broth (LB) medium and plate 100 μL of the cells on a pre-warmed (37 °C) ampicillin-containing LB-agar plate. Incubate the cells overnight at 37 °C.
- On the next day, pick four to six colonies and incubate them at 37 °C, 180 rpm, overnight in 3 mL of LB medium containing 100 μg⋅mL-1 ampicillin in a 15 mL tube.
- Isolate the plasmids from the picked colonies using a standard DNA MiniPrep kit.
- Verify the correctness of the plasmids by Sanger sequencing using the Cassette 2 reverse primer (Table 2).
Once a plasmid containing the first ORF has been identified, subclone the second ORF in that plasmid following the same steps as step 2.2 but using the PmeI and PacI enzymes.
Sequence the resulting plasmids using the Cassette 1 reverse primer (Table 2).
3. Testing of the Fusion Proteins
NOTE: The following instructions are for the dual inducible expression plasmids (Figure 2) and HeLa-11ht cells, a subclonal HeLa-CCL2 cell line, stably expressing the reverse tetracycline-controlled transcription activator rtTA-M2 and containing a locus for RMCE12. The growth medium for these cells is Dulbecco's Modified Eagle Medium (DMEM) containing 10% tetracycline-free fetal bovine serum (FBS). When using another cell type, exact seeding conditions and growth medium will need to be adapted.
- Transient transfection
- Seed cells at a concentration of 1 x 105 cells in 2 mL per well of a six-well plate the day before transfection and incubate the cells overnight at 37 °C, 5% CO2 in a cell culture incubator.
- On the day of transfection, remove the medium of each well and replace with 2 mL of fresh medium.
- Prepare the four transfection reactions according to Table 1. For each well to transfect, add 6 µg of polyethylenimine to 3 µg of plasmid DNA in a 1.5 mL sterile tube and fill to 500 µL with DMEM medium without serum.
- Incubate each transfection mix for at least 5 min at room temperature before adding drop wise to each well. Incubate the cells overnight at 37 °C, 5% CO2.
- Induction and proximity-labeling
- The day after transfection, remove the medium and replace with medium supplemented with biotin at 50 µM to stimulate biotinylation and doxycycline at 200 ng⋅mL−1 to induce the expression of the fusion proteins. Incubate the cells for at least 20 h at 37 °C, 5% CO2.
- To make a stock solution of biotin, dissolve biotin at 50 mg⋅mL-1 (correspond to ca. 200 mM) in 2 M ammonium hydroxide. Once it is completely dissolved, dilute it to 50 mM in 500 mM Hepes, pH 7.4, then adjust the pH to 7.4 with HCl. Aliquot and store the resulting 1,000x stock solution at -20 °C. Dissolve doxycycline at 10 mg⋅mL-1 in 70% ethanol and store in a screw cap microtube at -20 °C in the dark.
- Cell lysate preparation
- Wash the cells once with 1 mL of cold (4 °C) phosphate-buffered saline (PBS).
- To each well, add 100 µL of lysis buffer (50 mM Tris pH 7.4, 150 mM NaCl, 2 mM EDTA, 0.5% NP-40, 0.5 mM DTT and protease inhibitors).
- Harvest the cells with a cell scrapper and transfer to 1.5 mL tubes.
- Centrifuge the samples at 14,000 x g for 10 min at 4 °C to remove cell debris.
- Transfer the supernatants to fresh tubes and place on ice.
- Determine protein amounts with a Bradford assay.
- SDS-polyacrylamide gel electrophoresis (PAGE) and Western blotting
- Prepare a SDS-polyacrylamide gel.
- For each cleared lysate, prepare a PAGE sample of 30 µg (minimum 15 µg) protein in a total of 30 µL of SDS-loading buffer. Then load equal amounts of each sample (20 µL/well) on the SDS-polyacrylamide gel and proceed to electrophoresis.
- After electrophoresis, transfer the fractionated proteins to a low fluorescence PVDF blot membrane using any standard protocol. NOTE: For analyzing protein biotinylation, a 10 min transfer time with the "high MW" program of a fast transfer semi-dry Western blotting device usually works well.
- After the transfer, block the membrane in 5% dry milk in PBS for 30 min at RT.
- Incubate the membrane for 30–60 min at RT with fluorescent streptavidin conjugate diluted 1:15,000 in PBS containing 2% BSA and 0.1% Tween-20.
- Wash the membrane three times, each for 10 min, with PBS containing 0.1% Tween-20, and then once more with PBS.
- Scan the membrane on a fluorescence scanner imaging system. NOTE:A typical Western blot is shown on Figure 3. The above procedure describes a fluorescent-based detection but a luminescence-based detection using HRP-coupled streptavidin works equally well.
4. Split-BioID for Proteomics Studies
CRITICAL NOTE: For the final mass spectrometric analysis, all the following steps are to be performed in keratin-free conditions, all material and reagents should be as keratin-free as possible.
- Transient transfection
- The day before transfection, seed three to four 10 cm plates for each condition at a concentration of 8 x 105 cells in 10 mL per plate, and incubate the cells overnight at 37 °C, 5% CO2 in a cell culture incubator.
- On the next day, prepare a master transfection mix for each transfection condition: for three plates per condition, 36 µg of polyethylenimine, and 18 µg of plasmid DNA dissolved in 900 µL serum-free DMEM. Incubate each master mix for at least 5 min at RT.
- In the meantime, replace the medium of each dish with fresh medium, and then add 300 µL of the transfection mixtures drop wise to each plate.
- Incubate the cells overnight at 37 °C, 5% CO2.
- Induction and proximity-labeling
- The day after transfection, transfer the cells to 15 cm dishes. For each dish, remove the medium, wash the cells with 7 mL of PBS, add 1.5 mL of a trypsin-EDTA solution and incubate for 5 min at RT. Add 3.5 mL of growth medium to resuspend the cells and transfer the cell suspension to a 15 cm dish filled with 20 mL of growth medium supplemented with biotin at 50 µM to stimulate biotinylation and doxycycline at 200 ng⋅mL−1 (final concentrations) to induce the expression of the fusion proteins.
- Incubate the cells for at least 20 h at 37 °C, 5% CO2.
- Harvesting and storage of the cells
- Wash the cells twice with PBS, then add 1.5 mL of PBS to each plate and harvest the cells with a scrapper.
- Transfer the harvested cells corresponding to one condition to a 15 mL tube and harvest them by centrifugation at 1,200 x g, 5 min, 4 °C.
- Remove the supernatants and snap freeze the pellets in liquid nitrogen, then store at 80 °C until further processing. NOTE: Alternatively, cells can also be detached by trypsinization, harvested in growth medium, transferred to a 15 mL tube and washed three time with PBS before freezing.
- Preparation of cell lysates
- Resuspend the cell pellets in 1 mL of lysis buffer (50 mM Tris pH 7.4, 500 mM NaCl, 0.4% SDS, 5 mM EDTA, 1 mM DTT, 1x complete protease inhibitor) at RT. Pass the cells 10–20 times (five to ten strokes) through a 25 G needle.
- Sonicate the samples with a sonification device. NOTE: With the sonification device indicated in Table of Materials, the following program can be used: four cycles at high intensity, 30 s per cycle in a cold (4 °C) water bath. Any other sonification device is suitable but parameters might need to be adjusted accordingly.
- Add Triton X-100 to the recovered sonicated lysate to reach a final concentration of 2% (typically, add 100 μL of 20% Triton X-100 to 900 μL sonicated cell lysate), and then 2.3 mL of 50 mM Tris, pH = 7.4 per 1 mL of lysate to adjust the NaCl concentration to 150 mM before binding to streptavidin-coupled beads. Critical NOTE: Higher salt concentrations often result in much less efficient binding to the beads.
- Distribute the adjusted lysates in 1.5 mL tubes (ca. 1.1 mL in three tubes) and centrifuge them at 16,000 x g, 10 min, 4°C.
- Transfer the supernatants (ca. 3.2 mL) to a 15 mL tube, and keep 50–100 μL as INPUT material.
- Measure the concentration of each sample with a Bradford assay and use equivalent of 3 to 3.5 mg protein content for the streptavidin pulldown.
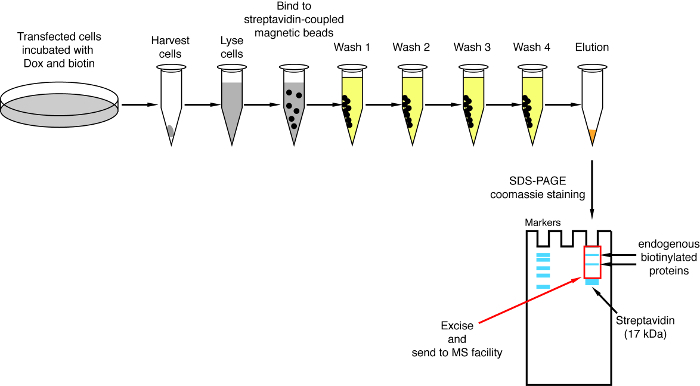
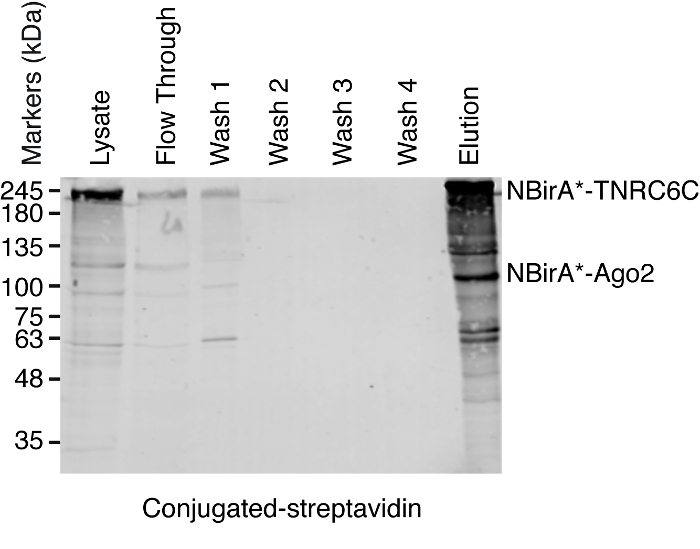
- Streptavidin pulldown NOTE: An overview of the pulldown procedure is shown on Figure 4. It is almost identical to the original protocol published by Roux and colleagues2. The first time the pulldown is performed, samples of flow through and wash 1–4 may be put aside for Western blot analysis to ensure the procedure worked properly (Figure 5).
- For each condition, transfer 200 μL of streptavidin-coupled magnetic beads suspension to a 1.5 mL tube. Place the tubes on a magnetic rack, wait until the beads stick to the side of the tubes (ca. 1 min) and remove the storage buffer.
- Wash the beads by gently mixing with 1 mL of equilibration buffer (50 mM Tris pH 7–4, 150 mM NaCl, 0.05% Triton X-100, and 1 mM DTT).
- Dispatch the equilibrated beads in equal amount in the necessary number of tubes (usually when starting with 3–3.5 mg protein content, the lysates from each condition can be dispatched in two to four 1.5 mL tubes) and place back on the magnetic rack.
- Remove the equilibration buffer and resuspend each set of beads with equal amounts of the corresponding cell lysates from step 4.4.7. Incubate overnight at 4 °C on a rotating wheel.
- On the next day, place the 1.5 mL tubes on a magnetic rack, wait until the beads stick to the side of the tubes and transfer the supernatants to a 15 mL tube labeled as Flow Through. NOTE: From now on, all the steps are performed at room temperature unless indicated otherwise.
- Resuspend the beads in each tube with 200 μL of wash buffer 1 (2% SDS in water), and combine each set of resuspended beads corresponding to one condition in 1.5 mL tubes.
- Wash the beads twice for 8 min on a rotation wheel with 1 mL of wash buffer 1.
- Wash the beads twice for 8 min on a rotation wheel with 1 mL of wash buffer 2 (50 mM HEPES pH 7.4, 1 mM EDTA, 500 mM NaCl, 1% Triton X-100, and 0.1% Na-deoxycholate).
- Wash the beads twice for 8 min on a rotation wheel with 1 mL of wash buffer 3 (10 mM Tris pH 8, 250 mM LiCl, 1 mM EDTA, 0.5% NP-40, and 0.5% Na-deoxycholate).
- Wash the beads twice for 8 min on a rotation wheel with 1 mL of wash buffer 4 (50 mM Tris pH 7.4, 50 mM NaCl, 0.1% NP-40).
- To make sure the wash buffer is completely removed after the last wash step, remove most of the supernatant, and then spin down the samples. Put them back on the magnetic rack, wait until the beads stick to the side of the tubes and then remove the remaining buffer.
- Add 30 μL of elution buffer (10 mM Tris pH 7.4, 2% SDS, 5% β-mercaptoethanol, and 2 mM Biotin) to the beads. Incubate at 98 °C for 15 min, then immediately remove the beads on a magnetic rack.
- Transfer the eluted sample to a fresh tube and store at -20 °C until further processing.
- SDS-PAGE and Western blotting NOTE: Prior to mass spectrometry analysis, it is recommended to assess the success of the biotinylation and pulldown by SDS-PAGE and Western blot. If no biotinylation pattern is observed one may consider that either dox or biotin was not added to the medium or that one of the two stock solutions is compromised.
- For each input sample, prepare a PAGE sample by mixing equal amounts of protein sample with the appropriate volume of 3x SDS-loading buffer in a total of 28 μL. Prepare PAGE samples by mixing 5 µL of each elution sample with 2.5 µL of 3x SDS-loading buffer.
- Load the samples (25 µL/well for inputs, 7 µL/well for elution samples) on an SDS-polyacrylamide gel. Proceed to electrophoresis and Western blotting as described in section 3.4.
- If performing the pulldown for the first time, prepare PAGE samples for each step of the pulldown (Input, flow through, Wash 1–4, elution) by mixing 20 µL of each sample (Input, flow through, Wash 1–4) with 10 µL of 3x SDS-loading buffer or 5 µL (elution) with 2.5 µL of 3x SDS-loading buffer and proceed as on step 4.6.4 (Figure 5).
- SDS-PAGE for MS analysis NOTE: To minimize potential keratin contamination, precast gels and commercial sample loading buffer may be used.
- Add 6.25 μL of 4x sample buffer to 18.75 μL of each elution sample and run the samples on a 4–20% precast SDS-gel until they migrate 2–3 cm into the gel.
- Stain the gel with colloidal Coomassie Brilliant Blue G250 staining13 in a 15 cm petri dish.
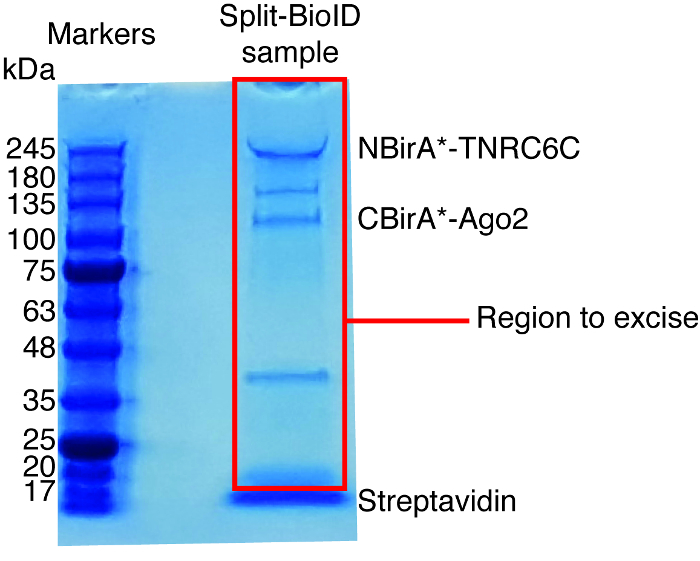
- Use a clean scalpel to excise the whole lanes for each sample, excluding the streptavidin band (running at ca. 17 kDa, Figure 6), and transfer the excised bands to 1.5 mL tubes.
- Send these samples to a proteomics facility for further analysis. NOTE: Typical MS results can be seen in reference 9 (Figure 3 and Figure 7, and supplementary tables). The whole datasets are available on the PRIDE repository (accession number PXD005005).
Representative Results
To illustrate how this method works, the open reading frames (ORFs) of the proteins Ago2, TNRC6C and Dicer (all involved in the miRNA-mediated gene silencing pathway) were cloned in split-BioID plasmids. Ago2 is known to interact with TNRC6C within a miRNA-induced silencing complex (miRISC) that represses translation and stimulate decay of target mRNAs14. Prior to assemble the miRISC, Ago2 interact with Dicer, the enzyme that produces mature miRNAs, within a complex in which it may get loaded with a miRNA15. Hence split-BioID was applied to either the Ago2/Dicer pair or the Ago2/TNRC6C pair. For each pair of tested proteins, Ago2 was either fused to NBirA* or CBirA* using our split-BioID plasmids (Figure 2), and Dicer and TNRC6C to the corresponding cognate BirA* fragment. In addition, each protein was fused to CBirA* and paired with an NBirA*-GFP fusion as a negative control. This results in testing four iterations for each pair of tested protein (Table 1).
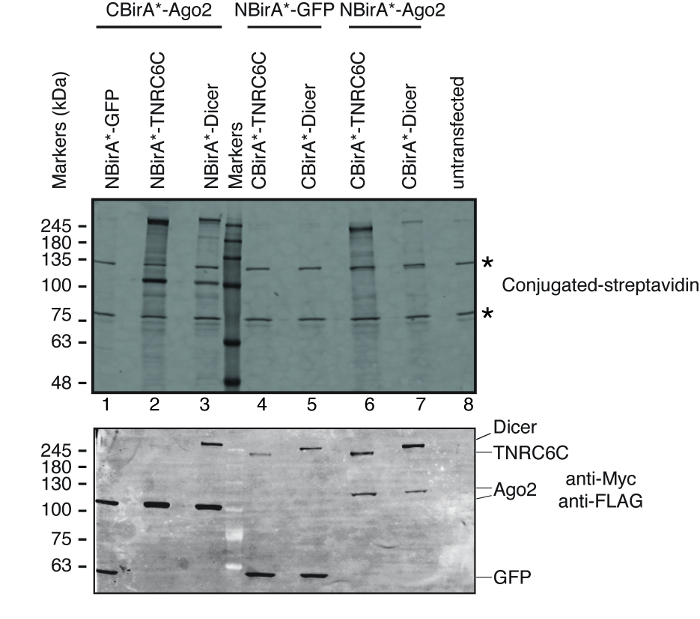
To test whether split-BioID is activated upon the interaction of the pair of tested proteins, we followed the scheme depicted on Figure 1. The plasmids were transiently transfected in a tet-system compatible HeLa cell line. The expression of the fusion proteins was induced with doxycycline (dox) and biotinylation was stimulated by adding excess biotin to the growth medium. Following a 20 h incubation time with dox and biotin, cells were lysed and analyzed by Western blotting using conjugated streptavidin to detect biotinylated proteins. In mammalian cells, two major bands are typically detected by the conjugated streptavidin in the untransfected sample (Figure 3, stars) and correspond to endogenously biotinylated proteins (most probably mitochondrial carboxylases). These two bands are present in all samples and can be conveniently used as internal loading controls, thus, the detection of a housekeeping protein to control the loading of equal protein amounts is superfluous. Typical for a BioID/split-BioID experiment, the additional major bands that can be observed are the fusion proteins that got self-biotinylated. Even if no other biotinylated protein is seen, detecting biotinylation of the fusion proteins at this stage already indicates that the two tested proteins interacted in the cells. In the experiment depicted on Figure 3, it is clear that having an NBirA*-Ago2 fusion protein paired with CBirA* fusions to TNRC6C or Dicer is more efficient than the opposite combinations in which CBirA*-Ago2 is paired to NBirA* fusions of the other two proteins (Figure 3, upper panel, compare the intensities of lanes 2-3 to lanes 6-7). Moreover, the activation was specific as none of the CBirA* fusions could activate the NBirA*-GFP control fusion protein to appreciable levels (Figure 3, compare lanes 1, 4-5 to lane 8 that corresponds to untransfected cells). Since in our plasmids, NBirA* has a myc tag and CBirA* has a FLAG tag (Figure 2), the expression levels of each fusion protein can be analyzed with antibodies against these two tags (Figure 3, bottom panel).
When interaction-induced biotinylation is observed, the experiment can be scaled up, and the biotinylated proteins isolated on streptavidin-coupled beads as indicated in paragraph 4 of the protocol (Figure 4). When performing the isolation the first time, all the steps of the purification may be analyzed by Western blotting (Figure 5). Typically, binding to the beads should be almost quantitative and virtually no leak through should be observed in the washes. Prior processing the samples for mass spectrometry, we recommend running a Western blot to ensure induced-biotinylation worked as expected and that the fusion proteins were expressed. The lack of expression of the fusion proteins is either due to poor transfection efficiency or faulty dox induction. If the fusion proteins were expressed but no biotinylation is observed, check if excess biotin (50 μM) was actually added to the medium and that the stock biotin is still active. When the eluted material is analyzed on a Coomassie-stained protein gel (Figure 6), typically, the strongest band to be observed runs at about 17 kDa and corresponds to monomeric streptavidin. Bands corresponding to the endogenous biotinylated proteins and the fusion proteins may also be observed. We typically excise the area of the sample lane above the streptavidin band up to the loading well (Figure 6). The excised band can be stored in a 1.5 mL tube and sent to a mass spectrometry facility. Alternatively, bound proteins may also be trypsin-digested on the streptavidin-coupled beads and the digested peptides eluted form the column. We routinely use the MaxQuant software16 (using mostly default parameters and adding lysine biotinylation as a possible post-translational modification, see reference 9 for more details and for typical MS results) to analyze the MS raw data and the Perseus suite17 for the subsequent statistical analysis, both are free software. Samples are typically run in three biological replicates. Using label-free quantification, specifically enriched proteins can be identified over control conditions. To filter for endogenously biotinylated proteins and for proteins that are non-specifically labeled by the BirA* enzyme, we only consider proteins that are significantly enriched over hits from six datasets generated with six unrelated proteins. In addition, we only consider hits that are enriched over a split-BioID dataset in which the NBirA* fusion proteins have been replaced by NBirA*-GFP. Other data analysis strategies have been proposed notably using stable isotope labeling with amino acids in cell culture (SILAC) for quantitative proteomics18. In addition, various strategies have been described for the direct isolation of biotinylated peptides using a streptavidin variant with weakened affinity to biotin18, special elution conditions using organic solvents19 or biotin-specific antibodies20,21. While not necessarily leading to the discovery of more proteins, the identification of biotinylation sites add more confidence as to the specificity of the hits and is useful when addressing the topology of an interaction.
Figure 1: Overview of the split-BioID procedure. Protein 1 interacts with protein 2 as part of Complex A, or with protein 3 as part of Complex B. To specifically probe the composition of Complex A, split-BioID can be applied to proteins 1 and 2. The photograph of the mass spectrometer is under a Creative Commons Attribution-Share Alike 3.0 Unported license and was downloaded from https://commons.wikimedia.org with file name of ThermoScientificOrbitrapElite.JPG. Please click here to view a larger version of this figure.
Figure 2: Expression cassettes of the split-BioID plasmids. We provide four plasmids to allow testing all combinations of NBirA* and CBirA* fusion proteins. The plasmids and complete maps are available at addgene.org under the indicated numbers. The plasmids have a tet-responsive element (7x tetO) and need to be used in a cell line that is compatible with the tet expression system. Also note that in all plasmids the ORFs of FKBP and FRB are fused to the NBirA* and CBirA* fragments respectively. These two proteins interact only in the presence of rapamycin and hence the plasmids can be used to quickly test the system in the presence or absence of this chemical9. The indicated restriction sites are unique. Please click here to view a larger version of this figure.
Figure 3: Typical Western blot for a split-BioID experiment. Upper panel: detection of biotinylated proteins with fluorescently labeled streptavidin. Lower panel: detection of the fusion proteins with anti-Myc and anti-FLAG antibodies. Two pairs of proteins were tested: Ago2/TNRC6C and Ago2/Dicer. In lanes 2 & 3, Ago2 was appended to the CBirA* fragment. In lanes 6 & 7, Ago2 was appended to the NBirA* fragment. No significant signal was observed when any of the three proteins were combined with NBirA*-GFP (lanes 1, 4-5). The stars indicate the bands corresponding to endogenously biotinylated proteins that can serve as internal loading controls. This figure is adapted from Figure 5B of Schopp et al.9 under a Creative Commons Attribution 4.0 International license. Please click here to view a larger version of this figure.
Figure 4: Overview of the streptavidin pulldown procedure. Major steps for the isolation of biotinylated proteins for mass spectrometry analysis are depicted. Please click here to view a larger version of this figure.
Figure 5: Typical Western blot for a streptavidin pulldown experiment. Equal volumes of each indicated sample were loaded on an SDS-polyacrylamide gel. Following Western blotting, biotinylated proteins were detected with HRP-coupled streptavidin. Bands corresponding to NBirA*-TNRC6C and CBirA*-Ago2 are indicated. Please click here to view a larger version of this figure.
Figure 6: Typical Coomassie-stained protein gel for mass spectrometry analysis. The eluted sample from streptavidin-coupled beads was loaded on a precast protein gel and run until the sample migrate 2-3 cm. The major band seen at about 17 kDa is streptavidin. The area directly above that band is excised and sent to a mass spectrometry facility. Bands corresponding to NBirA*-TNRC6C and CBirA*-Ago2 are indicated. Please click here to view a larger version of this figure.
| Transfection sample | Condition tested |
| 1 | NBirA*-protein1/CBirA*-protein2 |
| 2 | CBirA*-protein1/NBirA*-protein2 |
| 3 | NBirA*-GFP/CBirA*-protein1 |
| 4 | NBirA*-GFP/CBirA*-protein2 |
| 5 | no transfection |
Table 1: Typically tested conditions when applying split-BioID to two proteins.
| Sequencing primer | sequence |
| Cassette 1 reverse primer (CBirA* fusion) | TATACTTTCTAGAGAATAGGAAC |
| Cassette 2 reverse primer (NBirA* fusion) | GTGGTTTGTCCAAACTCATC |
Table 2: Sequencing primers for the split-BioID plasmids.
Discussion
The outlined procedure describes how to clone genes of interest into the split-BioID plasmids, how to test for interaction-induced biotinylation and how to isolate biotinylated proteins for mass spectrometry analysis. We describe here a procedure based on transient transfection. While the expression of the fusion proteins can be tuned by the amount of dox added to the medium, transient transfection may lead to non-homogenous protein expression with some cells that grossly overexpress the fusion proteins when compared to the endogenous counterparts. This may lead to distortions of the corresponding interactomes and to PPI that do not faithfully reflect the interactions that involve the endogenous proteins. It is thus generally advisable to construct stable cell lines once split-BioID has been established with the transient system. The plasmids are compatible with the Flp-mediated recombination system and place both genes of interest under the regulation of the same tet-responsive element. If needed, and when used with compatible mammalian cells, they allow the easy creation of stable inducible cell lines. For example, we use the HeLa-EM2-11 line that expresses the rtTA tetracycline-activated transcription activator and a unique targetable genomic locus from which tetracycline-mediated gene expression can be tightly regulated12. Using this cell line and Flp-mediated recombination, stable cell lines that contain only one copy of the transgene can be obtained within two-three weeks. Alternatively, one could also use current genome editing techniques to introduce the BirA* fragments in the native genomic loci of the genes of interest.
As in any assay that relies on tagging a protein, one needs to consider if the resulting fusion proteins are functional. Available data in which the proteins of interest were tagged (for example with GFP for imaging studies) and functionally tested are useful to decide if the BirA* fragments should be cloned upstream or downstream the genes of interest. If no such data are available, one should test N-terminally or C-terminally tagged proteins in a functional assay. For example, the activity of the fusion proteins can be tested in a cell line in which the endogenous protein has been knocked-out and compared to the wild type situation. If the proteins of interest tolerate both N- and C-terminal tags, both should be tested. Indeed, in BioID experiments, the orientation of the fusion protein can influence the efficiency of labeling22. In addition, we have observed that when applying split-BioID to a pair of proteins, which of the two proteins is appended to either the NBirA* or CBirA* fragment also influence the efficiency of labeling9. In the split-BioID plasmids, the 16 amino acid long glycine/serine rich linkers coupling the proteins of interest to the BirA* fragments were taken from another PCA23 and worked for us for all interacting proteins we have tested so far. However, one should consider that some protein pairs might work better with shorter or longer linkers. Of final note, another assay was described by the Bollen group24. In this assay, BirA* is split at another site (E140/Q141) than ours (E256/G257). We have tested both split-BioID flavors side-by-side and found that E256/G257, described in this protocol, leads to stronger re-activation when coupled to two interacting proteins9.
One general drawback of this method is the slow speed of labeling. Typically, 6 to 24 h incubation time with biotin is necessary to obtain appreciable biotinylation6, precluding the use of this technique for studying dynamic remodeling of protein complexes. While this assay partially addresses this caveat as it is only activated when two proteins interact, the slow speed of labeling preclude its use for studying response to very dynamic processes or to analyze short-lived proteins. The engineered peroxidase APEX2 is known to promote efficient labeling of proximal proteins within 1 min3. A PCA based on APEX2 might thus address the limitations of the slow labeling speed of BioID-derived assays. A proof-of-principle study described such a split-APEX2 assay25. However, although a homodimerizing protein was successfully biotinylated, whether the assay can also be used to label and identify proteins that assemble around a pair of interacting proteins remains to be demonstrated. Very recently, directed evolution was used to create TurboID and miniTurbo, two variants of BirA* with enhanced activity that allow much shorter labeling time windows, down to 10 min26. Adapting split-BioID to these new variants will further extend the use of this technique to a broader field of applications.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was financed by the German Research council (DFG) through the German excellence initiative (CellNetworks DFG-EXC 81) and a partial financing by the collaborative research centre SFB638.
References
- Scott DE, Bayly AR, Abell C, Skidmore J. Small molecules, big targets: drug discovery faces the protein-protein interaction challenge. Nat Rev Drug Discov. 2016;15(8):533–550. doi: 10.1038/nrd.2016.29. [DOI] [PubMed] [Google Scholar]
- Roux KJ, Kim DI, Raida M, Burke B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol. 2012;196(6):801–810. doi: 10.1083/jcb.201112098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam SS, et al. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat Methods. 2015;12(1):51–54. doi: 10.1038/nmeth.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HW, et al. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science. 2013;339(6125):1328–1331. doi: 10.1126/science.1230593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DI, et al. Probing nuclear pore complex architecture with proximity-dependent biotinylation. P Natl Acad Sci USA. 2014;111(24):E2453–E2461. doi: 10.1073/pnas.1406459111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DI, et al. An improved smaller biotin ligase for BioID proximity labeling. Mol Biol Cell. 2016;27(8):1188–1196. doi: 10.1091/mbc.E15-12-0844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JP, Tucholska M, Go C, Knight JD, Gingras AC. Proximity biotinylation and affinity purification are complementary approaches for the interactome mapping of chromatin-associated protein complexes. J Proteomics. 2015;118:81–94. doi: 10.1016/j.jprot.2014.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morriswood B, et al. Novel bilobe components in Trypanosoma brucei identified using proximity-dependent biotinylation. Eukaryot Cell. 2013;12(2):356–367. doi: 10.1128/EC.00326-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schopp IM, et al. Split-BioID a conditional proteomics approach to monitor the composition of spatiotemporally defined protein complexes. Nat Commun. 2017;8 doi: 10.1038/ncomms15690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béthune J, Artus-Revel CG, Filipowicz W. Kinetic analysis reveals successive steps leading to miRNA-mediated silencing in mammalian cells. EMBO Rep. 2012;13(8):716–723. doi: 10.1038/embor.2012.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue H, Nojima H, Okayama H. High efficiency transformation of Escherichia coli with plasmids. Gene. 1990;96(1):23–28. doi: 10.1016/0378-1119(90)90336-p. [DOI] [PubMed] [Google Scholar]
- Weidenfeld I, et al. Inducible expression of coding and inhibitory RNAs from retargetable genomic loci. Nucleic Acids Res. 2009;37(7):e50. doi: 10.1093/nar/gkp108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyballa N, Metzger S. Fast and sensitive colloidal coomassie G-250 staining for proteins in polyacrylamide gels. J Vis Exp. 2009. [DOI] [PMC free article] [PubMed]
- Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet. 2015;16(7):421–433. doi: 10.1038/nrg3965. [DOI] [PubMed] [Google Scholar]
- MacRae IJ, Ma E, Zhou M, Robinson CV, Doudna JA. In vitro reconstitution of the human RISC-loading complex. P Natl Acad Sci USA. 2008;105(2):512–517. doi: 10.1073/pnas.0710869105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26(12):1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Tyanova S, et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods. 2016;13(9):731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- Opitz N, et al. Capturing the Asc1p/Receptor for Activated C Kinase 1 (RACK1) Microenvironment at the Head Region of the 40S Ribosome with Quantitative BioID in Yeast. Mol Cell Proteomics. 2017;16(12):2199–2218. doi: 10.1074/mcp.M116.066654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackmull MT, et al. Landscape of nuclear transport receptor cargo specificity. Mol Syst Biol. 2017;13(12):962. doi: 10.15252/msb.20177608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DI, et al. BioSITe: A Method for Direct Detection and Quantitation of Site-Specific Biotinylation. J Proteome Res. 2017. [DOI] [PMC free article] [PubMed]
- Udeshi ND, et al. Antibodies to biotin enable large-scale detection of biotinylation sites on proteins. Nat Methods. 2017;14(12):1167–1170. doi: 10.1038/nmeth.4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapat C, et al. Cap-binding protein 4EHP effects translation silencing by microRNAs. P Natl Acad Sci USA. 2017;114(21):5425–5430. doi: 10.1073/pnas.1701488114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luker KE, et al. Kinetics of regulated protein-protein interactions revealed with firefly luciferase complementation imaging in cells and living animals. P Natl Acad Sci USA. 2004;101(33):12288–12293. doi: 10.1073/pnas.0404041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Munter S, et al. Split-BioID: a proximity biotinylation assay for dimerization-dependent protein interactions. FEBS Lett. 2017;591(2):415–424. doi: 10.1002/1873-3468.12548. [DOI] [PubMed] [Google Scholar]
- Xue M, et al. Optimizing the fragment complementation of APEX2 for detection of specific protein-protein interactions in live cells. Sci Rep. 2017;7(1):12039. doi: 10.1038/s41598-017-12365-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branon TC, et al. Directed evolution of TurboID for efficient proximity labeling in living cells and organisms. bioRxiv. 2017.