

Abstract
Traumatic brain injury (TBI) is a major clinical challenge with high morbidity and mortality. Despite decades of pre-clinical research, no proven therapies for TBI have been developed. This paper presents a novel method for pre-clinical neurotrauma research intended to complement existing pre-clinical models. It introduces human pathophysiology through the use of human induced pluripotent stem cell-derived neurons (hiPSCNs). It achieves loading pulse duration similar to the loading durations of clinical closed head impact injury. It employs a 96-well format that facilitates high throughput experiments and makes efficient use of expensive cells and culture reagents. Silicone membranes are first treated to remove neurotoxic uncured polymer and then bonded to commercial 96-well plate bodies to create stretchable 96-well plates. A custom-built device is used to indent some or all of the well bottoms from beneath, inducing equibiaxial mechanical strain that mechanically injures cells in culture in the wells. The relationship between indentation depth and mechanical strain is determined empirically using high speed videography of well bottoms during indentation. Cells, including hiPSCNs, can be cultured on these silicone membranes using modified versions of conventional cell culture protocols. Fluorescent microscopic images of cell cultures are acquired and analyzed after injury in a semi-automated fashion to quantify the level of injury in each well. The model presented is optimized for hiPSCNs but could in theory be applied to other cell types.
Keywords: Neuroscience, Issue 134, Stretch, neurotrauma, neuron, human induced pluripotent stem cell, silicone, cell culture, high content imaging
Introduction
TBI is a major cause of mortality and morbidity in the United States, causing around 52,000 deaths and 275,000 hospitalizations every year1. More than 30 clinical trials of candidate therapeutics for TBI have been conducted without a single success2. This uniform failure suggests that human-specific processes separate human TBI from the pathophysiology observed in commonly used pre-clinical rodent models.
The advent of hiPSCNs has created an opportunity to study neurotrauma in a human in vitro model. Drug screening with hiPSCN-based models may deliver results that are more predictive of clinical success than models employing rodent cells. Also, hiPSCNs can be genetically manipulated to isolate and study the effect of individual human genetic variants on pathology3.
The method described in this manuscript is designed to bring the unique advantages of hiPSCN-based disease modeling to neurotrauma. In vitro stretch injury models of neurotrauma are well established4,5,6 with primary rodent cells and human neural cancer cell lines. Most of these models generate stretch by pneumatically loading a silicone membrane. This approach is effective in a single well format but has proven difficult to scale up to a multi-well format7. As a result, there has never been a high throughput screen for agents to treat stretch injured neurons.
In this model, the membrane stretches due to indentation from underneath with a rigid indenter. This approach has been shown repeatedly to generate clinically relevant pathology in vitro in single well systems8,9,10. Our recent work has shown that it easily scales up to a 96-well format while maintaining pulse durations on the order of tens of milliseconds11, which is the time domain of closed head impact events12,13.
In summary, the key advantages of this in vitro injury model are the 96-well format, the use of hiPSCNs, and the clinically relevant time domain of the insult.
Protocol
1. Silicone Detoxification
Cut 254 µm thick, 30.48 cm x 30.48 cm silicone membranes into 7.5 cm x 11 cm rectangles using a razor blade and an acrylic template. 10 rectangular membranes can be made with each sheet. Save the paper that comes with the silicone.
Place the membranes in a tub of deionized (DI) water with glassware soap. One at a time, scrub the membranes vigorously with gloved fingertips for at least 20 s or until lathered.
Rinse the membranes under running DI water until the lathered soap is visibly removed. Lay the membranes on the paper (from their packaging) and autoclave on a gravity cycle.
- Soak each silicone membrane in 250 mL of 70% ethanol per membrane on an orbital shaker at 60 rpm for 24 h. Use a container made from a material that does not react with ethanol, such as polypropylene.
- If more than one membrane is placed in a single bin, or if the membranes stick to the bottom of the bin, separate the membranes from their environment with plastic pipette tips and pipette tip racks.
Transfer the silicone membranes with gloved hands to 250 mL of DI water per membrane and soak on the orbital shaker for 48 h at 60 rpm.
Lay the membranes back on the paper and place in a 93 °C glassware oven for 4 h.
Store the membranes on the paper, in a clean and dry place, covered with another sheet of paper to protect them from dust.
2. Plate Fabrication
Plasma treat a 96-well plate top with a 200 W plasma cleaner (see Table of Materials) for 60 s on high power, placing it bottom-side-up inside the plasma cleaner. Check for a purple glow visible through the window of the chamber, which indicates that an effective plasma has formed. This bonding technique is adapted from Sunkara et al.14
Within 60 s, place the plate top in 200 mL of 1.5% (3-Aminopropyl) triethoxysilane (APTES) in DI water for 20 min, bottom-side-down. This solution is unstable: prepare it no more than 60 s before introducing the plate. CAUTION: Work with APTES in a fume hood.
Plasma treat an extracted silicone membrane in the plasma cleaner for 60 s on high power. Time the plasma treatment such that it finishes no more than 5 min before the end of the 20 min period in step 2.2.
Use forceps to position the plasma treated membrane on top of a 7.5 x 11 cm2 parchment paper rectangle. Align a 7.5 cm x 11 cm x 0.5 cm aluminum slab on the lower portion of the plate fabrication clamp. Align the silicone membrane and parchment paper on the aluminum slab using forceps. NOTE: Computer-aided design (CAD) files for this device are provided in the supplemental materials as a supplementary code file 'Press Die - Generic 3D.STEP'. The associated bill of materials is supplied in Supplementary Table 1: Custom Built Devices - BOM.xlsx.
Remove the plate tops from the APTES bath and shake off excess solution. Dip into a 200 mL DI water bath for 5 s and shake off excess water. Dip into a different 200 mL DI water bath for 5 s and shake off excess water. Replace the water in these baths before dipping another plate top.
Use compressed air to completely dry the plate top.
Place the plate top in the upper portion of the plate fabrication clamp. Gently close the plate fabrication clamp to press the plate top and silicone together. Clamp for at least 1 h.
Allow the plate to cure for 24 h at room temperature before use, protected from dust.
3. Stretching a Plate
- Clean the indenters. NOTE: See Supplementary Figure 1.
- Prepare a 60 W bath-style sonicator with DI water at room temperature.
- Support the indenter block above the sonicator bath, inverted so that only the tops of the indenters are submerged to a depth of at least 1 mm. Sonicate the indenters for 8 min at 42 kHz.
- Blow dry the indenters with compressed air.
- Align the indenter block.
- Position a camera with a live feed above the stretching device so that it is looking down on the indenter array. Focus on the tops of the indenters. NOTE: The setup described in Section 4, "Characterizing Membrane Stretch," is one possible camera setup for this task.
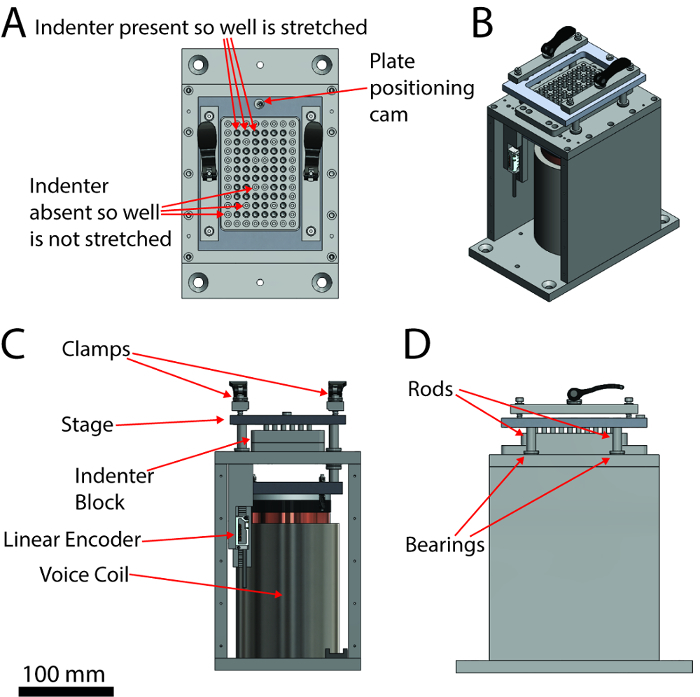
- Secure a plate on the stage of the injury device using the stage clamps (Figure 1) and place a dome light over the device. NOTE: See the Table of Materials for the instrument control software.
- Launch the instrument control software by clicking on the software icon on the desktop of the computer that is controlling the injury device. Run the "in_vitro_neurotrauma.lvproj" project by clicking on it in the launch window. From the project window, launch the motion control and position tracker virtual instruments (VIs), which are named 'motion_control.vi' and 'position_tracker.vi', respectively, by double-clicking on them.
- Close the cage surrounding the injury device. Press the 'arrow' button in the top left corner of the motion control VI to run the VI and click 'Near Bottom' to lower the stage to within 2 mm of contacting the indenters. Click the 'Stop' button to stop the VI. Caution: Keep hands clear of the stretcher when manipulating the camera in the cage. The cage should be fitted with a door switch that delivers power to the stretching device only when the cage door is closed.
- In the project window, right click on 'Axis 1'. Click on 'Interactive Test Panel'. In the window that opens, set the step size to 50 µm by entering '500' units in the 'Target Position' field.
- Click the green 'go' button at the bottom of the 'Interactive Test Panel' window repeatedly until the plate first makes contact with any indenters. Check for contact on the live image displayed by the camera. Note the vertical stage position reported in the top left of the 'Interactive Test Panel' window; this is the first contact position.
- Lower further (as described in step 3.2.6) until every well has made contact with the indenters. If necessary, move the camera to see the whole plate and move the stage up (by specifying a negative 'Target Position') and down. Note the vertical stage position reported in the top left of the window when all posts are in contact (this is the full contact position).
- Note the difference between the first contact and full contact positions. Close the 'Interactive Test Panel'. Run the 'motion control VI' (see step 3.2.4) and click on 'Top' to raise the stage. Stop the motion control VI with the 'Stop' button.
- Once the stage is at the top of its travel, open the door to deactivate the device. Adjust the set screws on the corners of the indenter block. Loosen the set screw on the corner that made contact first, to lower it and tighten the opposite screw to raise the corner that made contact last.
- Repeat the process of lowering the stage until the plate contacts the indenters, inspecting the contact images for tilt, raising the stage, and adjusting (step 3.2.9) the indenter block. When the stage and block are aligned, all indenters will make contact at once.
- Open the door to deactivate the device. Insert the tie-down screws into their holes on the indenter block and tighten them. Confirm that the block is level with the tie-down screws in place (steps 3.2.4-3.2.8). Take note of the stage position reported by the 'Interactive Test Panel' when the plate makes contact. This will be the zero position for indentation experiments.
- Lubricate the indenters.
- If the indenter block has just been set up and has not yet been lubricated, clean a solid rubber pad (7.5 cm x 11 cm x 1.5 mm) and a soft, closed-cell foam rubber pad (7.5 cm x 11 cm x 3 mm) with an ethanol-soaked lab wipe. Allow them to air dry.
- Soak a lab wipe in corn oil and spread it on the solid rubber pad to a dull shine.
- Place the foam pad onto the solid rubber pad to transfer oil to the foam pad. Place a 7.5 cm x 11 cm x 0.5 cm aluminum slab on top of the foam pad and load it with a ballast weight of approximately 360 g (e.g., 6 conical tubes loaded with 45 mL of water each) to ensure consistent transfer of oil from the solid rubber pad to the foam pad. Allow 10 s for the oil to transfer to the foam rubber pad.
- Move the foam rubber pad onto the indenter array. Place the aluminum slab and the ballast weight on top of it to ensure consistent transfer of the oil to the tips of the indenters. Allow 10 s for the oil to transfer to the indenters.
- If no plate has been stretched since the indenter block was set up and lubricated for the first time, stretch a test plate before beginning the experiment. Note: This will prevent any inconsistency between the first stretch and subsequent stretches from confounding the experiment. For subsequent stretches, repeat only steps 3.3.2-3.3.5 before each stretch.
- Stretch a plate.
- Lubricate the indenters as described in step 3.3.
- Secure the plate on the stage of the injury device with the stage clamps. If sterility is required, adjust the position of the lid to secure the plate without exposing the culture surface to room air.
- Lower the stage to the zero-position using the 'motion control VI' (see steps 3.2.4-3.2.6); zero-position is determined during step 3.2.
- Change the file name in the "file path" field in the 'position tracking VI' to a unique file name, then run it with the 'arrow' button in the top left corner of that window.
- Set the depth and duration of indentation (typically 1-4 mm and 30 ms, respectively, maximum depth 5 mm, minimum duration 15 ms) in the 'Injury (mm)' and 'Injury Duration (ms)' fields in the 'movement control panel VI' by clicking on the fields and typing in the desired values.
- Run the 'movement control panel VI' and click on 'Injure' to indent the plate.
- Move the stage up to the top of its travel by clicking on 'Top'. Then stop the VI with the 'Stop' button and deactivate the injury device by opening the door.
- Inspect the displacement history of the stage presented in the 'position tracker VI' to confirm that the specified maximum displacement was applied. Right click on the graph and click on 'export to Excel'. Click on 'File| Save' as to save the data.
4. Characterizing Membrane Stretch
Paint the recess at the top of each indenter white to provide a high contrast background for high speed videography. Caution: Do not paint the rims of the indenters where they make contact with plates.
3D print with poly(lactic acid) (PLA), or otherwise fabricate, a cylindrical stamp with which to make a dot in each well. Make the cylinder 5.9 mm in diameter and 12.2 mm high, with a cylindrical protrusion 1.5 mm diameter and 1.0 mm high centered on top. NOTE: A 3D printable model 'stamp.STL' is available as a supplementary file.
Plasma treat the plate right-side-up to activate the silicone cell culture surface in the wells for 60 s, as mentioned in step 2.1.
Place the plate on a rubber pad or other soft surface. Prime the small protrusion on the stamp (see step 4.2) with ink from a permanent marker pen. Insert the stamp into the well to be tested and tap to ensure good transfer of ink. Prime the stamp before every well.
Align the indenter block and lubricate the indenters of the injury device (see steps 3.2-3.3). Clamp the plate onto the stage of the injury device. Position a bright diffuse axial light above the injury device.
- Set up a high-speed camera on a boom stand over the injury device, facing straight downward, with the lens set to the smallest numbered f-stop, and turn it on. Launch the camera software on the computer connected to the camera. In the frame rate drop down menu, select 2,000 frames per second, and in the shutter drop down menu, select the fastest exposure time which yields high contrast images. Center over the dotted wells.
- Position the camera so that the field of view contains 12 wells in a 3 x 4 grid. NOTE: This field of view offers an optimal compromise between throughput and resolution for a 1,280 × 1,024 image.
Lower the plate to the zero-point (see steps 3.2.4-3.2.6). One-click the record button on the camera software so that it reads 'trigger in'. Initiate the position tracker VI (step 3.4.4).
Turn on the bright diffuse axial light. Indent the plate as described in step 3.4.6.
Turn off the bright diffuse axial light.
- On the camera control computer, find the 30-40 ms window of the recording in which the indentation occurs: drag the beginning and end arrows in the high-speed video play bar in the camera software. Click Save, set the name in the 'File Name' field, select 'TIFF' in the 'Format' field, and click on 'Save'.
- Look through the .TIF-files for images of the least stretched (beginning) and most stretched (peak indentation) states.
- To measure the height and width of the dots in both images, use Fiji to open the images of the least and peak stretch side-by-side. Using the default rectangular selection tool, click and drag to draw a box around a dot in the least stretch image. Measure the height and width with 'Analyze| Measure'.
- Repeat for the dot in the same well on the peak stretch image. Repeat for the rest of the wells.
Calculate the Lagrangian strain in the x and y directions as follows:
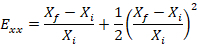
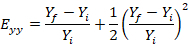 NOTE: Here, Exx is the Lagrangian strain in the x direction, Eyy is the Lagrangian strain in the y direction, X is the width of the dot, Y is the height of the dot, f denotes the final image (i.e., the peak indentation image), and i denotes the initial image (i.e., the pre-indentation image). Average the two values to determine the strain in that well.
NOTE: Here, Exx is the Lagrangian strain in the x direction, Eyy is the Lagrangian strain in the y direction, X is the width of the dot, Y is the height of the dot, f denotes the final image (i.e., the peak indentation image), and i denotes the initial image (i.e., the pre-indentation image). Average the two values to determine the strain in that well.
5. Plating the Cultured Cells
Autoclave the bins and ballast weights that will be used for sterilization.
Plasma treat the silicone-bottomed plates right-side-up for 60 s (see step 2.1). Immediately submerge the plates in sterile bins containing 70% ethanol for 15 min.
Submerge the plates in sterile phosphate buffered saline (PBS) in separate sterile bins for 30 min.
Aspirate the PBS from the wells. Only dry one plate at a time to prevent the wells from losing the plasma treatment. Caution: Silicone bottomed plates provide less tactile feedback than rigid plates when pipetting and are more likely to block the tip of a pipette.
Add 100 µL of 0.1 mg/mL poly-L-ornithine (PLO) per well to the sterilized plates. Incubate at room temperature for 1 h.
Rinse the wells twice with 100 µL sterile PBS, leaving the second wash on the wells until the cell suspension is ready.
Remove the vial of hiPSCNs from liquid nitrogen storage using safety gloves and place on dry ice. Quickly transport the vial to a 37 °C water bath and thaw for exactly 3 min. Do not swirl the vial.
In a sterile hood, gently transfer the contents of the vial to a 50 mL conical tube using a 1 mL serological pipette.
Rinse the empty cryogenic vial with 1 mL of room temperature complete maintenance medium (media base + supplement).
Transfer the 1 mL of media into the 50-mL tube drop-wise at about one drop per second. Gently swirl the tube while adding. Add 8 mL of room temperature complete maintenance medium to the 50 mL tube at about 2 drops/s.
Cap the tube and invert 2-3 times. Count the cells with a hemocytometer. Compute the volume of the additional media required to dilute the cell suspension to 225,000 cells/mL.
Gently pipette the amount of media (calculated above) into the tube of cell suspension using a 25 mL serological pipette.
Add 10 µL of 1 mg/mL laminin stock per 1 mL of cell suspension with a 1,000 µL micropipette to achieve 10 µg/mL of laminin in the cell suspension. Aspirate up and down once with the tip used for laminin, then cap the tube and invert the tube once.
Aspirate the PBS from the plates, one plate at a time. Use a multichannel pipette to add 100 µL of the hiPSCN cell suspension to each well. The wells have a culture area of 0.33 cm2, so the cell density per area is 67,500 cells/cm2.
Rest the plates at room temperature for 15 min after seeding to promote attachment. To avoid fan vibrations of sterile culture hood, place the covered plates on the lab bench. Incubate the cultures at 37 °C with 5% CO2.
Perform a full media change at 24 h, refilling the wells with 200 µL of complete maintenance media. Perform a half media change of 100 µL/well every 2-3 days.
6. Injuring Cultures
Align the indenter block and find the zero position as described in step 3.2. Lubricate the indenters as described in step 3.3.
Set the filename for the displacement history in the 'position tracker VI' (step 3.4.4). Set the injury parameters in the 'motion control VI' (step 3.4.5).
Take the plate to be injured out of the incubator and clamp it onto the stage. Adjust the position of the lid to secure the plate without exposing the cultures to room air.
Lower the plate to the zero-point (steps 3.2.4-3.2.6) using the 'motion control VI'. Start the 'position tracker VI' (step 3.4.4).
Use the movement control VI to indent the plate (as described in step 3.4.6). For sham stretches, skip only this step.
Return the stage to the top of its range of motion (see step 3.4.7). Return the plate to the incubator.
Inspect and save the displacement trace (as in step 3.4.8).
7. Microscopy
Prepare a 10x staining solution with 2 µg/mL Hoechst 33342 and 5 µg/mL Calcein AM in maintenance media.
Stain each well with a 20 µL spike of 10x staining solution.
Incubate for 15 min with stain at 37 °C.
Acquire wide field fluorescent images. Use conventional FITC and DAPI filter sets to image the Calcein AM and Hoechst 33342 signals, respectively. If the multi-well imaging sequence will take more than 10 min, enclose the plate in a stage top incubator to maintain the health of the cultures. NOTE: A 10X, 0.30 NA lens provides sufficient detail for determination of cell viability and morphology. Adjust gains to ensure good visualization of the neurites, even if this causes some saturation of the much brighter soma.
Segment live cell images in the green Calcein AM channel to identify soma and neurites. Use nuclear images in the blue Hoechst 33342 channel to assist with the identification of the soma.
Representative Results
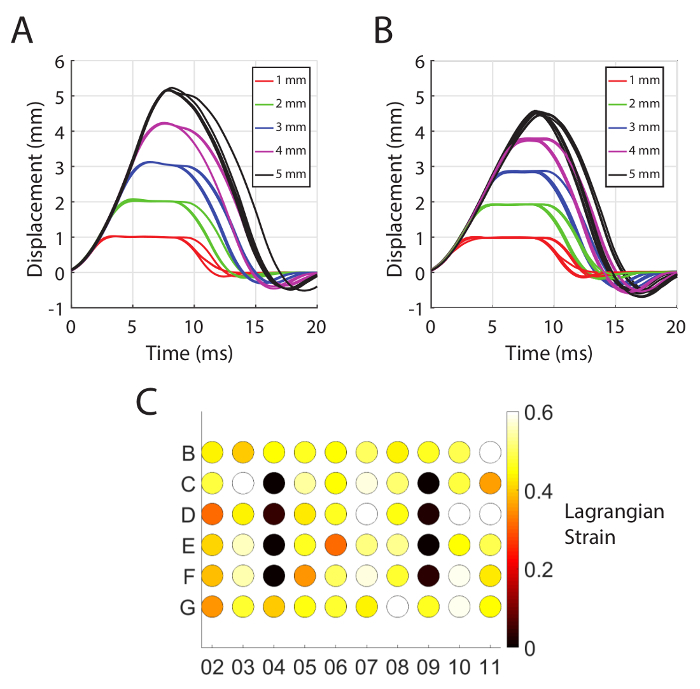
The stretcher device is capable of moving the stage repeatably with pulse durations as short as 10-15 ms depending on the amplitude of the pulse (Figure 2A). The pulse amplitudes are highly repeatable, but the pulse duration varies by approximately 1 ms between repetitions. The actual pulse amplitude diverges from the prescribed pulse amplitude when a large number of wells are loaded, and the prescribed amplitude is high (see Figure 2B). As the amplitude of stage displacement is increased beyond 3 mm, the actual displacement amplitude increasingly falls short of the prescribed displacement amplitude (see Figure 2B). Careful alignment of the post block eliminates any trend in the membrane strain across rows or columns (Figure 2C). At the 3.5 mm prescribed stage displacement amplitude (3.3 mm actual displacement amplitude) with 52 wells indented, the mean Lagrangian strain across all well locations was 0.451 (standard deviation of means for all locations = 0.051, mean of standard deviations for all locations = 0.065, n = 5 measurements per well). These results are presented here for completeness although some of them have already been reported11.
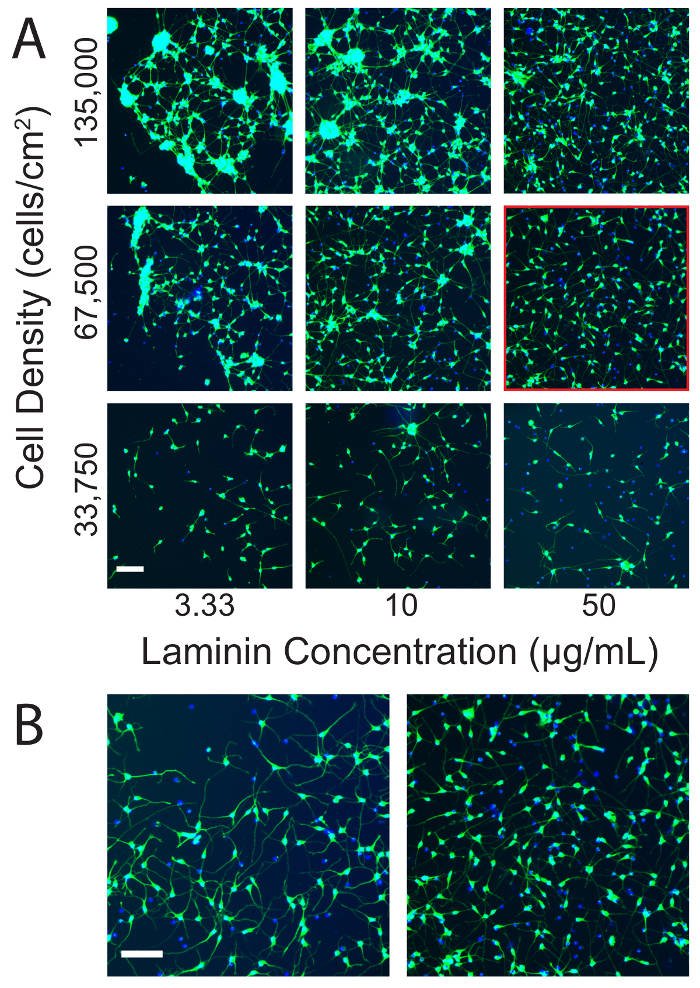
An optimal, uninjured culture will have few if any clumps of more than 5 cells. Neurites will be individual, long, slender, and curved with little or no sign of tension or beading (Figure 3A). Under ideal conditions, the viability of the cultures should closely approach the viability specified in the manufacturer's data sheet (typically 60-70%) and cultures on silicone should resemble those maintained on conventional rigid culture substrates (Figure 3B). Neurites may or may not be visible on a low power bright field microscope. Laminin concentration and cell density both influence cultures at baseline and after injury. Increasing the cell density increased the number and size of clumps that formed in culture. Increasing the laminin concentration often counteracted this effect (Figure 3A). However, increasing the laminin concentration too much blunted the sensitivity of the cultures to injury (Figure 4). The optimal laminin concentration for uninjured cultures was 50 µg/mL of laminin (Figure 3), but the optimal separation between the sham and stretch injured populations was obtained at 10 µg/mL of laminin (Figure 4). Higher laminin concentrations reduced the sensitivity of the cultures to injury at short time points (Figure 4), but also improved baseline cell viability at longer time points (e.g., 7 days). In summary, it is worthwhile to optimize the laminin concentration for each experimental scenario.
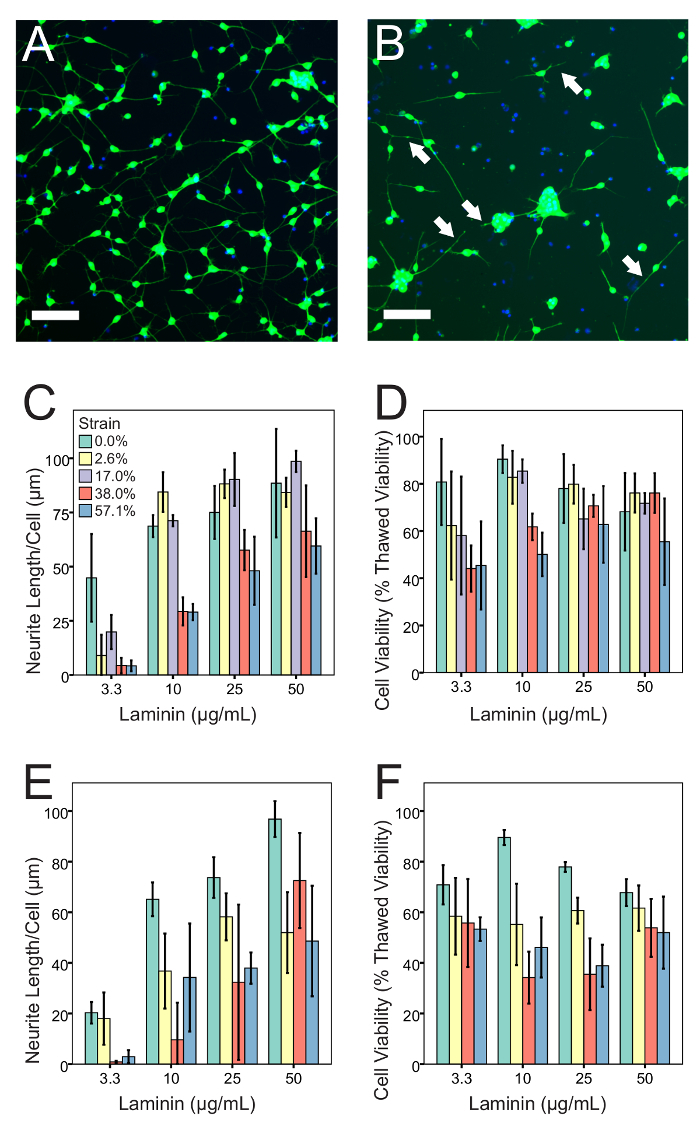
Membrane strain, post-injury imaging time point, laminin concentration, and cell density all exerted a highly statistically significant main effect on the neurite length per cell (ANOVA, p <0.001). The effect of membrane strain on neurite length per cell had highly statistically significant interactions with post-injury imaging time point and laminin concentration (ANOVA, p <0.001) and a statistically significant interaction with cell density (ANOVA, p <0.05). Similarly, membrane strain, post-injury imaging time point, cell density, and laminin concentration all exerted a highly statistically significant main effect on cell viability (ANOVA, p <0.001). The effect of the membrane strain on cell viability had a highly statistically significant interaction with the post-injury imaging time point (ANOVA, p <0.001) and a statistically significant interaction with the cell density (ANOVA, p <0.05). These results prove that timing, cell density, and laminin concentration exert an important influence on the relationship between the applied insult and experimental outcomes, so each should be optimized carefully.
Low cell viability and beaded neurites, along with stunted neurite growth, indicate toxic culture conditions that can arise from improperly prepared silicone. Skipping or shortening the water soak or the oven dry can leave absorbed ethanol or water in the membrane, respectively, which can diffuse into the media and stress the cells. Well-injured cultures will have reduced cell viability, shortened or missing neurites, beaded neurites, and neurites that look taut or tensioned. Injury may induce clumping in cell cultures that were well-dispersed pre-injury. Large clumps can confound the morphological analysis. For morphological analysis, the injury level should be tuned such that noticeable changes occur, but cells are still present with some neurites.
Figure 1: A labeled schematic of the injury device. (A) Top view, (B) isometric view, (C) front view, (D) right side view. The scale bar applies to the orthographic views (A, B and D). Please click here to view a larger version of this figure.
Figure 2: Kinematics of the mechanical insult. (A) The stage displacement histories over 5 pulses at a range of prescribed amplitudes (prescribed amplitudes are listed in the legend) when no wells are loaded. (B) The stage displacement histories over 10 pulses at a range of prescribed amplitudes (prescribed amplitudes are listed in the legend) when 52 wells are loaded. (C) The average strain in each well with stage displacement amplitude of 3.3 mm (n = 5 measurements per well, average standard error per well = 0.029). Note that C4-F4 and C9-F9 are unstretched control wells. This figure has been modified from Sherman et al.11 Please click here to view a larger version of this figure.
Figure 3: Optimization of culture conditions on silicone. (A) The effect of varying cell density and laminin concentration on hiPSCN cultures on silicone. Clumping increases with increasing cell density and decreasing laminin concentration. Cell density and laminin concentration must be optimized to achieve mono-dispersed cultures. Mono-dispersed cultures are less vulnerable to artifacts during quantification. Note that the dynamic range has been adjusted to optimize visualization of the neurites. As a consequence, the much brighter soma are saturated. This presentation is preferred to the alternative of optimizing the dynamic range with respect to the soma, which renders the much dimmer neurites almost invisible. The condition highlighted by the red square was deemed to be optimal for in vitro stretch injury experiments. (B) Under optimal conditions, cultures on silicone membranes appear similar to cultures on conventional rigid substrates. The left panel shows hiPSCNs cultured at 33,750 cells/cm2 with 3.3 µg/mL of laminin on a conventional, rigid 96-well plate (the cell culture substrate is a tissue culture treated cyclic olefin co-polymer). The right panel reproduces the panel outlined in red from (A). Scale bars = 100 µm. Please click here to view a larger version of this figure.
Figure 4: The injury phenotype and its dependence on laminin concentration. (A) Healthy culture, using 10 µg/mL laminin and 67,500 cells/cm2. Neurites are long with no beads. There are few dead nuclei, and few clumps. (B) Culture using the same culture conditions, injured with 57% peak strain and imaged after 4 h. Neurites are shortened or missing, and some have beads (indicated by arrows). There are fewer Calcein AM-positive cells and more Calcein AM-negative (i.e., dead) nuclei. Injury has increased clumping among the surviving cells. (C) 4 h after injury, neurite length per cell declines with increasing strain in a manner that depends on the laminin concentration. (D) 4 h after injury, cell viability declines with increasing strain in a manner that depends on the laminin concentration. (E) 24 h after injury, neurite length per cell declines with increasing strain in a manner that depends on the laminin concentration. (F) 24 h after injury, cell viability declines with increasing strain in a manner that depends on the laminin concentration. (n = 4 per bar, error bars are ± 1 standard deviation, Scale bars = 100 µm). Strain values are deduced from stage displacement using data from a prior publication by Sherman et al.11 Please click here to view a larger version of this figure.
Supplementary Figure 1: Technical drawing of the indenter. Please click here to download this figure.
Supplementary Table 1: Custom Built Devices. Please click here to download this table.
Supplementary Table 2: 96 Well Plate-loader Pinout Wiring Diagram. Please click here to download this table.
Supplementary Code File 1: Computer-aided design drawings of the injury device. Please click here to download this file.
Supplementary Code File 2: Computer-aided design drawings of the plate fabrication clamp. Please click here to download this file.
Supplementary Code File 3: 3D representation of the stamp geometry, suitable for use with a 3D printer. Please click here to download this file.
Supplementary Code File 4: SubVI for MuStLiMo_si_initialize.vi, which is a SubVI for motion_control.vi. Converts entries in dialog boxes into parameters for motion. Please click here to download this file.
Supplementary Code File 5: SubVI for Multiple Straight Line Moves_simplified.vi, which is a SubVI for motion_control.vi. Converts entries in dialog boxes into parameters for motion. Please click here to download this file.
Supplementary Code File 6: SubVI for position_tracker.vi. Counter tracks displacement input from linear encoder. Please click here to download this file.
Supplementary Code File 7: Base LabVIEW Project. Please click here to download this file.
Supplementary Code File 8: Top level VI that moves the device. Please click here to download this file.
Supplementary Code File 9: SubVI for motion_control.vi. Executes the rapid displacement that stretches the plate. Please click here to download this file.
Supplementary Code File 10: SubVI for motion_control.vi. Executes the slow displacement that moves the stage. Please click here to download this file.
Supplementary Code File 11: SubVI for motion_control.vi. Plots a (usually undersampled) displacement history in the motion_control.vi control panel. Please click here to download this file.
Supplementary Code File 12: Top level VI that records the displacement history. Please click here to download this file.
Supplementary Code File 13: Contains Variable2, which communicates between motion_control.vi and position_tracker.vi. Please click here to download this file.
Supplementary Code File 14: Schematic for printed circuit board. Please click here to download this file.
Supplementary Code File 15: Layout for printed circuit board. Please click here to download this file.
Discussion
The key to obtaining a consistent, biofidelic phenotype in this model is applying a consistent biofidelic mechanical insult. This model can generate pulse durations as short as 10-15 ms, which are similar to the pulse durations for human head impacts according to cadaveric experiments12,13. The consistency of this insult depends on the alignment of the plate with the indenter block and consistent lubrication of the indenters. When the indenter block is well aligned, there is no trend in the applied strain across rows or columns (Figure 2C). A thin layer of lubricant typically creates less friction than a thick layer, and viscous greases are not recommended because they foul the silicone and obstruct the passage of light during microscopy. The actual stage displacement amplitude can fall substantially short of the prescribed displacement amplitude when many indenters are used, and the prescribed stage displacement amplitude is large (> 3 mm). However, while the actual displacement is less than the prescribed displacement at large amplitudes, it remains repeatable (Figure 2B). Therefore, large, actual displacements amplitudes can be reliably obtained by entering a prescribed value in excess of the desired value. Displacement amplitude matters only because it is an easily recorded proxy for the peak membrane strain, which directly measures the mechanical insult that induces pathology. Therefore, the procedure described for determining membrane strain from stage displacement is critical. This process should be repeated if any major changes are made to the system that affect the interaction between the plate and the indenters, for example if different diameter indenters, different indenter materials or coatings, or different types of silicone bottomed plate are used. The process of realigning the indenter block and determining the zero position should be repeated at the start of each experiment. A schematic of the stretching device is shown in Figure 1. CAD models required to reproduce the device are provided as supplemental materials: 'Injury Device - FULL ASSEMBLY - Generic 3D.STEP'; the associated bill of materials provided as 'Supplementary Table 1: Custom Built Devices - BOM.xlsx. Also see Supplementary Table 2 96 Well Plate _loader - Pinout Wiring Diagram.xlsx, which describes the cabling connections that connect various components of the systems. 'Interconnector_circuit_board.dip' describes a circuit board that interconnects the cables.
If the device is deactivated with the stage near the middle of its travel, the stage will move after the power is cut off because it is spring-loaded. When the power is restored, the feedback loop will detect a large difference between the last known prescribed position and the actual position. This will cause the stage to move suddenly to the position it was in when the device was deactivated. This sudden motion can cause errors in the output of the encoder, so care should be taken to deactivate the device only when it is in its unpowered resting position at the top of its travel.
The fabrication clamp is designed to bring the plate body and silicone bottom together in a manner that allows optimal bonding. To this end, there are three key features in the design presented in the supplemental file 'Press Die - Generic 3D.STEP'. First, the clamp plate body holder is parallel to the silicone bottom. If this is properly built, it will require no adjustment after the initial setup. Second, the layer of foam rubber in the clamp provides a small amount of compliance under the plate, as a completely rigid system would theoretically experience a sudden increase from zero clamping force to infinite clamping force when the clamp was closed. The position of the crossbar and set screw of the clamp are adjustable so that the distance between the two sides of the clamp can be fine-tuned.
Every effort should be made to provide a bright, white background behind the dot on the well bottom during strain characterization experiments. The better the contrast in these images, the easier it will be to automate the process of measuring the height and width of the dot, which can become tedious for a human operator analyzing a large experiment. High-speed videography of the bottom of a well in a 96-well plate presents challenges because the walls of the well tend to cast shadows. The use of a dome light or diffuse axial light that can illuminate along the line of sight of the camera without obscuring the image eliminates shadows or specular reflections that would arise with a conventional light source. The brightest available light source should be used because bright illumination allows images to be acquired with a short exposure time. Short exposure times minimize motion blur. Upgrading the light emitting diodes (LEDs) in the diffuse axial light allows shorter exposure times during high-speed video acquisition. The LEDs can be upgraded by opening the diffuse axial light, removing the stock LEDs, mounting 4 high power LED arrays to the back pane using LEDs holders, connecting them to a constant current power supply, and reassembling the diffuse axial light (see Table of Materials for catalog numbers). The disadvantage of upgrading the LEDs is that the passively cooled LEDs cannot be kept on for more than a few seconds due to the risk of overheating. Therefore, a different light is needed for alignment of the post-block and camera adjustment.
The presented method of quantifying membrane strain by measuring the dilation of a dot stamped on to the membrane is relatively crude, but it can scale up to multiple wells in a robust manner. The strain field across the well bottom can be characterized in more detail using digital image correlation. This technique involves spraying a speckled pattern onto the base of the well and then imaging it at high-speed during deformation. Commercial software can then be used to quantify strain at every point in the image by tracking the evolution of the speckled pattern.
This protocol produces a multi-faceted, clinically relevant, stretch injury phenotype in hiPSCNs. Cell death, neurite degeneration, and neurite beading are all well-documented sequelae of TBI in humans and animal models15. The key to success in this model is establishing and maintaining healthy cultures. Generally speaking, a cell culture protocol developed with conventional rigid plates is a worthwhile starting point for stretchable plate culture. However, the possibility that the cells in question may respond differently on silicone must always be considered. This is particularly true of hiPSCNs, which are very sensitive to culture conditions. Some examples of optimizing cell density and laminin concentration are supplied in the Representative Results section (Figure 3, Figure 4). Activation of the silicone with plasma treatment is vital. Silicone is hydrophobic and unreactive; in its natural state, it will not bind to laminin or other molecules used to promote cell attachment. Plasma treatment renders the surface hydrophilic and exposes reactive groups. These changes allow adhesion molecules to bind to the silicone and promote cell attachment. It is important to note that the plasma treatment effect dissipates within minutes unless the surface is submerged in liquid, and so procedures that involve drying the activated surface should be performed as quickly as possible. A simple way to check if the effect of plasma treatment has worn off is to place a droplet of water on the surface. On untreated silicone, the droplet will bead up whereas on plasma treated silicone, it will spread out. With the hiPSCNs that we used (see Table of Materials), the manufacturer recommends adding the laminin with the cell suspension rather than pre-coating. This protocol has incorporated this approach successfully. While segmentation can, in theory, be accomplished with open source software or general-purpose programming languages, a high degree of proficiency with these tools is required to obtain good results. Neurites are frequently difficult to distinguish from background signal because they are so slender. Therefore, we recommend the use of commercial software tools distributed by high content microscopy companies with dedicated modules for segmentation and quantification of neurons, if they are available. Even with commercial software, it is wise to export images of the segmentation to visually verify accuracy.
There are some limitations associated with working in stretchable plates compared to working with conventional, rigid plates. Stretchable plates can be imaged as normal with air objectives. However, imaging with immersion objectives is very difficult. Lens oil may damage the silicone. Additionally, the objective exerts pressure on the silicone membrane as it moves upwards. This pressure displaces the membrane vertically, making it difficult to bring the sample into focus. The silicone membranes currently used in fabricating the plates are approximately 250 µm thick. This thickness exceeds the focal distance of many high power, immersion objectives. Special care must be taken to lay the membranes perfectly flat before clamping to achieve the flatness required for microscopy. Autofocus systems can compensate for deviations in the flatness of the finished plate to some extent. Future versions of the protocol may pre-tension the membrane before it is bonded to the plate top to ensure flatness. The adhesive-free procedure for bonding the silicone membrane to the plate top14 is considered an important strength of the current protocol. It eliminates the risk of neurotoxicity from the adhesive as well as any deviations in flatness due to non-uniform thickness of the adhesive layer.
Multi-electrode arrays are commonly used in experiments with hiPSCNs to assess their maturity and functionality. Unfortunately, these systems are incompatible with this model because the cell culture substrate is rigid. It is possible to create a stretchable multi-electrode array, although this has so far only been demonstrated in a single well format16,17. Note that indenters can be removed individually from the indenter block so that some wells are not indented and can serve as shams. Removing the indenter prevents indentation but does not completely eliminate mechanical loading since there is still inertial motion of the fluid in the wells while the stage is moving. It is worth comparing these wells to wells in plates that were never subject to stage motion to measure any pathological influence of fluid motion. Also, the array of indenters in the block should be bisymmetric (symmetrical from front to back and side to side). This precaution ensures that the plate is evenly loaded during indentation, so that the stage does not tilt sideways and cause the rods to bind in their bearings.
One of the primary challenges to therapeutic innovation in neurotrauma is the complexity and heterogeneity of the condition. Trauma applies multi-modal stress to every cell type in the central nervous system simultaneously. Neurons have been reliably generated from human induced pluripotent stem cells (hiPSCs) and are now widely available from commercial vendors. Innovation is proceeding quickly in this field, and other neural cell types such as astrocytes18 and microglia19 are also being derived from hiPSCs. It may soon be possible to isolate the cell-autonomous responses of each of these cell types to trauma in vitro and then to co-culture different cell types to understand how they communicate after trauma. In this way, it may ultimately be possible to recreate the clinical challenge from the bottom up to thoroughly understand it in a human system. This approach is distinct from the conventional approach relying on rodent models and has the potential to generate novel insights that lead to the first therapies for this common, devastating, and intractable condition.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work is supported in part by a grant from the National Institutes of Health (R21NS098129). We would like to acknowledge excellent technical assistance from SueSan Chen, Jonathan Tan, Courtney Cavanaugh, Shi Kai Ng and Feng Yuan Bu, who designed and built a structure to support lights used during high speed imaging experiments described in this manuscript.
References
- Faul M, L Xu, Wald MM, Coronado V. Traumatic brain injury in the United States: emergency department visits, hospitalizations, and deaths, 2002-2006. 2010.
- Kabadi SV, Faden AI. Neuroprotective strategies for traumatic brain injury: improving clinical translation. Int J Mol Sci. 2014;15(1):1216–1236. doi: 10.3390/ijms15011216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiskinis E, et al. Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell. 2014;14(6):781–795. doi: 10.1016/j.stem.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DH, Wolf JA, Lusardi TA, Lee VM, Meaney DF. High tolerance and delayed elastic response of cultured axons to dynamic stretch injury. J Neurosci. 1999;19(11):4263–4269. doi: 10.1523/JNEUROSCI.19-11-04263.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf JA, Stys PK, Lusardi T, Meaney D, Smith DH. Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. J Neurosci. 2001;21(6):1923–1930. doi: 10.1523/JNEUROSCI.21-06-01923.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusardi TA, Wolf JA, Putt ME, Smith DH, Meaney DF. Effect of acute calcium influx after mechanical stretch injury in vitro on the viability of hippocampal neurons. J Neurotrauma. 2004;21(1):61–72. doi: 10.1089/089771504772695959. [DOI] [PubMed] [Google Scholar]
- Magou GC, et al. Engineering a high throughput axon injury system. J Neurotrauma. 2011;28(11):2203–2218. doi: 10.1089/neu.2010.1596. [DOI] [PubMed] [Google Scholar]
- Morrison B, 3rd, Cater HL, Benham CD, Sundstrom LE. An in vitro model of traumatic brain injury utilising two-dimensional stretch of organotypic hippocampal slice cultures. J Neurosci Methods. 2006;150(2):192–201. doi: 10.1016/j.jneumeth.2005.06.014. [DOI] [PubMed] [Google Scholar]
- Cater HL, et al. Stretch-induced injury in organotypic hippocampal slice cultures reproduces in vivo post-traumatic neurodegeneration: role of glutamate receptors and voltage-dependent calcium channels. J Neurochem. 2007;101(2):434–447. doi: 10.1111/j.1471-4159.2006.04379.x. [DOI] [PubMed] [Google Scholar]
- Kang WH, Morrison B., 3rd Functional tolerance to mechanical deformation developed from organotypic hippocampal slice cultures. Biomech Model Mechanobiol. 2015;14(3):561–575. doi: 10.1007/s10237-014-0622-4. [DOI] [PubMed] [Google Scholar]
- Sherman SA, et al. Stretch Injury of Human Induced Pluripotent Stem Cell Derived Neurons in a 96 Well Format. Sci Rep. 2016;6:34097. doi: 10.1038/srep34097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy WN, et al. Investigation of Head Injury Mechanisms Using Neutral Density Technology and High-Speed Biplanar X-ray. Stapp Car Crash J. 2001;45:337–368. doi: 10.4271/2001-22-0016. [DOI] [PubMed] [Google Scholar]
- Hardy WN, et al. A study of the response of the human cadaver head to impact. Stapp Car Crash J. 2007;51:17–80. doi: 10.4271/2007-22-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunkara V, et al. Simple room temperature bonding of thermoplastics and poly(dimethylsiloxane) Lab Chip. 2011;11(5):962–965. doi: 10.1039/c0lc00272k. [DOI] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH. Axonal pathology in traumatic brain injury. Exp Neurol. 2013;246:35–43. doi: 10.1016/j.expneurol.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacour SP, et al. Flexible and stretchable micro-electrodes for in vitro and in vivo neural interfaces. Med Biol Eng Comput. 2010;48(10):945–954. doi: 10.1007/s11517-010-0644-8. [DOI] [PubMed] [Google Scholar]
- Yu Z, Morrison B., 3rd Experimental mild traumatic brain injury induces functional alteration of the developing hippocampus. J Neurophysiol. 2010;103(1):499–510. doi: 10.1152/jn.00775.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tcw J, et al. An Efficient Platform for Astrocyte Differentiation from Human Induced Pluripotent Stem Cells. Stem Cell Reports. 2017;9(2):600–614. doi: 10.1016/j.stemcr.2017.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muffat J, et al. Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat Med. 2016;22(11):1358–1367. doi: 10.1038/nm.4189. [DOI] [PMC free article] [PubMed] [Google Scholar]