

Abstract
Cancer cells can resist the effects of DNA-damaging therapeutic agents via utilization of DNA repair pathways, suggesting that DNA repair capacity (DRC) measurements in cancer cells could be used to identify patients most likely to respond to treatment. However, the limitations of available technologies have so far precluded adoption of this approach in the clinic. We recently developed fluorescence-based multiplexed host cell reactivation (FM-HCR) assays to measure DRC in multiple pathways. Here we apply a mathematical model that uses DRC in multiple pathways to predict cellular resistance to killing by DNA-damaging agents. This model, developed using FM-HCR and drug sensitivity measurements in 24 human lymphoblastoid cell lines, was applied to a panel of 12 patient-derived xenograft (PDX) models of glioblastoma to predict glioblastoma response to treatment with the chemotherapeutic DNA-damaging agent temozolomide. This work showed that, in addition to changes in O6-methylguanine DNA methyltransferase (MGMT) activity, small changes in mismatch repair (MMR), nucleotide excision repair (NER), and homologous recombination (HR) capacity contributed to acquired temozolomide resistance in PDX models and led to reduced relative survival prolongation following temozolomide treatment of orthotopic mouse models in vivo. Our data indicate that measuring the combined status of MMR, HR, NER, and MGMT provided a more robust prediction of temozolomide resistance than assessments of MGMT activity alone.
Introduction
Therapy resistance is a universal problem in cancer treatment, and our understanding of its origins remains incomplete. Many cancers can initially be treated effectively with chemotherapy and radiation. However, some cancers are innately resistant to therapy, and most cancers acquire resistance to therapy over the course of treatment. Consequently, patients who do not benefit from treatment are needlessly subjected to the severe and sometimes lasting side effects of chemotherapy and radiotherapy, including an increased risk of second primary cancers. Moreover, precious time is lost, during which alternative and potentially more effective therapy could have been applied.
Most cancer therapy regimens include chemotherapy and radiotherapy, both of which damage DNA, killing cancer cells by inducing damage that interferes with DNA replication and transcription, ultimately activating cell death pathways. Efficient repair of DNA damage can render cells resistant to killing. Each drug may induce several different types of DNA damage, and at least 6 major DNA repair pathways, as well as numerous sub-pathways, are involved in the repair of damage (1–3). Accordingly, the DNA repair pathways involved in acquired resistance mechanisms vary from drug to drug, and all of the major DNA repair pathways can alter the sensitivity of cancer cells to DNA damaging agents.
Glioblastoma is an aggressive cancer with a median survival of 12 to 15 months under the standard of care treatment of aggressive surgery, radiation, and chemotherapy. Almost all patients succumb to this disease, reflecting intrinsic or acquired resistance to frontline treatment with radiotherapy combined with the alkylating agent temozolomide (4). Temozolomide is one of several chemotherapeutic SN1-type methylating agents, including dacarbazine and procarbazine, which induce a similar spectrum of methylated DNA bases, among which O6-methylguanine (O6-MeG) is the predominant cytotoxic DNA lesion (5). Promoter hypermethylation of the MGMT gene, encoding a protein that repairs O6-MeG, has been used to identify patients whose tumors are O6-methylguanine DNA methyltransferase (MGMT)-deficient and are thus more likely to benefit from temozolomide therapy (6). However, multiple DNA repair pathways affect the sensitivity of cells to killing with SN1-type methylating agents (1), suggesting that successful personalized therapy based on DRC in glioblastoma will require multiplexed measurements of repair activity. Progress toward this goal has been significantly hindered by a lack of quantitative, high-throughput assays for multiple DNA repair pathways, and the challenges of predicting phenotypes from genetic or other `omics approaches (7).
Here we apply recently developed fluorescence-based multiplexed host cell reactivation (FM-HCR) assays to measure DNA repair capacity (DRC) for multiple repair pathways in a set of human cell lines (8). We present mathematical models that predict sensitivity to DNA damaging agents from DRC data. We then apply FM-HCR assays to assess global DRC in patient-derived xenograft (PDX) models of glioblastoma. Our mathematical model predicts the effectiveness of temozolomide therapy both for killing glioblastoma cells in vitro and for prolonging the survival of glioblastoma tumor-bearing mice upon temozolomide treatment in vivo. The model integrates DRC across four repair pathways, and outperforms predictions made based on tumor MGMT activity alone. This work lays the foundation for using functional assays to make quantitative, personalized predictions concerning the effectiveness of cancer therapy.
Materials and Methods
PDX models and cell culture
Lymphoblastoid cells, from the Coriell Cell Repository, were maintained in log phase in RPMI media supplemented with 15% FBS plus penicillin and streptomycin (8). Cells were obtained in 2001. The Coriell Cell Repository authenticates and tests cell cultures for contamination with mycoplasma, bacteria, and fungi; cells have not been authenticated by the authors. For cell cultures derived from glioblastoma PDX lines, flank glioblastoma tumor xenografts were excised, dissociated into single-cell suspensions, and cultured in DMEM with 10% FBS as previously described (9).
MMS and temozolomide sensitivity assays
Lymphoblastoid cell lines were assayed for sensitivity to BCNU (reported previously in ref. 10) and temozolomide usinga cell proliferation assay that measures quenching of DNA-bound Hoechst 33258 fluorescence by BrdUrd incorporated during DNA synthesis (10, 11). Cells were suspended in 300 μL serum free media at 4.5 × 105 cells/mL, and treated at a final concentration 240 mmol/L temozolomide for 1 hour at 37oC. Cells were centrifuged for 5 minutes at 450 g, and washed once with 200 mL PBS. Next, cells were suspended in 280 mL complete media, and subsequently cultured in flat bottom 96-well plates. After two doubling times, BrdUrd was added to achieve a final concentration of 45 mmol/L. After two additional doubling times, cells were stained with Hoechst 33258 and analyzed as previously reported (10). Sensitivity to MNNG and sensitivity to MMS were measured using a previously described cell proliferation assay (12). Briefly, logarithmically growing cells were treated with 0.4 mmol/L MMS or left untreated. After 72 hours, % control growth was calculated from the ratio of the total number of viable-treated cells divided by the total number of viable untreated cells using a Vi-CELL cell viability analyzer with trypan blue staining to exclude dead cells.
FM-HCR assays
FM-HCR assays in lymphoblastoid cell lines were carried out as previously described (8). For FM-HCR assays in PDX lines, glioblastoma cells were seeded at 50,000 cells per well in a 24-well tissue culture plate 24 hours before transfection. 0.5 mg of plasmid mixtures composed of previously described reporter plasmids for the MGMT, mismatch repair (MMR), nucleotide excision repair (NER), non-homologous end joining (NHEJ), and homologous recombination (HR) pathways were transfected into cells using “Lipofectamine 2000 LTX with plus reagent” (Thermo Fisher). At 24 hours posttransfection, cells were analyzed for fluorescent reporters by flow cytometry and DRC for both lymphoblastoid cells and glioblastoma cells was calculated in terms of % reporter expression as previously described (8).
Mathematical modeling
The overall modeling approach is summarized in Supple- mentary Fig. S1. Following log-transformation of FM-HCR data, Z-scores for relative DRC among the 24 lymphoblastoid cell lines were generated for each pathway using Eq. 1:
| 1 |
where Zij is the Z-score for a DNA repair pathway in cell line i in pathway j, xij is the DRC for a given pathway j in cell line i, xij is the mean value of the DRC in pathway j over the 24 lympho-blastoid cell lines (i = 1–24), and 𝛔j is the standard deviation of the DRC in pathway j. Z-scores were also generated for relative DRC among the 12 PDX models of glioblastoma using the same approach. Because there is an inverse relationship between % reporter expression and DRC for the MGMT reporter (larger signal represents lower activity), Z-scored MGMT data were multiplied by 1. Simple linear regression was used to generate the models #1 and #4, denoted as “MGMT” in Table 1. These models relate MGMT activity to sensitivity to killing with temozolomide and MNNG. A graphical interpretation of the parameters associated with model #1 can be generalized to the multiple linear models, and is detailed in Supplementary Fig. S2. MATLAB scripts (provided in the Supplementary Information) were run to generate linear models using DRC in five pathways in the lymphoblastoid cell lines as the predictor variables and sensitivity to cell killing (measured as % control growth) with the DNA damaging agents temozolomide, MNNG, MMS, or BCNU as the response variable. First, multiple linear regression (MLR) models were fit to all five DNA repair pathways; these models are listed as “MLR” models in Table 1. Then, LASSO regression was performed to select optimal combinations of DRC predictor variables for incorporation into MLR models of sensitivity to cell killing with each DNA dam-aging agent (13). LASSO regression was carried out in MATLAB to return least squares regression coefficients for a set of 100 values of the regularization coefficient l, which determines the size of the penalty for coefficients remaining in the model. The value of λ is gradually increased until the least squares regression coefficients for all five predictor variables take a value of zero, resulting in a family of combinations of predictor variables. MLR was performed and evaluated with exhaustive leave-one-out cross validation using the combination of predictor variables selected by LASSO at each value of λ; those for which LASSO returned least squares regression coefficients equal to zero were excluded from MLR analysis. R2 parameters were calculated for the relationship between predicted sensitivities obtained from cross-validation versus the known sensitivity for each cell line, and the model yielding the largest R2 values were selected. Plots of sensitivity predicted from leave-one-out cross-validation versus observed sensitivity are presented in Supplementary Fig. S3. MLR models using the combination of predictor variables selected using LASSO are listed as “MLRL” in Table 1.
Table 1.
Linear models relating sensitivity to DNA-damaging agents to DNA repair capacity in multiple pathways
| Agent | # | Model | Equation | R2FIT | R2LOO | AUC |
|---|---|---|---|---|---|---|
| MNNG | 1 | MGMT | 13.3 * MGMT + 59 | 0.41 | 0.33 | 0.54 |
| 2 | MLR | 13.8 * MGMT − 9.3 * MMR + 6.8 * HR + 4.4 * NER − 2.3 * NHEJ + 59 | 0.68 | 0.49 | 0.89 | |
| 3 | MLRL | 13.3 * MGMT + 8.1 * HR − 7.0 * MMR + 59 | 0.63 | 0.50 | 0.74 | |
| TMZ | 4 | MGMT | 19.0 * MGMT + 71 | 0.61 | 0.50 | 0.54 |
| 5 | MLR | 17.5 * MGMT + 8.5 * NER + 8.3 * HR − 3.6 * MMR − 0.2 * NHEJ + 71 | 0.90 | 0.81 | 0.86 | |
| 6 | MLRL | 17.4 * MGMT + 8.5 * NER + 8.2 * HR − 3.5 * MMR + 71 | 0.90 | 0.84 | 0.86 | |
| BCNU | 7 | MLR | 10.4 * MGMT + 2.5 * NER + 2.2 * NHEJ − 1.3 * MMR + 0.3 * HR + 33 | 0.54 | 0.19 | 0.80 |
| 8 | MLRL | 10.6 * MGMT + 33 | 0.49 | 0.40 | 0.54 | |
| MMS | 9 | MLR | 6.9 * HR + 5.8 * NHEJ + 5.6 * NER + 5.2 * MGMT − 1.5 * MMR + 44 | 0.38 | 0.06 | 0.71 |
| 10 | MLRL | 8.9 * HR + 44 | 0.17 | 0.07 | 0.71 |
NOTE: The models are numbered under column heading “#”. R2FIT is the goodness of fit for each model; R2FIT can adopt values between 0 (no fit at all) and 1 (perfect fit). R2LOO is a measure of the predictive accuracy for each model and is calculated from exhaustive leave-one-out cross-validation presented graphically in Supplementary Fig. S2. Once again, R2LOO can adopt values between 0 (no fit at all) and 1 (perfect fit). AUC is a statistic that measures the performance of the models when they are used to classify the PDX models as sensitive versus resistant. AUC can adopt values between 0 (all samples are classified incorrectly) and 1 (all samples are classified correctly). AUC is calculated from ROC curves (Fig. 5) generated using the sensitivity of PDX models calculated from the respective models and the actual status of each glioblastoma model (sensitive or resistant)
Receiver operating characteristic (ROC) analysis was carried out in MATLAB using PDX sensitivities calculated from mathematical models and the actual sensitivity status. DRC data were input into the models in Table 1 to calculate the predicted sensitivity for each PDX model. To evaluate the performance of the models, ROC curves were generated, and the area under the curve (AUC) computed (Table 1). ROC analysis provides a quantitative estimate and graphical representation of the accuracy of predictive assays (14), and is particularly useful when the test result, reported on a continuous scale, is to be used to predict a binary variable, as in the present case. The ROC curve shows the relationship between the true positive rate and the false-positive rate as the discrimination threshold is varied. For an ideal assay, AUC = 1.0, indicating that the discrimination threshold can be set in such a way that all true positives can be correctly called without calling any false positives; the distributions of values given by the assay for the positive and negative populations are completely separated. By contrast, for an assay that only performs as well as random chance, the distributions are indistinguishable, and AUC = 0.5, because there are equal chances of calling a positive or a negative at all discrimination thresholds. The true positive rate is equivalent to assay sensitivity, and the false positive rate is calculated from 1 – (assay specificity).
Results
Models relating DRC to alkylation-induced cell killing in human lymphoblastoid cell lines
Twenty-four lymphoblastoid cell lines derived from genetically diverse, apparently-healthy individuals were used as a model system (15). These cell lines have been previously shown to exhibit a broad range of sensitivity to killing by the SN1-type alkylating agent MNNG (12), and a DNA cross-link forming alkylating agent, BCNU (10). We have now addition-ally measured the sensitivity of these cell lines to MMS, an SN2-type alkylating agent, for which the predominant cytotoxic lesions induced are 3-methyladenine and 7-methylguanine, and the sensitivity of a subset of the 24 cell lines to the SN1-type alkylating chemotherapy agent temozolomide. Temozolomide is regarded as a functional analog of MNNG because it methylates DNA via the same reactive methyldiazonium ion intermediate (16), and temozolomide sensitive cells are also sensitive to MNNG (17).
A broad and continuous range of sensitivity to MNNG (12), temozolomide, BCNU (10), and MMS is observed across the 24 lymphoblastoid cell lines (Fig. 1A and Supplementary Table S1). The cell lines were organized in order of decreasing MNNG sensitivity and assigned to color-coded quartiles to facilitate comparisons with respect to temozolomide, BCNU, and MMS sensitivity. Although MNNG and temozolomide sensitivity were strongly correlated (R=0.81) as expected, MNNG sensitivity was less strongly correlated to BCNU sensitivity (R = 0.54) and MMS sensitivity (R =0.49). Thus, consistent with clinical observations, sensitivity to one agent does not necessarily predict sensitivity to other agents, especially when they induce a different spectrum of DNA damage (18, 19).
Figure 1.
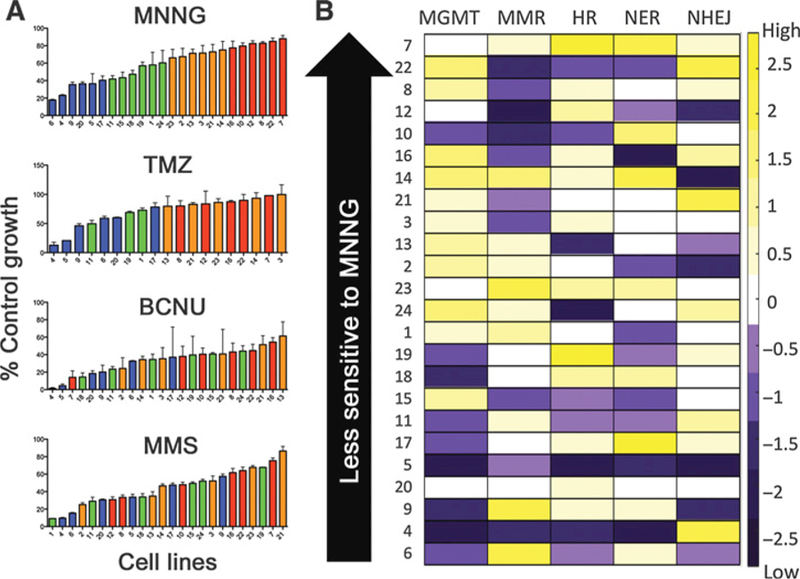
DRC and sensitivity data for Coriell lymphoblastoid cell line panel. A, Twenty-four lymphoblastoid cell lines organized (from left to right) in descending order of sensitivity to four different DNA-damaging agents. The colors assigned to bars for each cell line in the top panel (MNNG) by quartile were maintained in the temozolomide (TMZ), BCNU, and MMS panels. Sensitivity to MNNG and MMS was measured at 72 hours. Sensitivity to BCNU and temozolomide was measured at four doubling times post-exposure. B, Heat map representing Z-scores for relative DNA repair capacity among the 24 lymphoblastoid cell lines (rows) in the five pathways (columns). Purple, cell lines with repair capacity below the mean; yellow, cell lines with repair capacity above the mean; white, cell lines with repair capacity at or near the mean. Z-units are reported at the right side of the figure. Cell lines are ordered by sensitivity to MNNG.
The relationships among sensitivities to the four DNA dam-aging agents and previously measured repair capacity in five pathways: NHEJ, HR, MMR, NER, and MGMT were evaluated in the lymphoblastoid cell lines (Fig. 1B and Supplementary Tables S1 and S2; ref. 8). The data in Fig. 1B have been Z-scored to emphasize repair capacity variation among the 24 cell lines. No statistically significant linear correlations were observed among repair capacities for the five reporters (Supplementary Tables S3 and S4), and each cell line has a unique constellation of repair capacities. MGMT activity has previous-ly been shown to correlate with MNNG sensitivity in the 24 cell lines (12). This study, with MGMT activity measured by FM-HCR, confirms this result, and further finds that MGMT activity correlates significantly with sensitivity to killing by temozolomide and BCNU (Supplementary Fig. S3). Pairwise linear correlations between DRC in each individual pathway and sensitivity to killing with DNA damaging agents were evaluated based on P-values calculated for linear trends, as well as the value of the correlation coefficient R (Supplementary Tables S5 and S6). A majority of the correlations were found to be stronger when calculated for sensitivity versus the logarithm of DRC. All modeling work was thus carried out using the base 10 logarithm of repair capacities as the predictor variable.
Mathematical models were developed to test whether DRC in multiple pathways is correlated to sensitivity to DNA damaging agents (Supplementary Fig. S1). A simple linear model (Y = mx b) relates MNNG sensitivity to MGMT activity (model #1 in Table 1). The positive slope in this model indicates that a 1 standard deviation increase in MGMT activity leads to a 13 point increase in % control growth (Supplementary Fig. S2). This relationship was expected based on the inverse correlation between MGMT activity and MNNG sensitivity that has been observed previously (12), and presently in Supplementary Tables S4 and S5 and Supplementary Fig. S3. A linear model relating MGMT activity to temozolomide sensitivity similarly indicated that an increase in MGMT activity leads to an increase in temozolomide resistance (model #4 in Table 1). A MATLAB script, available in the supplemental information, was used next to generate a MLR model relating Z-scored DRC data in all five DNA repair pathways to cytotoxicity induced by each of the four agents, MNNG, temozolomide, BCNU, and MMS. In these models (#2, #5, #7, and #9 in Table 1), DRC values for each pathway in the 24 cell lines in Fig. 1B served as the five (independent) predictor variables, and the sensitivity of the same 24 cell lines (reported as % control growth in Fig. 1A) was the (dependent) response variable. The resulting MLR models take the form given below in Eq. 2,
| 2 |
where Y represents the response variable (MNNG sensitivity) reported as % control growth; xj are the DRC predictor variables for the five substrates reported as z-scored log % reporter expression, mi are the slopes along the corresponding dimensions, and b is a constant that represents the Y intercept. These models (#2, #5, #7, and #9) are denoted as “MLR” in Table 1. The slopes and y-intercepts of these models are interpreted in the same manner as described above for model #1. For all models, the goodness of fit is reported as the R2 parameter, and predictive accuracy is reported as the R2LOO parameter for predicted sensitivity versus observed sensitivity, calculated from exhaustive leave-one-out cross-validation (Table 1 and Supplementary Fig. S4). An additional parameter in Table 1, AUC, reports how accurately each model predicts temozolomide induced cell killing from DRC for a panel of PDX models of glioblastoma. The application of the models to predict temozolomide sensitivity in glioblastoma is discussed in more detail below.
The models were further refined using a second MATLAB script (available in the Supplementary Information) running the LASSO algorithm (13) to select combinations of DRC predictor variables to be used for building multiple linear regression (MLR) models of the form given in Eq. 3.
| 3 |
The number and identity of predictor variables (n [max 5]) used to build the model is established from the results of the LASSO algorithm, which minimizes overfitting by excluding variables that do not improve model performance under cross-validation (13). Models obtained using this approach (#3, #6, #8, and #10) for each DNA damaging agent are denoted as “MLRL” in Table 1. The temozolomide MLRL retained four pathways (MGMT, HR, MMR, and NER); the MNNG MLRL model retained three path-ways (MGMT, HR, and MMR). The MLRL models for BCNU and MMS retained only a single pathway (MGMT and HR, respectively). As expected, for each alkylating agent, the predictive accuracy of the MLRL models was improved relative to the predictive accuracy of the MLR models; this is reflected in higher R2LOO values (Table 1), which correspond to better fits in plots of predicted sensitivity versus observed sensitivity for the 24 lymphoblastoid cell lines under leave-one-out cross-validation (Supplementary Fig. S4).
The use of Z-scored DRC data for generating the mathematical models allows for a straightforward interpretation of the slopes (mi) that are associated with each model: the steeper the slope, the more strongly the model responds to a given number of standard deviations about the mean for that particular pathway. A slope of 0 would indicate a model in which drug sensitivity is entirely independent of the DNA repair pathway in question.
Sensitivity models for the SN1-type alkylating agents temozolomide and MNNG
The MLR models for the SN1 alkylating agents MNNG and temozolomide (#2 and #5) are similar in several respects (Table 1). First, the coefficients for MGMT and HR are large and positive in both models, indicating that the models are relatively sensitive to small changes in repair capacity in these pathways, and higher MGMT or HR repair capacity leads to a lower sensitivity to killing with either agent. Second, MMR takes a negative coefficient in both models, indicating that higher MMR capacity leads to higher sensitivity to killing. This aspect of the modeling is borne out in the raw data; higher MMR activity in cell line #6 versus cell line #10 without major differences in other pathways is associated with higher MNNG sensitivity in cell line #6 (Fig. 1). Third, the NHEJ coefficient is the smallest in both models, suggesting that this pathway plays the smallest role in sensitivity to SN1 type alkylating agents. Finally, the NER coefficient is positive in both models, indicating that higher NER capacity is associated with lower sensitivity to SN1 alkylating agents. Both the MNNG and temozolomide MLRL models (#3 and #6) exclude the NHEJ pathway, further indicating that this pathway does not contribute significantly to sensitivity to either agent for this panel of cells. The MLRL models also both retain MGMT, MMR, and HR, but differ in that the temozolomide model also includes NER. To test whether the differences in MLRL models (#3 and #6) were due to the inclusion of five additional cell lines in model #6, an MNNG sensitivity model was generated for the same subset of 19 cell lines for which the temozolomide models were generated (not shown). Like model #3, this MLRL model also did not include the NER pathway. We infer that the differences in models #3 and #6 are not due to the use of a subset of cell lines as the training population for the temozolomide model. As detailed in the Supplemental Information, these models are consistent with previously established roles for the MGMT, MMR, HR, and NER pathways affecting the sensitivity of cells to killing with SN1-type alkylating agents (Fig. 2), although the quantitative contributions of each pathway have not previously been determined.
Figure 2.
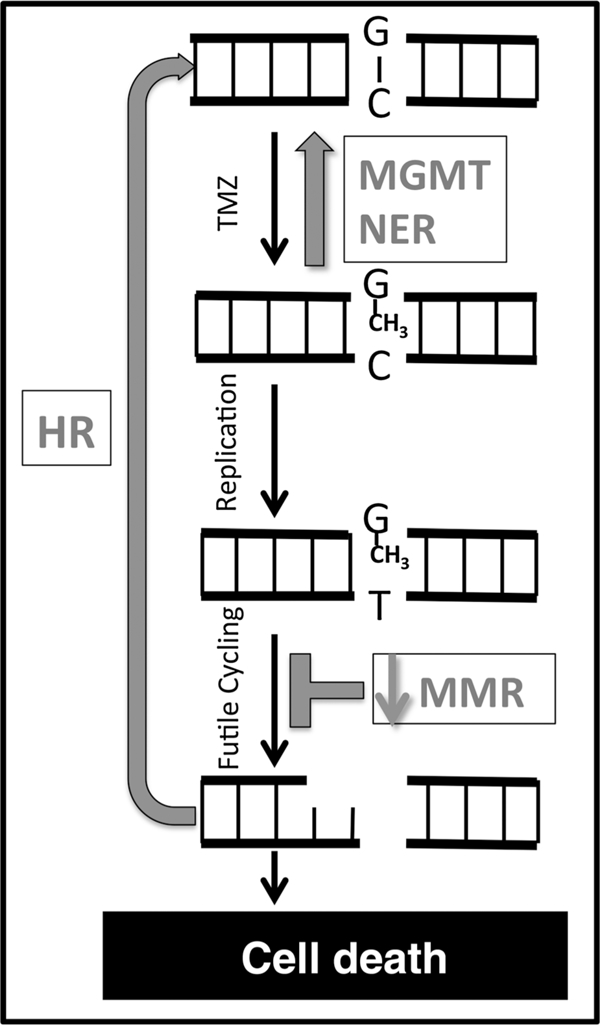
Model for temozolomide (TMZ) resistance mechanisms mediated by DNA repair capacity. Double-stranded DNA is represented by parallel black lines. A normal G:C base pair is indicated at the top of the figure. Strand breaks are indicated as gaps in the black lines. Gray arrows and text indicate DNA repair processes that mediate sensitivity and resistance to DNA methylation damage.
Sensitivity models for BCNU and MMS
The BCNU and MMS sensitivity models are distinguished in several respects from the SN1 alkylating agent sensitivity models. For both the BCNU and MMS MLR models (#7 and #9), the MMR coefficient was small (Table 1), and was not retained in the MLRL models (#8 and #10), indicating that MMR capacity does not play a major role in determining cellular sensitivity in the models. Both MLRL models retain only a single DNA repair pathway, and the MMS MLRL model is the only model that excludes the MGMT pathway. The MLRL models indicate that HR plays a significant role in determining MMS sensitivity, whereas MGMT activity is the major contributor to BCNU sensitivity. As with the temozolomide and MNNG sensitivity models, models #7–10 are consistent with previously established roles for DRC affecting the cellular MMS and BCNU sensitivity (see Supplemental Information).
Measuring DRC in glioblastoma
The temozolomide and MNNG models suggest that relatively small changes in MGMT, MMR, HR, and NER could be responsible for changes in sensitivity to killing with SN1-type alkylating agents in cancer cells. To test whether relationships between DRC and temozolomide sensitivity in lymphoblastoid cell lines also apply to glioblastoma, we measured changes in DRC that accompany temozolomide resistance acquired in vivo for several paired sensitive and resistant PDX models of glioblastoma (Fig. 3 and Supplemental Table S7), as well as one PDX model (GBM46) innately temozolomide-resistant model (9). The parental PDX models (GBM12, GBM14, GBM22, GBM39, GBM59, and GBM46) were previously classified as “resistant” or “sensitive” to temozolomide based on whether temozolomide treatment produces a statistically significant survival benefit in vivo (20), and statistically significant increases in temozolomide resistance in the PDX models (GBM12TMZ_3080, GBM12TMZ_5476, GBM14TMZ, GBM39TMZ, and GBM59_TMZ) have been measured in vitro either by neurosphere formation or by cell proliferation assays (21).
Figure 3.
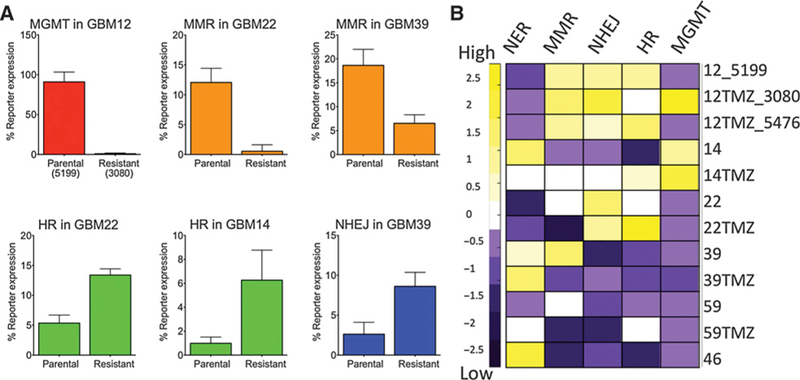
DNA repair capacity measured using FM-HCR in glioblastoma cells. A, Statistically significant changes in DRC in PDX models of acquired temozolomide resistance. Note that for the MGMT assay, lower percentage of reporter expression corresponds to higher MGMT activity. Error bars represent the standard deviation of at least three measurements. B, A heat map representing Z-scored DNA repair capacity in five pathways (columns) for all 12 glioblastoma models (rows; numbers refer to the parental GBM lines and the “TMZ” suffix refers to acquired temozolomide resistance lines labeled “Resistant” in panel A). Purple, cell lines with repair capacity below the mean; yellow, cell lines with repair capacity above the mean; white, cell lines with repair capacity at or near the mean.
Strikingly, we observe statistically significant changes in DRC that are consistent with the relationship between DRC and SN1-type alkylating agent sensitivity suggested by the MNNG and temozolomide sensitivity models (Table 1). In cell lines that acquired temozolomide resistance, we observed increased MGMT activity (GBM12/GBM12TMZ_3080), increased HR capacity (GBM14/GBM14TMZ and GBM22/GBM22TMZ), or decreased MMR capacity (GBM22/GBM22TMZ and GBM39/ GBM39TMZ) relative to the temozolomide-sensitive parental cell lines (Fig. 3A). Statistically significant changes in NHEJ (GBM39/GBM39TMZ) were also observed (Fig. 3A). Although the differences were not statistically significant, NER activity trended higher in all of the acquired resistance models relative to their corresponding parental lines, except for GBM14. A trend toward reduced MMR and elevated HR capacity was also observed in GBM59TMZ relative to GBM59; however, these apparent changes likewise did not reach statistical significance (Fig. 3B). Overall, the data indicate that the relationship between DRC and sensitivity to temozolomide and MNNG in lymphoblastoid cell lines (Table 1) are applicable to temozolomide sensitivity in glioblastoma.
Applying mathematical modeling to predict sensitivity to temozolomide in glioblastoma
The ability of mathematical models to classify PDX models of glioblastoma as temozolomide sensitive or temozolomide-resistant was tested. ROC plots for a subset of the models are presented in Fig. 4A. The MLRL model trained using temozolomide sensitivity in lymphoblastoid cell lines (#6) is the best predictor of temozolomide sensitivity in glioblastoma (AUC=0.86). The MLRL model generated from DRC for sensitivity to MNNG (#3) yielded an AUC of 0.74. Predictions using a model for MMS sensitivity yielded an AUC = 0.71 (model #10), and a model generated for BCNU sensitivity (model #8) barely outperformed random chance for this panel of PDX models (AUC = 0.54).
Figure 4.
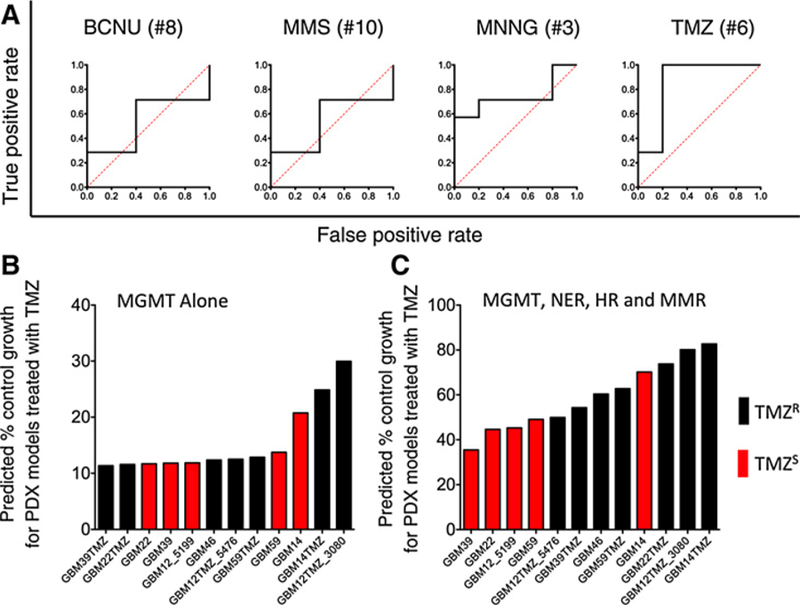
Classification of glioblastoma PDX models as temozolomide (TMZ) sensitive or temozolomide resistant. A, Plots showing ROC curves (black lines) for sensitivity models for BCNU (model #8); MMS (#10); MNNG (#3); temozolomide (#6). The numbers refer to models detailed in Table 1. AUC is calculated from the area under the ROC curves and can adopt values between 0 (all samples are classified incorrectly) and 1 (all samples are classified correctly). The dotted red line represents a theoretical ROC curve for a model that classifies samples randomly (AUC = 0.5). B, PDX models organized in ascending order of predicted percent control growth as calculated from model #4. Lower predicted percent control growth represents higher predicted sensitivity to temozolomide. C, PDX models organized in ascending order of predicted percent control growth as calculated from model #6. Temozolomide sensitivity was determined previously for the PDX models; black bars, PDX models that temozolomide resistant (TMZR); red bars, PDX models that are TMZ sensitive (TMZS). A Mann–Whitney U test indicated that the two populations (TMZR and TMZS) were significantly different in C (P = 0.03), but not in B.
The best model (#6) also correctly predicts temozolomide resistance in the acquired resistance models relative to the corresponding parental cell lines in all of the paired PDX models (Fig. S5). Importantly, using DRC data for multiple pathways in glioblastoma cell lines improves the predictive accuracy for temozolomide sensitivity models versus those that include only the MGMT pathway. MGMT activity measured by FM-HCR was correlated to methylation status (R = 0.61 and Supplementary Fig. S6), but the extent of promoter methylation varied over a ⁓10-fold range among glioblastoma lines that all had very low MGMT activity, consistent with previous observations indicating that widely used MGMT tumor promoter methylation assays may not accurately predict MGMT transcript levels and activity (22). When cells are ranked according to temozolomide sensitivity as predicted by model #4, which includes only MGMT activity (Fig. 4B), two temozolomide-resistant cell lines, GBM22TMZ and GBM39TMZ, are predicted to be the most sensitive, with predicted % control growth 10%, and AUC = 0.54. By contrast, when PDX models are ranked according to temozolomide sensitivity predicted by model #6, which takes MGMT, MMR, HR, and NER into account, the predicted response across the glioblastoma cell lines is much more accurate relative to previously measured sensitivity (20, 21); the improved accuracy can be appreciated both qualitatively (Fig. 4C) and quantitatively (AUC = 0.86).
Using DRC-based modeling to predict effectiveness of therapy with temozolomide in vivo
The ultimate goal of predicting the sensitivity of glioblastoma cells to SN1 alkylating agents is to generate a metric that could potentially guide cancer therapy decisions. This goal requires that repair capacity in vitro predicts therapeutic response in vivo, where the sensitivity of cancer cells to DNA damaging therapies can be strongly influenced by tumor microenvironment (23). The PDX models for which we have measured DRC profiles have previously been shown to exhibit a broad range of responsiveness to in vivo temozolomide therapy, measured by relative survival prolongation of tumor bearing mice treated with temozolomide versus tumor bearing mice receiving placebo (21). Relative survival prolongation is calculated by dividing the median survival time for treated mice by the median survival time for untreated mice; larger values reflect longer survival in treated mice relative to untreated mice.
To test whether the DRC profiles measured using FM-HCR in cultured cells predict temozolomide therapy effectiveness in vivo, relative survival prolongation in temozolomide-treated PDX mouse models was compared against temozolomide sensitivities predicted using the models in Table 1. Sensitivities predicted using a model that includes MGMT activity alone (model #4) are only weakly correlated to relative survival prolongation, and PDX models with low MGMT expression (black dots in Fig. 5A) range from highly responsive to completely refractory to treatment with temozolomide. A stronger correlation is observed between survival prolongation and predicted sensitivity calculated for the PDX models using a temozolomide sensitivity model that includes MGMT, MMR, HR, and NER (model #6; Fig. 5B). These results indicate that relatively small changes in HR, MMR, and NER capacity collectively contribute to changes in glioblastoma temozolomide sensitivity, and that the resistance mechanisms detailed in Fig. 2 operate in vivo. When the analysis is restricted to MGMT-deficient PDX models, the correlation between relative survival prolongation and the combined DRC score is even stronger (Fig. 5C). The strong correlation in MGMT-deficient cells underscores the importance of DNA repair pathways other than MGMT in determining the effectiveness of temozolomide therapy. Importantly, these results demonstrate that DRC measurements carried out in cells in vitro can at least partially predict therapeutic response in vivo.
Figure 5.
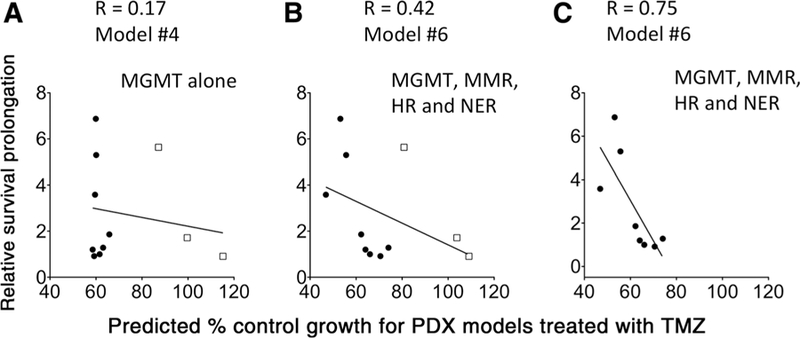
Correlation between relative survival prolongation for PDX mouse models treated with temozolomide (TMZ) and predicted percent control growth for PDX models treated with temozolomide calculated from model #4 (A) or model #6 (B and C). In C, PDX models were restricted to those expressing low levels of MGMT (black dots). Cells with low MGMT levels were defined as those for which the average percentage of reporter expression for the MGMT assay was at least 50%; this value of percentage of reporter expression was observed previously for a cell line that is known to be MGMT deficient (TK6). PDX models with higher levels of MGMT reporter expression are represented with open squares.
Discussion
The ability to choose the optimal therapy for each patient would have tremendous ramifications for cancer treatment. Advance knowledge of whether a particular therapy will be effective would increase the likelihood of achieving the primary goal of killing cancer cells, and avoid subjecting patients to unnecessary treatments that cause serious side effects and increase the risk of additional cancers (24–26). DRC continues to emerge as an important determinant for the response of cancer cells to therapy (27), making accurate DRC assessments a key goal for precision medicine (7).
The potential to predict temozolomide sensitivity from DRC in multiple pathways underscores need to measure MGMT, HR, NER, and MMR activity in order to make the most robust DNA repair-based predictions regarding the effectiveness of temozolomide therapy. However, direct measurements of DNA repair activity in clinical samples has been limited because previous methods were cumbersome, time consuming, incompatible with multiplexing, and in most cases did not report on the functional status of repair (7). Tumor MGMT status is considered a strong predictor of therapeutic response in glioblastoma (28), and clinicians use MGMT promoter methylation assays or in some cases immunohistochemical analysis of excised tumor tissue to infer MGMT activity in glioblastoma (29). Nevertheless, concerns remain about standard clinical MGMT assays that unlike FM-HCR, do not directly measure MGMT activity; immunohisto-chemical analysis is subject to individual bias (30), and although MGMT activity was, as expected, inversely correlated to MGMT promoter methylation in the glioblastoma lines in this study (Supplementary Fig. S6), the high degree of variability in the extent of promoter methylation among MGMT-deficient glioblastoma lines adds to existing evidence that promoter methylation status is not always a reliable measure of tumor MGMT activity (9, 22). Similarly, microsatellite instability assays are used to assess MMR status in clinical samples, but this approach is not quantitative and may not report minor changes in MMR capacity that are sufficient for temozolomide resistance (31, 32). Several approaches have been taken to estimate HR capacity in cancer cells (33), but they rely on transcriptional profiling and mutational signatures that may not predict the functional status of the HR pathway, and are unlikely to detect small differences in functional activity. Finally, unscheduled DNA synthesis has long been used as a method of estimating NER capacity in patient cells (34), but is relatively laborious. By contrast, FM-HCR assays provide a rapid functional assessment of DRC in key pathways that we have shown are useful predictors of temozolomide sensitivity in lymphoblastoid and PDX lines.
This work has been focused on modeling temozolomide sensitivity in glioblastoma, but the fundamental principles governing resistance to DNA-damaging agents, particularly those driven by relative changes in DRC, are expected to be applicable to multiple agents and multiple cell types. Strikingly, a model trained using DRC and temozolomide sensitivity data for lymphoblastoid cells in culture accurately predicted temozolomide sensitivity of glioblastoma cells treated in mouse PDX models. Additional cell-autonomous therapy resistance mechanisms, including changes in DNA damage signaling (35), damage detoxification mechanisms (36), and drug efflux pumps are expected to complement DNA repair-based resistance mechanisms (37). Single cell-based assays for processes that contribute to resistance, such as FM-HCR, hold the major advantage of being amenable to identifying small, therapy-resistant subpopulations within a tumor, which can expand and emerge as a resistant tumor following treatment (38). Therefore, models integrating FM-HCR assays for DNA repair-based resistance mechanisms with cell-based functional assessments of other therapy resistance mechanisms hold promise for further enhancing predictions of therapy response and advancing personalized cancer treatment.
In summary, our results indicate that temozolomide resistance in cancer cells is associated with differences in repair capacity in at least four DNA repair pathways (MMR, HR, NER, and MGMT), and that a combined DNA repair score derived from FM-HCR assays provides a more robust prognostic biomarker than MGMT activity alone for temozolomide therapy.
Supplementary Material
Acknowledgments
The authors thank Rebecca Fry, Brad Hogan, and Meriem Sefta (MIT) for help with measuring MMS sensitivity and temozolomide sensitivity in the Coriell Cell lines. The authors further acknowledge Mark Schroeder and Brett Carlson (Mayo Clinic) for propagating xenograft lines.
Grant Support
This work was supported by the NIH (DP1-ES022576 and U54-CA112967). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Disclosure of Potential Conflicts of Interest
J.N. Sarkaria reports receiving a commercial research grant from Novartis, Basilea, Cavion, Curtana, Genentech, Sanofi, Beigene, Lilly, Glaxo-Smith-Kline, Peloton, Glionova, and Bristol-Myers Squibb Pharmaceuticals. No potential conflicts of interest were disclosed by the other authors.
References
- 1.Fu D, Calvo JA, Samson LD. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat Rev Cancer 2012;12:104–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lindahl T, Wood RD. Quality control by DNA repair. Science 1999;286: 1897–905. [DOI] [PubMed] [Google Scholar]
- 3.Hoeijmakers JHJ.Genome maintenance mechanisms for preventing cancer. Nature 2001;411:366–74. [DOI] [PubMed] [Google Scholar]
- 4.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005;352:987–96. [DOI] [PubMed] [Google Scholar]
- 5.Colvin M, Alkylating agents and platinum antitumor compounds. In:Kufe DW, Pollock RE, Weichselbaum RR, Bast RC, Gansler TS, Holland JF, et al. editors. Holland-Frei Cancer Medicine 6th ed. Vol. 2 Hamilton, Ontario, Canada: BC Decker; 2003. [Google Scholar]
- 6.Pegg AE. Multifaceted roles of alkyltransferase and related proteins in DNA repair, DNA damage, resistance to chemotherapy, and research tools. Chem Res Toxicol 2011;24:618–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagel ZD, Chaim IA, Samson LD. Inter-individual variation in DNA repair capacity: a need for multi-pathway functional assays to promote transla- tional DNA repair research. DNA Repair 2014;19:199–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nagel ZD, Margulies CM, Chaim IA, McRee SK, Mazzucato P, Ahmad A, et al. Multiplexed DNA repair assays for multiple lesions and multiple doses via transcription inhibition and transcriptional mutagenesis. Proc Natl Acad Sci U S A 2014;111:E1823–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kitange GJ, Carlson BL, Schroeder MA, Grogan PT, Lamont JD, Decker PA, et al. Induction of MGMT expression is associated with temozo- lomide resistance in glioblastoma xenografts. Neuro-Oncol 2009;11: 281–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Valiathan C, McFaline JL, Samson LD. A rapid survival assay to measure drug-induced cytotoxicity and cell cycle effects. DNA Repair 2012;11: 92–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sefta M. A study of inter-individual differences in the DNA damage response. Cambridge, MA: Massachusetts Institute of Technology; 2012. [Google Scholar]
- 12.Fry RC, Svensson JP, Valiathan C, Wang E, Hogan BJ, Bhattacharya S, et al. Genomic predictors of interindividual differences in response to DNA damaging agents. Genes Dev 2008;22:2621–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tibshirani R. Regression shrinkage and selection via the Lasso. J Royal Statist Soc Series B-Methodol 1996;58:267–88. [Google Scholar]
- 14.Hajian-Tilaki K. Receiver operating characteristic (ROC) curve analysis for medical diagnostic test evaluation. Caspian J Int Med 2013;4:627–35. [PMC free article] [PubMed] [Google Scholar]
- 15.Collins FS, Brooks LD, Chakravarti A. A DNA polymorphism discovery resource for research on human genetic variation. Genome Res 1998;8: 1229–31. [DOI] [PubMed] [Google Scholar]
- 16.Newlands ES, Stevens MF, Wedge SR, Wheelhouse RT, Brock C. Temozo- lomide: a review of its discovery, chemical properties, pre-clinical devel- opment and clinical trials. Cancer Treat Rev 1997;23:35–61. [DOI] [PubMed] [Google Scholar]
- 17.Briegert M, Kaina B. Human monocytes, but not dendritic cells derived from them, are defective in base excision repair and hypersensitive to methylating agents. Cancer Res 2007;67:26–31. [DOI] [PubMed] [Google Scholar]
- 18.Fischer DS, Knobf MT, Durivage HJ. The cancer therapy handbook St. Louis (MO: ): Mosby; 1997. [Google Scholar]
- 19.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med 2008; 359:492–507. [DOI] [PubMed] [Google Scholar]
- 20.Carlson BL, Grogan PT, Mladek AC, Schroeder MA, Kitange GJ, Decker PA, et al. Radiosensitizing effects of temozolomide observed in vivo only in a subset of O6-methylguanine-DNA methyltransferase methylated glioblas- toma multiforme xenografts. Int J Rad Oncoly Biol Phys 2009;75:212–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kitange GJ, Mladek AC, Carlson BL, Schroeder MA, Pokorny JL, Cen L, et al. Inhibition of histone deacetylation potentiates the evolution of acquired temozolomide resistance linked to MGMT upregulation in glioblastoma xenografts. Clin Cancer Res 2012;18:4070–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kreth S, Thon N, Eigenbrod S, Lutz J, Ledderose C, Egensperger R, et al. O- methylguanine-DNA methyltransferase (MGMT) mRNA expression predicts outcome in malignant glioma independent of MGMT promoter methylation. PLoS ONE 2011;6:e17156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tredan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst 2007;99:1441–54. [DOI] [PubMed] [Google Scholar]
- 24.Leone G, Voso MT, Sica S, Morosetti R, Pagano L. Therapy related leukemias: susceptibility, prevention and treatment. Leuk Lymphoma 2001;41:255–76. [DOI] [PubMed] [Google Scholar]
- 25.Wood ME, Vogel V, Ng A, Foxhall L, Goodwin P, Travis LB. Second malignant neoplasms: assessment and strategies for risk reduction. J Clin Oncol 2012;30:3734–45. [DOI] [PubMed] [Google Scholar]
- 26.Oeffinger KC, Bhatia S. Second primary cancers in survivors of childhood cancer. Lancet (London, England) 2009;374:1484–5. [DOI] [PubMed] [Google Scholar]
- 27.Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer 2008;8:193–204. [DOI] [PubMed] [Google Scholar]
- 28.Hegi ME, Diserens A, Gorlia T, Hamou M, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 2005;352:997–1003. [DOI] [PubMed] [Google Scholar]
- 29.Stupp R, Brada M, van den Bent MJ, Tonn JC, Pentheroudakis G. High- grade glioma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 2014;25 Suppl 3:iii93–101. [DOI] [PubMed] [Google Scholar]
- 30.Preusser M, Janzer RC, Felsberg J, Reifenberger G, Hamou MF, Diserens AC, et al. Anti-O6-methylguanine-methyltransferase (MGMT) immunohis- tochemistry in glioblastoma multiforme: observer variability and lack of association with patient survival impede its use as clinical biomarker. Brain Pathol 2008;18:520–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McFaline-Figueroa JL, Braun CJ, Stanciu M, Nagel ZD, Mazzucato P, Sangaraju D, et al. Minor changes in expression of the mismatch repair protein MSH2 exert a major impact on glioblastoma response to temo- zolomide. Cancer Res 2015;75:3127–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shin KH, Park JG. Microsatellite instability is associated with genetic alteration but not with low levels of expression of the human mismatch repair proteins hMSH2 and hMLH1. Eur J Cancer 2000;36:925–31. [DOI] [PubMed] [Google Scholar]
- 33.Timms KM, Abkevich V, Hughes E, Neff C, Reid J, Morris B, et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res 2014;16:475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kelly CM, Latimer JJ. Unscheduled DNA synthesis: a functional assay for global genomic nucleotide excision repair. Methods Mol Biol 2005;291: 303–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- 36.Rocha CR, Garcia CC, Vieira DB, Quinet A, de Andrade-Lima LC, Munford V, et al. Glutathione depletion sensitizes cisplatin- and temozolomide- resistant glioma cells invitro and invivo. Cell Death Dis 2014;5:e1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Noguchi K, Katayama K, Sugimoto Y. Human ABC transporter ABCG2/ BCRP expression in chemoresistance: basic and clinical perspectives for molecular cancer therapeutics. Pharmacogenomics Personalized Med 2014;7:53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012;481:506–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.