

Abstract
The intrinsically disordered protein β-synuclein is known to inhibit the aggregation of its intrinsically disordered homologue, α-synuclein, which is implicated in Parkinson’s disease. While β-synuclein itself does not form fibrils at the cytoplasmic pH 7.4, alteration of pH and other environmental perturbations are known to induce its fibrilization. However, the sequence and structural determinants of β-synuclein inhibition and self-aggregation are not well understood. We have utilized a series of domain-swapped chimeras of α-synuclein and β-synuclein to probe the relative contributions of the N-terminal, C-terminal and the central Non-Amyloid-β Component (NAC) domains to the inhibition of α-synuclein aggregation. Changes in the rates of α-synuclein fibril formation in the presence of the chimeras indicate that the NAC domain is the primary determinant of self-association leading to fibril formation, while the N- and C-terminal domains play critical roles in the fibril inhibition process. Our data provide evidence that all three domains of β-synuclein together contribute to providing effective inhibition, and support a model of transient, multi-pronged interactions between IDP chains in both processes. Inclusion of such multi-site inhibitory interactions spread over the length of synuclein chains may be critical for the development of therapeutics that are designed to mimic the inhibitory effects of β-synuclein.
Keywords: Parkinson’s disease, protein-protein interactions, chimeras, fluorescence, intrinsically disordered proteins
Graphics Abstract
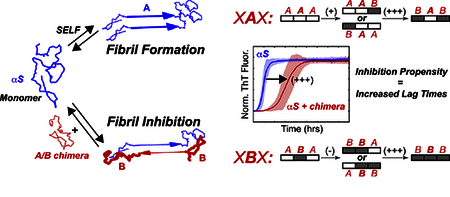
1. Introduction
Intrinsically disorder proteins (IDPs) lack stable secondary or tertiary structure, yet are important for many biological processes, such as intra-cellular and inter-cellular signaling and transcription regulation [1–3]. Much work has been done to try to describe the conformations of IDPs, which exist as ensembles of interconverting conformers, through a variety of techniques including nuclear magnetic resonance (NMR) spectroscopy, single-molecule fluorescence, small-angle scattering, and molecular dynamics computational modeling [4–12]. Beyond their natural physiological functions, IDPs have also been implicated in the progression of several debilitating illnesses, such as Alzheimer’s, Parkinson’s, and prion diseases, through a mechanism of self-association to form toxic oligomers and amyloid fibrils that accumulate in the brain [1, 13, 14]. Due to their conformational heterogeneity and dynamics, IDPs are also difficult drug targets, and few promising therapeutic strategies against neurodegenerative diseases involve binding to IDPs [15, 16]. One reason for the lack of effective drugs that target aberrant IDP self-association is that design strategies that are typically applied to folded proteins are challenging to implement in the context of dynamic IDP ensembles [16].
The 140-amino acid α-synuclein (αS) is a monomeric, intrinsically disordered protein that is involved in the progression of Parkinson’s disease, with a physiological function that is not yet fully understood [17–19]. It self-associates and eventually forms insoluble fibrils of parallel, in-register, β-sheet structure [20–23] which form Lewy Bodies (LBs), a pathophysiological hallmark of the disease progression [24]. It can also adopt a partial helical structure in the presence of a membrane [25, 26]. The mechanism by which αS self-associates has been extensively investigated [27–37], yet effective therapeutic strategies that inhibit αS aggregation processes and prevent the progression of Parkinson’s disease do not exist to date. However, several small molecules that interact with αS and alter the aggregation process, including phthalocyanine tetrasulfonates [38], epigallocatechin gallate [39], N-(4-Fluorophenyl)benzenesulfonamide [40], curcumin [41], CLR01 [42], gallic acid [43], and nortriptyline [44] have been identified. Another approach shown to modulate aggregation behavior of αS and other IDPs is to use immuno-based therapies of natural or designed antibodies ([45–47], also see reviews [48, 49]).
β-Synuclein (βS), an intrinsically disordered protein that co-localizes with αS [19], has been suggested to be a ‘natural inhibitor’ of αS aggregation [50]. It is a homologous, 134-amino acid IDP that is expressed at similar levels to αS, and has been shown in vitro to delay aggregation, and in vivo animal studies to diminish inclusion body formation [18, 27, 29, 50–52]. We have recently shown that while αS forms fibrils under neutral physiological conditions, βS does not, and only forms fibrils under mildly acidic conditions [53]. Other studies have also shown that βS forms fibrils under altered environmental conditions [27, 54]. Given the observed co-localization of βS and αS in vivo, an interesting therapeutic alternative may be to directly target the disease-associated αS IDP with its βS IDP partner, as a means to address the issue of targeting the natural plasticity of IDP ensembles. Proposed mechanisms for how βS delays fibril formation of αS include early-stage inhibitory head-to-tail interactions between αS and βS monomers [29, 55], and/or late-stage competitive βS binding to secondary nucleation sites [52]. However, effective therapeutic strategies require a deeper understanding of the molecular basis of αS self-aggregation versus αS/βS co-aggregation, and in particular, elucidation of the sequence regions that direct self-versus co-aggregation.
In the current work, we probe the contributions of different sequence regions (three domains) of βS towards the interaction with, and inhibition of, αS fibril formation. We previously developed a library of αS/βS domain-swapped chimeras [53]. Comparison of the rates of fibril formation by these chimeric constructs with and without co-incubation with αS allows the delineation of the contribution of the three synuclein domains to self-association and fibrilization inhibition. We find that αS self-fibrilization is directed primarily by its central NAC domain, whereas inhibition by the highly homologous βS requires all three domains of βS. Thus, molecular design strategies that seek to mimic βS-based inhibition may require inclusion of similar multi-pronged interactions spread over the entire IDP chain for effective inhibition.
2. Results
The synuclein family of proteins are composed of three domains: a 60-residue N-terminal domain, a 24–35 residue NAC domain, and a 31–50 residue C-terminal domain. The N-terminus is highly conserved between αS and βS, with only 6 amino acid differences (Fig. 1a). The NAC domain forms the hydrophobic core of each synuclein, with the largest difference being that the βS NAC is 11 residues shorter than αS. The C-terminal domain is the least conserved between αS and βS, but share similar characteristics, with both being comparatively more acidic and proline-rich than the N and NAC domains. In the remainder of this work, we use a system of XXX to define the domains (Fig. 1a) of each synuclein chimera, where each X indicates either an αS-domain (A) or a βS-domain (B), with the domains specified in a N-terminal, NAC, and C-terminal order. For example, BAA refers to a chimera with an N-terminal βS-domain sequence, a NAC αS-domain sequence and a C-terminal αS-domain sequence. The AAA or BBB chimeras refer to the wild-type αS or βS respectively.
Figure 1.
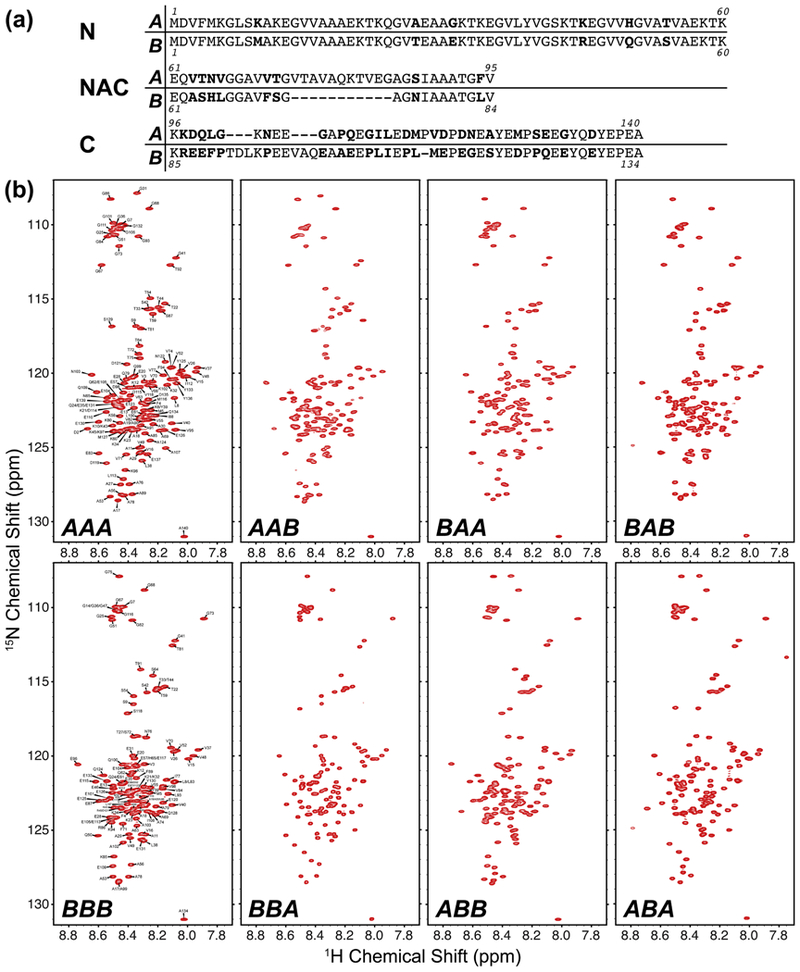
(a) Comparison of the primary sequences of αS and βS, broken down by the N-terminal, non-amyloid β component (NAC) and C-terminal domains. The sequences were aligned using EMBOSS Stretcher [78]. Residues that are different between the two aligned sequences are indicated in bold font. (b) 1H-15N HSQC spectra of the synuclein chimeras at pH 6 (20 mM MES, 100 mM NaCl), at 288 K. The small chemical shift dispersion and narrow linewidths indicate that all of the chimeras are intrinsically disordered.
Chimeras of αS and βS Exist as Intrinsically Disordered Proteins
To assess whether the domain swapped chimeras maintain the identity of an unfolded, intrinsically disordered protein, we measured the 1H-15N HSQC spectrum of each chimera and compared them to the HSQC spectra of WT αS and βS. Full backbone assignments of αS and βS have been performed previously [56, 57], and both spectra show the sharp peaks and a narrow chemical shift distribution characteristic of an IDP (Fig. 1b). The 6 domain-swapped chimeras all show the same spectral characteristics as the wild-type synucleins, confirming that they also exist as intrinsically disordered ensembles in solution. The chimeras all show minimal chemical shift perturbations relative to their respective domains from the wild-type AAA and BBB proteins, suggesting that they may retain some of the conformational characteristics of these αS and βS ensembles, but further analysis of the motion and dynamics of the chimeras is needed for detailed comparison between these disordered proteins.
Co-incubation of αS with βS NAC Domain-Containing Chimera Delays αS Aggregation
We utilized a thioflavin T (ThT) fluorescence assay to monitor the amyloid fibril formation rates in order to determine the fibril formation propensities and inhibitory potential of the chimeras. Our ThT fluorescence assays were performed under conditions of constant agitation, which promotes fragmentation of fibrils and aggregates, a mechanism that contributes to an accelerated fibril formation fluorescence profile relative to non-seeded fibrilization under quiescent conditions [28, 58]. Our previous work showed that the NAC domain was the primary determinant of self-aggregation of synuclein constructs: XBX chimeras did not form fibrils at pH 7.3, whereas XAX chimeras formed fibrils at the same pH [53]. To determine the contributions of the three βS domains to inhibition, we investigated the ability of βS NAC-domain containing chimeras (XBX) to affect αS fibril formation when present in a 5:1 stoichiometric excess (chimera : αS) under aggregation promoting conditions. We chose to use a 5-fold excess of chimera in our assays since previous work had shown that this ratio provides a maximal effect on modulating fibril formation behavior [29], and would provide the best chance to distinguish differences between the chimeras. The ThT fluorescence curves of amyloid fibril formation follow the form of a general logistic function, and have three parts: a lag phase, a growth phase, and an equilibrium phase. The lag phase can be described by a lag time (tlag), which is a reflection of the rate of the initial nucleation of monomers to form growth competent aggregates and eventually ThT active amyloid fibrils. The growth phase generally describes the rate of fibril elongation, as monomers are added to the ends of existing fibrils, which allows for more ThT binding and fluorescence. The maximal ThT fluorescence intensity at the plateau is proportional to the amount of amyloid fibrils formed as the fibril formation reaction reaches equilibrium. However, because of the variability of the absolute fluorescence intensities due to the differential ThT binding to the various chimeras, we chose to normalize the ThT curves to the maximal intensity and observe the lag times of fibril formation. Thioflavin T fluorescence curves show varying degrees of inhibition of fibril formation by XBX chimeras co-incubated with WT αS at neutral pH (Fig. 2a, red), conditions under which XBX chimeras do not form fibrils (Fig. 2a, black). The WT βS (BBB) construct appears to be the best inhibitor as indicated by the largest difference in lag time (Δtlag approximately 9 hours) for fibril formation under the conditions of our assay. Mixtures of αS with constructs BBA, ABA and ABB, in contrast, feature shorter lag times (Δtlag time between 3–5 hours), indicating that while the presence of βS NAC domain is still inhibitory, the identity of flanking domains modulates the inhibitory ability of the chimeric protein. Notably, the ability of βS-NAC containing constructs to inhibit fibril formation is retained at pH 5.8, where these constructs also form fibrils (with relatively longer tlag compared to αS). At both pH values the BBB construct is consistently a robust inhibitor of αS aggregation, indicating that all three βS domains contribute to the inhibitory potential of βS.
Figure 2.
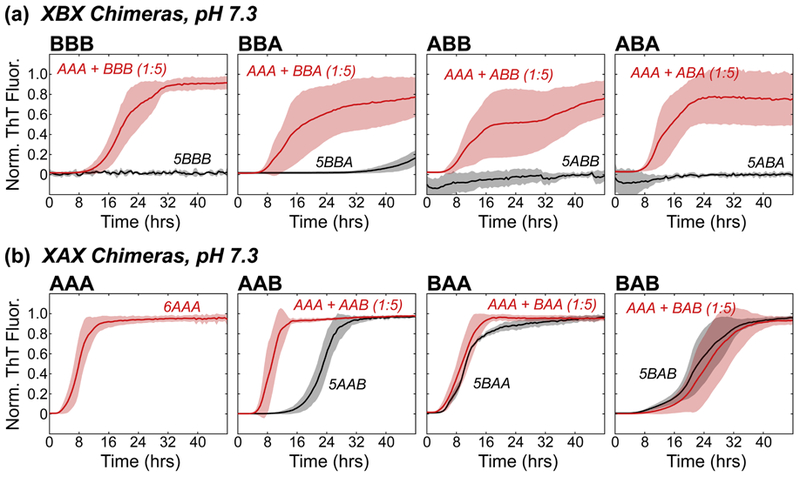
Normalized ThT fluorescence of self-incubated (black) or co-incubated (red) fibril formation of (a) XBX chimeras and (b) XAX chimeras at pH 7.3. Each fluorescence trace is an average of at least three measurements, and the standard deviation is reported. For the sake of clarity, traces that showed no increase in ThT fluorescence from the baseline were normalized and shifted to appear near zero by subtracting 1 from the normalized intensities (5ABA, 5ABB, 5BBB). The stoichiometry for each assay is indicated in parentheses for co-incubated data sets, with a 1 indicating a protein concentration of 1 mg/mL, and a 5 indicating a 5 mg/mL concentration. In the case of self-incubated data, the stoichiometry is indicated before the chimera name (e.g. 5XXX, 5 mg/mL protein concentration).
As all co-incubated mixtures eventually formed fibrils (as evidenced by saturation of ThT fluorescence), we investigated the incorporation of the inhibitor constructs in the fibrils by mass spectrometry. Strikingly, BBB and other XBX chimera were sparingly incorporated in the fibrils (Table 1) in spite of initially being present in 5:1 (chimera : αS) excess. Taken together, lag times and mass spectrometry results indicate that interactions between αS and βS-NAC containing chimeras (and WT βS) compete with the fibrilization-promoting self-interactions between αS chains.
Table 1.
Ratios of WT α-synuclein and chimeras obtained from mature fibrils taken from the endpoint of the co-incubated ThT assays, as determined by ESI-MS analysis of the dissolved fibrils.
| pH 7.3 | pH 5.8 | |
|---|---|---|
| αS : AAA | - | - |
| αS : BAA | 1 : 3.4 | 1 : 3.4 |
| αS : AAB | 1 : 2.4 | 1 : 4.8 |
| αS : BAB | 1 : 1.5 | 1 : 1.5 |
| αS : ABA | 3.4 : 1 | 1 : 1 |
| αS : BBA | 1 : 1 | 1 : 5 |
| αS : ABB | 4 : 1 | 3.5 : 1 |
| αS : BBB | 4 : 1 | 1.2 : 1 |
| αS : βΕ61Α | 1.6 : 1 | - |
Inhibitory Potential of the βS N-Terminal and C-Terminal Domains
To delineate the contribution of synuclein N- and C-terminal domains to amyloid formation, we next investigated the inhibitory ability of XAX chimeras on αS fibril formation with identical experimental conditions under which the βS-NAC domain containing constructs act as inhibitors. As these constructs themselves form fibrils (Fig. 2b, black), albeit with longer tlag compared to αS, two-plateau ThT-fluorescence curves are expected if self-interactions dominate over chimera-αS interactions. On the other hand, if significant chimera-αS interactions exist, a single-plateau is expected in the ThT-fluorescence curves. Indeed, we observed single plateau fluorescence curves of XAX chimeras, indicating the presence of significant chimera-αS interactions in the mixtures.
Further examination of the fibril formation rates at pH 7.3 reveals that for all αS-chimera mixtures, tlag either decreases or stays the same relative to the tlag of chimera self-incubation (Fig. 2, 3a). For example, the co-incubated AAB chimera has a significantly shorter tlag, decreasing by 11 hrs from the self-incubated state (Fig. 2b). In stark contrast, the BAB chimera shows the greatest resistance to fibril formation in the presence of αS with an apparent tlag that is approximately equal to its self-association. Thus, while the presence of αS NAC domain promotes fibril formation in the presence of at least one other αS domain, the presence of two flanking βS domains (BAB) is sufficient to overcome the effect of αS-NAC domain. These results highlight the significant inhibitory potential of βS N- and C-terminal domains especially when both are simultaneously present in the inhibitor IDP.
Figure 3.
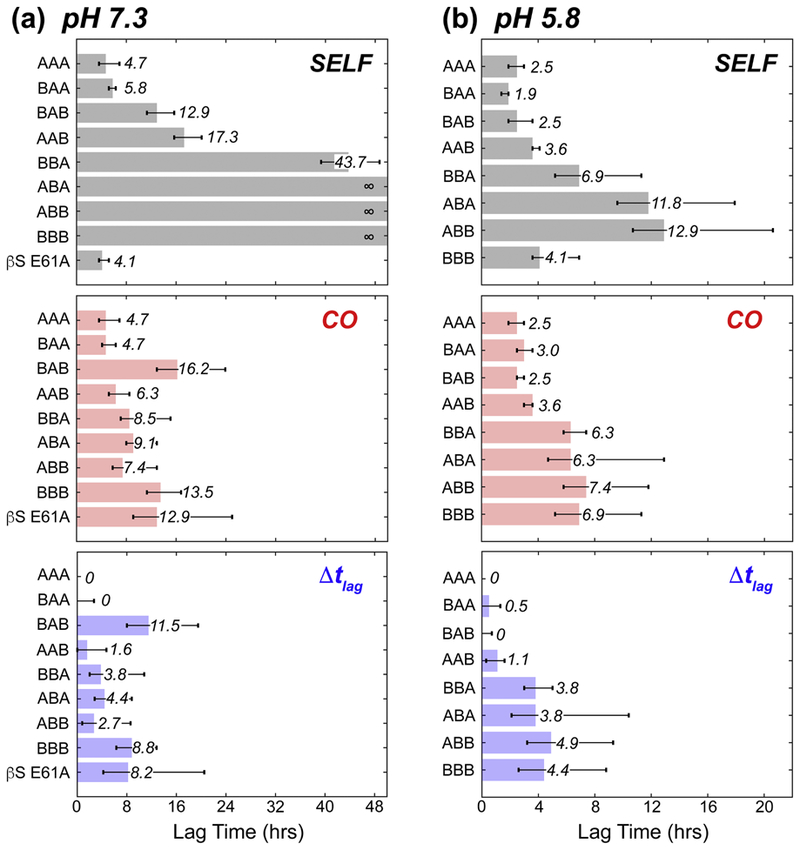
Comparison of the time it takes the fluorescence intensity to reach 10% of its normalized maximum (ie. lag time, tlag) for the chimeras at (a) pH 7.3 and (b) pH 5.8. The lag times of the control AAA are subtracted from the co-incubated (middle, red) lag times to give the lag time differences (Δtlag) shown in blue at the bottom. In each panel, the lag time is presented as a bar with text label, and the error bars shown represent the range of times at which the standard deviation reaches 10% normalized fluorescence intensity.
At pH 5.8, the ThT fluorescence profiles of the mixtures of αS and XAX chimeras are very similar to those observed for the chimeras and αS individually (Fig. S1), and there is little difference in the tlag between the self-incubated and co-incubated assays (Fig. 3b). Thus, the inhibitory potential of the BAB chimera is significantly diminished under mildly acidic conditions, and fibril-promoting interactions induced by the αS NAC domain, a well-known determinant of αS aggregation [32, 53], prevail over inhibitory ones. As such, at low pH, Δtlag times follow a NAC-determined dichotomy, with the XAX chimeras showing essentially no effect while the XBX chimeras show a 4–5 hour increase in tlag. These trends are also reflected in the relative amounts of the chimeric protein detected in the fibrils by mass spectrometry. At neutral pH, the XAX chimeras are detected as being in near-stoichiometric excess over αS in the fibrils, in contrast with fibrils obtained from mixtures of αS with the XBX chimeras where αS is in excess. The XAX chimeras, as well as the ABB chimera, are obtained in stoichiometric ratios in fibrils formed by mixtures at pH 5.8, where fibrilization of the chimeras is induced by αS. Taken together, results obtained with XAX chimeras highlight how N-terminal and C-terminal domains of βS contribute to fibrilization inhibition.
Co-Incubated βS E61A Shows a Large Increase in Lag Time
At pH 7.3, the BAB construct leads to the longest delay in αS fibril formation under co-incubation conditions, but the fibril formation rates of the αS-ΒΑΒ mixture closely follow that of BAB self-aggregation. To investigate the relationship between self-aggregation and inhibitory potential, we next utilized a βS variant with a single substitution in the NAC domain, E61A. This substitution at the interface between the N-terminal and NAC domains in βS, renders its fibril formation insensitive to environmental pH: WT βS does not form fibrils at neutral pH, but βS E61A does so at a rate similar to WT αS (Fig. 4a) [53]. Thus, we reasoned that the inhibitory behavior of βS E61A, with a self-fibrilization-promoting αS-like NAC domain, should allow the delineation of the relationship between inhibitory potential and self-associated fibril formation. If inhibition by a variant is largely a reflection of its own fibril formation propensity, a mixture of βS E61A and αS should show a small (αS-like) tlag. On the other hand, if inhibition and self-association are decoupled, given the high similarity between βS E61A and the BBB construct, a long tlag (indicative of inhibition) is expected. Indeed, when βS E61A is co-incubated with WT αS at pH 7.3, the resulting ThT fluorescence curve has a tlag comparable to both co-incubated BBB and BAB chimeras (Fig. 4b), and shows the largest increase in tlag (~9 hrs) between self-incubated and co-incubated mixtures of any of the protein constructs observed here (Fig. 3a). The Δtlag time shows a comparable increase on par with BBB, however the fluorescence curves show more complex behavior relative to BBB (Fig. 4b). The increase in Δtlag of βS E61A, along with that of BAB and BBB, further supports the view that the N- and C-terminal regions enhance the inhibition potential independent of the fibril formation propensity of the chimeric inhibitor.
Figure 4.
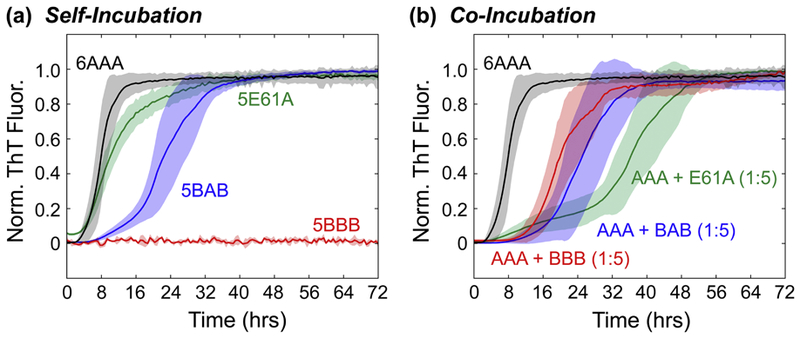
Normalized ThT fluorescence of βS with the E61A mutation (green), showing (a) self-incubated or (b) co-incubated fibril formation at pH 7.3. AAA (black), BBB (red), and BAB (blue) are shown for comparison. The stoichiometry for each assay is indicated in parentheses for co-incubated data sets, with a 1 indicating a protein concentration of 1 mg/mL, and a 5 indicating a 5 mg/mL concentration. In the case of self-incubated data, the stoichiometry is indicated before the chimera name (e.g. 5XXX, 5 mg/mL protein concentration).
Fibril Morphologies of Co-Incubated Chimeras from AFM
Atomic force microscopy (AFM) was used to gather information on the overall morphology of the mature fibrils formed at the end points of the ThT assays. The representative AFM images (Fig. S2) show that BBB, ABB and ABA co-incubated fibrils at pH 7.3 have a similar morphology to the control AAA, with long fibrils which are not clumped together. In contrast, the AAB, BAA, BAB, and BBA co-incubated fibrils show long fibril morphologies that are clumped together.
At low pH (Fig. S3), the control AAA fibrils are much shorter and more clumped relative to the high pH AAA image, as observed previously [53]. The fibrils formed by AAB and BBA chimeras co-incubated with αS share a similar morphology as the low pH AAA. In contrast, the BAA and ABA co-incubated chimeras show only globular oligomeric species under AFM. The observed ThT intensity increases for these samples (Fig. S1) indicate that these globular oligomeric species are likely composed of non-fibrillar aggregates that retain the ability to bind ThT [59, 60], or that the fibrils present in the sample did not adhere to the mica surface. AFM images of the ABB co-incubated sample shows both long, thin fibrils as well as globular oligomeric species, while the BBB and BAB co-incubated samples share similar morphology to the BBB pH 7.3 co-incubated fibrils.
These AFM imaged fibril morphologies correlate with the amount of chimera retained in the mature co-incubated fibril at pH 7.3; the appearance of smaller fibrils, oligomers, and more fibril clumping roughly correlate with chimeras being retained at ratios greater than or equal to 50% (Table 1). However, co-incubated fibril morphology does not seem to indicate the ability of a chimera to inhibit αS aggregation, since BBB and BAB have distinctly different morphologies yet share a similar tlag in the pH 7.3 ThT assays.
3. Discussion
Previous studies have shown that the IDP βS interacts with its close homolog αS and is capable of delaying or preventing αS fibril formation both in vitro and in vivo. However, the molecular-level determinants of the inhibition by βS are not well understood. Unlike folded protein-protein interactions that are determined by select “hot-spot” residues (which contribute disproportionately to the binding free energy [61]), IDP-based protein-protein interactions are generally spread over stretches of contiguous residues, and determined primarily by composition of these segmental domains [62]. The importance of determining the sequence regions that mediate protein-protein interactions has been highlighted in other investigations of the islet-amyloid polypeptide (IAPP) [63–66], β2-microglobulin [67, 68], and tau and Αβ peptides from Alzheimer’s disease [66, 69, 70], where co-polymerization between distinct species can have drastic effects on aggregation [71]. Therefore, we used αS/βS chimeric constructs and the E61A βS variant to probe the determinants of the observed inhibition of αS fibril formation by βS. Our results enable the delineation of the contribution of the three βS domains to its inhibitory effects, and provide support for a model in which fibrilization of αS (or chimeric constructs) and its inhibition by βS (or chimeric constructs) involves a balance between homotypic self-association and heterotypic co-association between IDP chains, both on- and off- the fibril formation pathway (Fig. 5a). At physiological pH, the self-associated fibril formation propensities of chimeric constructs are primarily determined by their NAC domains, with constructs containing βS NAC domains being largely resistant to forming fibrils, while constructs with αS NAC domains are prone to fibril formation. However, the inhibition potential of different chimeric constructs is determined to a large extent by the presence of βS N-terminal and C-terminal domains as evidenced by (1) the largest tlag being observed for mixtures of BBB and BAB chimera with αS, and that (2) the βS E61A mutant, which has an altered, self-fibrilization promoting NAC domain but wild type N- and C-terminal domains, continues to be a robust inhibitor of αS fibrilization. A comparison of the effects of single-domain swaps in the N-terminal and C-terminal domains (Fig. 5b) relative to a fixed NAC domain indicates that the contribution of each domain is context-dependent, and the greatest inhibitory potential is observed for constructs featuring both these domains simultaneously (BAB and BBB), suggesting that their effects may be cooperative. While the NAC domain appears to primarily determine self-association propensity, it also determines the amount of chimeric proteins incorporated in the mature fibrils. Chimeras featuring a βS NAC domain are significantly less abundant in the co-incubated fibril than those with the αS NAC domain. Thus, our results suggest that all three synuclein domains contribute to the balance of stabilities between homotypic interactions, leading to fibril formation, and heterotypic interactions, leading to inhibition of fibril formation.
Figure 5.
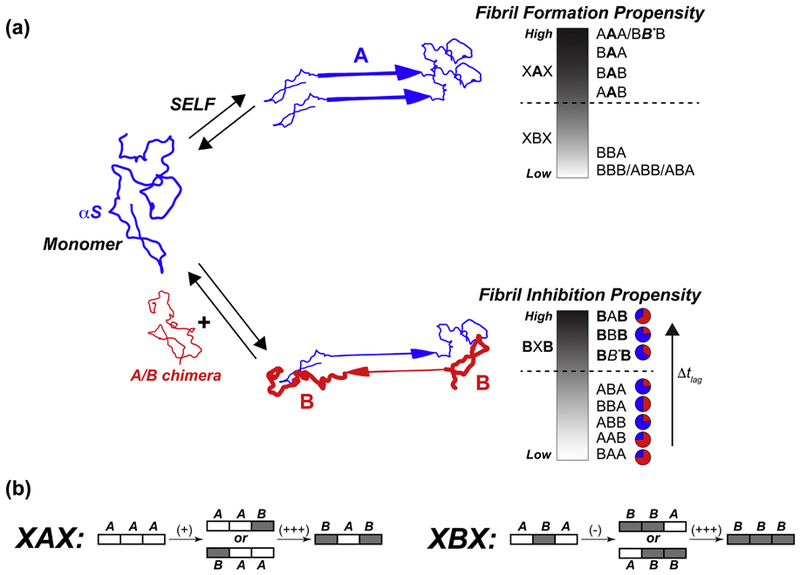
Schematic representation of synuclein aggregation pathways (a), in the cases of self-incubation (top) and co-incubation (bottom). Synuclein exists as an intrinsically disordered monomer in solution. In the case of self-incubation, monomers can interact in a predominately head-to-head configuration, which can lead to fibril formation. The propensity of the different chimeras to form fibrils falls along a continuum which can be generally separated by the NAC domain: XAX chimeras tend to quickly form fibrils at neutral pH, while XBX chimeras do not form fibrils (or are very resistant). When co-incubated together at neutral pH, the αS and chimera monomers can transiently interact in either a head-to-head orientation, which leads to on-pathway fibril formation, or in an off-pathway, head-to-tail orientation, which acts to kinetically trap the αS monomers and increase the time it takes to form fibrils. The propensity to inhibit, or slow down (i.e. increase Δtlag), fibril formation again falls along a continuum: inhibition is very high for BXB chimeras (large Δtlag), and lower for all others (small Δtlag). The relative proportions of αS (blue) and chimera (red) found in mature, co-incubated fibrils at pH 7.3 are shown as pie charts, and are summarized in Table 1. The BB*B chimera in this figure refers to the βS E61A chimera, indicating the fact that this mutant has exactly the same domains as the BBB chimera except for the single mutation in the NAC region. (b) Schematic of single-domain swaps and their effects on inhibition propensity, relative to A-NAC (left) or B-NAC (right) domains.
What is the underlying structural basis for the observed aggregation promoting and aggregation inhibiting effects in our study? One possibility is that the βS N-terminal and C-terminal domains may stabilize the off-pathway head-to-tail dimers between inhibitors and αS, whereas αS NAC promotes on-pathway head-to-head association to yield fibrils. For the BAB construct, the off-pathway head-to-tail interactions may be stronger than the head-to-head configurations otherwise stabilized by the αS NAC, and thereby inhibit fibril formation to a greater extent. Evidence for βS N-terminal to αS C-terminal transient interactions was previously observed in solution NMR experiments with these IDPs [29], and more recently single-molecule fluorescence experiments have shown the importance of early αS-βS monomer interactions in inhibiting the assembly and propagation of αS fibrils [55]. Our inhibition data, which shows that multiple domains are important for efficient inhibition, are consistent with these scenarios where multi-pronged, weak interactions spread over the synuclein chain contribute to its inhibitory potential. However, future NMR investigations of the interactions between our chimeric constructs and αS will provide more detailed insight into the residue-level contributions to the observed inhibition induced by the different βS domains.
Another key observation from our study is that inhibition by the WT βS protein and chimeric constructs is abolished at a moderately low pH 5.8, a condition we have previously shown allows βS to fibrillate [53]. Taken together, these results suggest that pH decrease along the endosomal-lysosomal pathway, which can induce fibrilization of both synucleins, may serve as a mechanism for homeostasis in the cell by removing these synucleins from the soluble protein pool and inducing their degradation in the lysosomal compartment. Indeed, evidence from recent work [72] suggests that cells may utilize small pH changes to induce phase separation of proteins as a mechanism for storing, protecting, or inactivating proteins.
It is also worth noting that the timescales of synuclein aggregation in vivo and in vitro are significantly different; βS presumably exerts its inhibitory effect for decades in healthy individuals, whereas under the conditions of our assay, fibril formation is observed within a few days. However, our biochemical assays are performed under aggregation promoting experimental conditions, yet the relative effects of various chimeric proteins relative to βS may indicate their relative physiological inhibitory potential. Previous studies on SOD1 aggregation found a remarkable correlation between disease progression in vivo and biochemical kinetics of aggregation [73]. Trends in aggregation delay induced by different chimeras (i.e. tlag) in vitro and their inhibitory potential in vivo will need to be elucidated for deriving a more quantitative relationship between these factors.
As the inhibitory potential of βS against αS fibril formation has long been recognized, several therapeutic strategies have sought to mimic its binding effects using molecules such as βS-derived peptides [74, 75] or small molecules [38–42, 44] as leads. Our results with chimeric constructs highlight the challenges in designing effective inhibitors by peptide/small-molecule mimicry of βS: the inhibition by βS is not localized to a single domain, such as the NAC domain, but arises due to multiple interactions spread throughout the three domains of the protein. Therefore, low-molecular weight compounds are unlikely to mimic and improve upon these interactions, and protein-based inhibitors are likely to be more promising mimics of inhibitory βS interactions with αS.
4. Materials and Methods
Protein Expression and Purification
Domain swapped αS/βS chimeras were obtained via a Gibson assembly protocol as described previously [53]. Plasmids were transformed into Escherichia coli BL21 DE3 for expression in LB media, or M9 minimal media containing 15NH4C1 for 15N labeling. Purification followed a “harsh” protocol as described previously [76]. Briefly, cultured cells were pelleted at 4.5k rpm for 30 minutes, suspended in 10 mM PBS buffer (pH 7.4), and homogenized three times at 10–15k psi. Cell lysates were pelleted (20k rpm, 30 minutes), and streptomycin sulfate was added to the supernatant (10 mg/mL) and mixed for 10–15 minutes at 4 °C. This mixture was pelleted (20k rpm, 30 minutes), and ammonium sulfate was added to the supernatant (0.361 g/mL) and mixed for 60 minutes at 4 °C, then centrifuged (20k rpm, 30 minutes). The pellet was suspended in 10 mM PBS buffer (pH 7.4), boiled for 15 minutes and cooled to room temperature. After final centrifugation (20k rpm, 30 minutes), the supernatant was dialyzed (6–8 kDa MWCO, Spectrum Laboratories Inc.) against 15 mM Tris overnight. The dialyzed protein solution was then loaded onto a HiTrap Q HP anion-exchange column (GE Life Sciences, Pittsburgh, PA) pre-equilibrated at pH 7.4 (25 mM Tris-HCl) on an Akta pure FPLC system (GE Life Sciences, Pittsburgh, PA), and eluted with 200–300 mM NaCl gradient. The purified protein was dialyzed against 15 mM ammonium biocarbonate, with four buffer changes, then lyophilized and stored at −20 °C until use. Purity and identity of the protein was checked via SDS-PAGE and ESI-MS.
Thioflavin T Fluorescence Assay
Lyophilized protein powder was dissolved in the desired buffer, either at pH 5.8 (20 mM phosphate, 100 mM NaCl) or pH 7.3 (10 mM PBS). Any large oligomers were removed by using a 50 kDa filter, and the protein solution was concentrated using a 3 kDa filter (Millipore Sigma, St. Louis, MO). Samples were diluted to either 1 mg/mL or 5 mg/ml protein concentration, mixed with 20 μΜ ThT (Acros Organics, Pittsburgh, PA), and loaded into 96 well plates (Corning, Corning, NY). A single Teflon bead (3 mm, Saint-Gobain N.A., Malvern, PA) was added to each well, and the wells were sealed with tape. Plates were shaken at 37°C and 600 rpm for at least 72 hours, and the increase in ThT fluorescence intensity (480 nm) was measured every 33 minutes by a POLAR Star Omega plate reader (BMG Labtech, Cary, NC). At least 3 replicates of the ThT assay were recorded for each sample. Each fluorescence trace was normalized to the intensity at 72 hours, and the average and standard deviation of these traces are presented.
Analysis of Fibrils by ESI-MS
Mature fibril samples were obtained from the end points of the ThT assay, and washed with 10 mM PBS buffer at least five times to remove residual, un-fibrilized monomer. A single wash step consisted of pelleting the fibrils by centrifugation at 14k rpm for 2 hours, removing the supernatant, and then re-suspending the fibrils in 1mL of fresh PBS buffer. After washing, fibrils were pelleted a final time by centrifugation at 14k rpm for 2 hours. The supernatant was removed, and the pelleted fibrils were then dissolved in 1 mL of 4 M guanidine hydrochloride (Millipore Sigma, St. Louis, MO) and incubated at room temperature for at least 12 hrs, in order to obtain the fibril’s component monomers. The protein solution was dialyzed (3.5 kDa MWCO) overnight against 50 mM ammonium acetate with 0.1% formic acid. The sample was diluted to a final protein concentration of 10 μΜ for electrospray ionization MS.
AFM Sample Preparation Protocol
Mature fibril samples were obtained from the end points of the ThT assay. Each fibril sample (20 μL) was coated onto freshly cleaved mica (Ted Pella Inc., Redding, CA) and incubated for 15 minutes. The mica surface was then washed with water (200 μL) three times to remove any un-adsorbed protein species and salt, and air-dried for 1 hour before imaging. All AFM images were obtained with a Park Systems NX-10 (Suwon, South Korea) using PPP-NCHR tips (Nanosensors, Neuchatel, Switzerland).
NMR Sample Preparation and Experiments
Lyophilized protein powder was dissolved in buffer at pH 6 (20 mM MES, 100 mM NaCl). The protein was filtered through a 100 kDa filter to remove higher order oligomers, and then concentrated using a 3 kDa filter (Millipore Sigma, St. Louis, MO). Samples were diluted to a final protein concentration of 200–300 μΜ with 10 % D2O added. 1H-15N HSQC spectra were recorded on Varian Inova or Bruker Avance III spectrometers at 600 MHz 1Η Larmor frequency. Spectra were processed using NMRPipe [77]
Supplementary Material
Highlights:
α-Synuclein aggregation is inhibited by β-synuclein.
α/β-Synuclein chimeras reveal domain contribution to inhibition.
The NAC domain is the primary, but not only, determinant of self-aggregation.
β-Synuclein N- and C-terminal domains critical for α-synuclein inhibition.
All three β-synuclein domains work in concert to provide effective inhibition.
Acknowledgements
We thank Dr. Gina Moriarty for helpful discussions. This work was supported by National Institutes of Health Grant GM110577 (J.B.). T.B.A. was supported by a Graduate Assistance in Areas of National Need (GAANN) fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of Interest: none
References
- [1].Uversky VN, Oldfield CJ, Dunker AK. Intrinsically disordered proteins in human diseases: Introducing the D2 concept. Annu Rev Biophys. 2008;37:215–46. [DOI] [PubMed] [Google Scholar]
- [2].Wright PE, Dyson HJ. Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Biol. 2015;16:18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Oldfield CJ, Dunker AK. Intrinsically disordered proteins and intrinsically disordered protein regions. Annu Rev Biochem. 2014;83:553–84. [DOI] [PubMed] [Google Scholar]
- [4].Brookes DH, Head-Gordon T. Experimental inferential structure determination of ensembles for intrinsically disordered proteins. J Am Chem Soc. 2016;138:4530–8. [DOI] [PubMed] [Google Scholar]
- [5].Kragelj J, Blackledge M, Jensen MR. Ensemble calculation for intrinsically disordered proteins using NMR parameters. Adv Exp Med Biol. 2015;870:123–47. [DOI] [PubMed] [Google Scholar]
- [6].Salvi N, Abyzov A, Blackledge M. Multi-timescale dynamics in intrinsically disordered proteins from NMR relaxation and molecular simulation. J Phys Chem Lett. 2016;7:2483–9. [DOI] [PubMed] [Google Scholar]
- [7].Chong S-H, Chatterjee P, Ham S. Computer simulations of intrinsically disordered proteins. Annu Rev Phys Chem. 2017;68:117–34. [DOI] [PubMed] [Google Scholar]
- [8].Lee T, Moran-Gutierrez CR, Deniz AA. Probing protein disorder and complexity at single-molecule resolution. Semin Cell Dev Biol. 2015;37:26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Aznauryan M, Delgado L, Soranno A, Nettels D, Huang J, Labhardt AM, et al. Comprehensive structural and dynamical view of an unfolded protein from the combination of single-molecule FRET, NMR, and SAXS. Proc Natl Acad Sci USA. 2016;113:E5389–E98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kikhney AG, Svergun DI. A practical guide to small angle X-ray scattering (SAXS) of flexible and intrinsically disordered proteins. FEBS Lett. 2015;589:2570–7. [DOI] [PubMed] [Google Scholar]
- [11].Watson MC, Curtis JE. Probing the average local structure of biomolecules using small-angle scattering and scaling laws. Biophys J. 2014;106:2474–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ferreon ACM, Moran CR, Gambin Y, Deniz AA. Single-molecule fluorescence studies of intrinsically disordered proteins. Methods Enzymol. 2010;472:179–204. [DOI] [PubMed] [Google Scholar]
- [13].Hartl FU. Protein misfolding diseases. Annu Reb Biochem 2017;86:21–6. [DOI] [PubMed] [Google Scholar]
- [14].Chiti F, Dobson CM. Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu Rev Biochem. 2017;86:27–68. [DOI] [PubMed] [Google Scholar]
- [15].Habchi J, Arosio P, Perni M, Costa AR, Yagi-Utsumi M, Joshi P, et al. An anticancer drug suppresses the primary nucleation reaction that initiates the production of the toxic Αβ42 aggregates linked with Alzheimer’s disease. Sci Adv. 2016;2:e1501244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Joshi P, Vendruscolo M. Druggability of intrinsically disordered proteins. Adv Exp Med Biol. 2015;870:383–400. [DOI] [PubMed] [Google Scholar]
- [17].Yu S, Ueda K, Chan P. α-Synuclein and dopamine metabolism. Mol Neurobiol. 2005;31:243–54. [DOI] [PubMed] [Google Scholar]
- [18].Murphy DD, Rueter SM, Trojanowski JQ, Lee VMY. Synucleins are developmentally expressed, and α-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci. 2000;20:3214–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jakes R, Spillantini MG, Goedert M. Identification of two distinct synucleins from human brain. FEBS Lett. 1994;345:27–32. [DOI] [PubMed] [Google Scholar]
- [20].Tuttle MD, Cornelias G, Nieuwkoop AJ, Coveil DJ, Berthold DA, Kloepper KD, et al. Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat Struct Mol Biol. 2016;23:409–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].de Oliveira GAP, Marques MdA, Cruzeiro-Silva C, Cordeiro Y, Schuabb C, Moraes AH, et al. Structural basis for the dissociation of α-synuclein fibrils triggered by pressure perturbation of the hydrophobic core. Sci Rep. 2016;6:37990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Piccirilli F, Plotegher N, Ortore MG, Tessari I, Brucale M, Spinozzi F, et al. High-pressure-driven reversible dissociation of α-synuclein fibrils reveals structural hierarchy. Biophys J. 2017;113:1685–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Flynn JD, McGlinchey RP, Walker RL, Lee JC. Structural features of α-synuclein amyloid fibrils revealed by raman spectroscopy. J Biol Chem. 2018;293:767–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Baba M, Nakajo S, Tu P-H, Tomita T, Nakaya K, Lee VMY, et al. Aggregation of α-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am J Pathol. 1998;152:879–84. [PMC free article] [PubMed] [Google Scholar]
- [25].Sung Y-H, Eliezer D. Secondary structure and dynamics of micelle bound β- and γ-synuclein. Prot Sci. 2006;15:1162–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Maltsev AS, Ying J, Bax A. Impact of N-terminal acetylation of α-synuclein on its random coil and lipid binding properties. Biochemistry. 2012;51:5004–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Uversky VN, Li J, Souillac P, Millett IS, Doniach S, Jakes R, et al. Biophysical properties of the synucleins and their propensities to fibrillate. J Biol Chem. 2002;277:11970–8. [DOI] [PubMed] [Google Scholar]
- [28].Buell AK, Galvagnion C, Gaspar R, Sparr E, Vendruscolo M, Knowles TPJ, et al. Solution conditions determine the relative importance of nucleation and growth processes in α-synuclein aggregation. Proc Natl Acad Sci USA. 2014;111:7671–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Janowska MK, Wu K-P, Baum J. Unveiling transient protein-protein interactions that modulate inhibition of alpha-synuclein aggregation by beta-synuclein, a pre-synaptic protein that co-localizes with alpha-synuclein. Sci Rep. 2015;5:15164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Giasson BI, Murray IV, Trojanowski JQ, Lee VM. A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assebly. J Biol Chem. 2001;276:2380–6. [DOI] [PubMed] [Google Scholar]
- [31].Zibaee S, Jakes R, Fraser G, Serpell LC, Crowther RA, Goedert M. Sequence determinants for amyloid fibrillogenesis of human alpha-synuclein. J Mol Biol. 2007;374:454–64. [DOI] [PubMed] [Google Scholar]
- [32].Rivers RC, Kumita JR, Tartaglia GG, Dedmon MM, Pawar A, Vendruscolo M, et al. Molecular determinants of aggregation behavior of alpha- and beta-synuclein. Protein Sci. 2008;17:887–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Theillet FX, Binolfi A, Bekei B, Martorana A, Rose HM, Stuiver M, et al. Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature. 2016;530:45–50. [DOI] [PubMed] [Google Scholar]
- [34].Roodveldt C, Andersson A, De Genst EJ, Labrador-Garrido A, Buell AK, Dobson CM, et al. A rationally designed six-residue swap generates comparability in the aggregation behavior of α-synuclein and β-synuclein. Biochemistry. 2012;51:8771–8. [DOI] [PubMed] [Google Scholar]
- [35].Koo HJ, Lee HJ, Im H. Sequence determinants regulating fibrillation of human alpha-synuclein. Biochem Biophys Res Commun. 2008;368:772–8. [DOI] [PubMed] [Google Scholar]
- [36].Kang L, Janowska MK, Moriarty GM, Baum J. Mechanistic insight into the relationship between N-terminal acetylation of α-synuclein and fibril formation rates by NMR and fluorescence. PLoS ONE. 2013;8:e75018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Xu L, Nussinov R, Ma B. Coupling of the non-amyloid-component (NAC) domain and the KTK(E/Q)GV repeats stabilize the α-synuclein fibrils. Eur J Med Chem. 2016;121:841–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lee E-N, Cho H-J, Lee C-H, Lee D, Chung KC, Paik SR. Phthalocyanine tetrasulfonates affect the amyloid formation and cytotoxicity of α-synuclein. Biochemistry. 2004;43:3704–15. [DOI] [PubMed] [Google Scholar]
- [39].Xu Y, Zhang Y, Quan Z, Wong W, Guo J, Zhang R, et al. Epigallocatechin gallate (EGCG) inhibits alpha-synuclein aggregation: A potential agent for Parkinson’s disease. Neurochem Res. 2016;41:2788–96. [DOI] [PubMed] [Google Scholar]
- [40].Toth G, Gardai SJ, Zago W, Bertoncini CW, Cremades N, Roy SL, et al. Targeting the intrinsically disordered structural ensemble of α-synuclein by small molecules as a potential therapeutic strategy for Parkinson’s disease. PLOS One. 2014;9:e87133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ahmad B, Lapidus LJ. Curcumin prevents aggregation in α-synuclein by increasing reconfiguration rate. J.Biol Chem. 2012;287:9193–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Acharya S, Safaie BM, Wongkongkathep P, Ivanova MI, Attar A, Klarner F-G, et al. Molecular basis for preventing α-synuclein aggregation by a molecular tweezer. J Biol Chem. 2014;289:10727–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ardah MT, Paleologou KE, Lv G, Abul Khair SB, Kazim AS, Minhas ST, et al. Structure activity relationship of phenolic acid inhibitors of α-synuclein fibril formation and toxicity. Front Aging Neurosci. 2014;6:197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Collier TJ, Srivastava KR, Justman C, Grammatopoulous T, Hutter-Paier B, Prokesch M, et al. Nortriptyline inhibits aggregation and neurotoxicity of alpha-synuclein by enhancing reconfiguration of the monomeric form. Neurobiol Dis. 2017;106:191–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Sormanni P, Aprile FA, Vendruscolo M. Rational design of antibodies targeting specific epitopes within intrinsically disordered proteins. Proc Natl Acad Sci USA. 2015; 112:9902–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lee CC, Julian MC, Tiller KE, Meng F, DuConge SE, Akter R, et al. Design and optimization of anti-amyloid domain antibodies specific for β-amyloid and islet amyloid polypeptide. J Biol Chem. 2016;291:2858–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Sahin C, Lorenzen N, Lemminger L, Christiansen G, Moller IΜ, Vesterager LB, et al. Antibodies against the C-terminus of α-synuclein modulate its fibrillation. Biophys Chem. 2017;220:34–41. [DOI] [PubMed] [Google Scholar]
- [48].Brundin P, Dave KD, Kordower JH. Therapeutic approaches to target alpha-synuclein pathology. Exp Neurol. 2017;298:225–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Chan DKY, Xu YH, Chan LKM, Braidy N, Mellick GD. Mini-review on initiatives to interfere with the propagation and clearance of alpha-synuclein in Parkinson’s disease. Trans Neurodegen. 2017;6:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Hashimoto M, Rockenstein E, Mante M, Mallory M, Masliah E. β-Synuclein inhibits α-synuclein aggregation: A possible role as an anti-Parkinsonian factor. Neuron. 2001;32:213–23. [DOI] [PubMed] [Google Scholar]
- [51].Tsigelny IF, Bar-On P, Sharikov Y, Crews L, Hashimoto M, Miller MA, et al. Dynamics of α-synuclein aggregation and inhibition of pore-like oligomer development by β-synuclein. FEBS J. 2007;274:1862–77. [DOI] [PubMed] [Google Scholar]
- [52].Brown JWP, Buell AK, Michaels TCT, Meisl G, Carozza J, Flagmeier P, et al. β-Synculein suppresses both the initiation and amplification steps of α-synuclein aggregation via competitive binding to surfaces. Sci Rep. 2016;6:36010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Moriarty GM, Olson MP, Atieh TB, Janowska MK, Khare SD, Baum J. A pH-dependent switch promotes β-synuclein fibril formation via glutamate residues. J Biol Chem. 2017;292:16368–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Yamin G, Munishkina LA, Karymov MA, Lyubchenko YL, Uversky VN, Fink AL. Forcing nonamyloidogenic β-synuclein to fibrillate. Biochemistry. 2005;44:9096–107. [DOI] [PubMed] [Google Scholar]
- [55].Leitao A, Bhumkar A, Hunter DJB, Gambin Y, Sierecki E. Unveiling a selective mechanism for the inhibition of α-synuclein aggregation by β-synuclein. Int J Mol Sci. 2018;19:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Wu K-P, Kim S, Fela DA, Baum J. Characterization of conformational and dynamic properties of natively unfolded human and mouse α-synuclein ensembles by NMR: Implication for aggregation. J Mol Biol. 2008;378:1104–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Bertoncini CW, Rasia RM, Lamberto GR, Binolfi A, Zweckstetter M, Griesinger C, et al. Structural characterization of the intrinsically unfolded protein β-synuclein, a natural negative regulator of α-synuclein aggregation. J Mol Biol. 2007;372:708–22. [DOI] [PubMed] [Google Scholar]
- [58].Gaspar R, Meisl G, Buell AK, Young L, Kaminski CF, Knowles TPJ, et al. Secondary nucleation of monomers on fibril suface dominates α-synuclein aggregation and provides autocatalytic amyloid amplification. Q Rev Biophys. 2017;50:1–12. [DOI] [PubMed] [Google Scholar]
- [59].Khurana R, Coleman C, Ionescu-Zanetti C, Carter SA, Krishna V, Grover RK, et al. Mechanism of thioflavin T binding to amyloid fibrils. J Struct Biol. 2005;151:229–38. [DOI] [PubMed] [Google Scholar]
- [60].Biancalana M, Koide S. Molecular mechanism of thioflavin-T binding to amyloid fibrils. Biochim Biophys Acta. 2010;1804:1405–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Clackson T, Wells JA. A hot spot of binding energy in a hormone-receptor interface. Science. 1995;267:383–6. [DOI] [PubMed] [Google Scholar]
- [62].Hughes MP, Sawaya MR, Boyer DR, Goldschmidt L, Rodriguez JA, Cascio D, et al. Atomic structures of low-complexity protein segments reveal kinked β sheets that assemble networks. Science. 2018;359:698–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Young LM, Cao P, Raleigh DP, Ashcroft AE, Radford SE. Ion mobility spectrometry-mass spectrometry defines the oligomeric intermediates in amylin amyloid formation and the mode of action of inhibitors. J Am Chem Soc. 2014;136:660–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Young LM, Mahood RA, Saunders JC, Tu L-H, Raleigh DP, Radford SE, et al. Insights into the consequences of co-polymerisation in the early stages of IAPP and Αβ peptide assembly from mass spectrometry. Analyst. 2015;140:6990–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Young LM, Tu L-H, Raleigh DP, Ashcroft AE, Radford SE. Understanding co-polymerization in amyloid formation by direct observation of mixed oligomers. Chem Sci. 2017;8:5030–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Yan L-M, Velkova A, Tatarek-Nossol M, Andreetto E, Kapurniotu A. IAPP mimic blocks Αβ cytotoxic self-assembly: Cross-suppression of amyloid toxicity of Αβ and IAPP suggests a molecular link between Alzheimer’s disease and type II diabetes. Angew Chem Int Ed. 2007;46:1246–52. [DOI] [PubMed] [Google Scholar]
- [67].Karamanos TK, Kalverda AP, Thompson GS, Radford SE. Visualization of transient protein-protein interactions that promote or inhibit amyloid assembly. Mol Cell. 2014;55:214–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Sarell CJ, Woods LA, Su Y, Debelouchina GT, Ashcroft AE, Griffin RG, et al. Expanding the repertoire of amyloid polymorphs by co-polymerization of related protein precursors. J Biol Chem. 2013;288:7327–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, et al. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science. 2003;300:636–40. [DOI] [PubMed] [Google Scholar]
- [70].Guo J-P, Arai T, Miklossy J, McGeer PL. Αβ and tau from soluble complexes that may promote self aggregation of both into the insoluble forms observed in Alzheimer’s disease. Proc Natl Acad Sci USA. 2006;103:1953–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Sarell CJ, Stockley PG, Radford SE. Assessing the causes and consequences of co-polymerization in amyloid formation. Prion. 2013;7:359–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Franzmann TM, Jahnel M, Pozniakovsky A, Mahamid J, Holehouse AS, Nuske E, et al. Phase separation of a yeast prion protein promotes cellular fitness. Science. 2018;359:eaao5654. [DOI] [PubMed] [Google Scholar]
- [73].Lang L, Zetterstrom P, Brannstrom T, Marklund SL, Daneilsson J, Oliveberg M. SOD1 aggregation in ALS mice shows simplistic test tube behavior. Proc Natl Acad Sci USA. 2015;112:9878–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Jha NN, Ranganathan S, Kumar R, Mehra S, Panigrahi R, Navalkar A, et al. Complexation of NAC derived peptide ligands with C-terminus of α-synuclein accelerates its aggregation. Biochemistry. 2018;57:791–804. [DOI] [PubMed] [Google Scholar]
- [75].Shaltiel-Karyo R, Frenkel-Pinter M, Egoz-Matia N, Frydman-Marom A, Shalev DE, Segal D, et al. Inhibiting α-synuclein oligomerization by stable cell-penetrating β-synuclein fragments recovers phenotype of Parkinson’s disease model flies. PLoS ONE. 2010;5:el3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Kang L, Moriarty GM, Woods LA, Ashcroft AE, Radford SE, Baum J. N-terminal acetylation of α-synuclein induces increased transient helical propensity and decreased aggregation rates in the intrinsically disordered monomer. Prot Sci. 2012;21:911–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–93. [DOI] [PubMed] [Google Scholar]
- [78].McWilliam H, Li W, Uludag M, Squizzato S, Park YM, Buso N, et al. Analysis Tool Web Services from the EMBL-EBI. Nucleic Acids Res. 2013;41:W597–W600 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.