

Abstract
Nutrient deprivation induces autophagy through inhibiting TORC1 activity. We describe a novel mechanism in Drosophila by which TORC1 regulates RNA processing of Atg transcripts and alters ATG protein levels and activities via the cleavage and polyadenylation (CPA) complex. We show that TORC1 signaling inhibits CDK8 and DOA kinases, which directly phosphorylate CPSF6, a component of the CPA complex. These phosphorylation events regulate CPSF6 localization, RNA binding, and starvation-induced alternative RNA processing of transcripts involved in autophagy, nutrient, and energy metabolism, thereby controlling autophagosome formation and metabolism. Similarly, we find that mammalian CDK8 and CLK2, a DOA ortholog, phosphorylate CPSF6 to regulate autophagy and metabolic changes upon starvation, revealing an evolutionarily conserved mechanism linking TORC1 signaling with RNA processing, autophagy and metabolism.
Keywords: TORC1, CPSF6, RNA processing, autophagy, metabolism
In Brief
Tang et al. investigate the mechanisms of how TORC1 regulates autophagy and cell metabolism. They demonstrate that CDK8 and DOA, two kinases downstream of TORC1 signaling, directly phosphorylate CPSF6 to regulate alternative RNA polyadenylation and splicing and mediate TORC1-dependnt physiological functions.
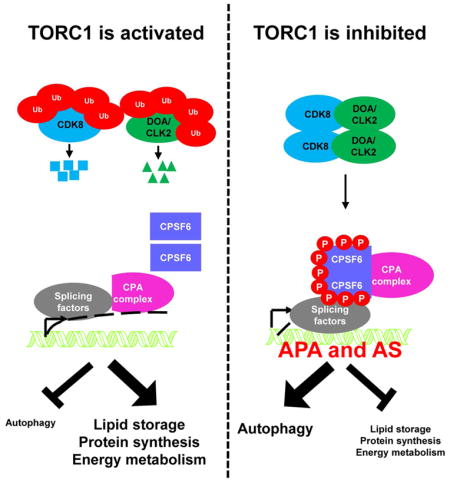
Introduction
Autophagy is a conserved process by which cells recycle macromolecules or organelles engulfed by specialized double-membraned vesicles called autophagosomes. Autophagosomes fuse with lysosomes, where their inner membranes and contents are degraded by hydrolases. This degradative process can provide energy or resources to maintain homeostasis during various environmental stresses, and is implicated in many biological processes and diseases (Jiang and Mizushima, 2014). More than 30 autophagy-related (ATG) genes were identified in yeast, and most of them are conserved in mammal. These ATG proteins can be classified into six functional units and among them the ATG1 kinase complex (ATG1/ATG13/FIP200) is the central autophagic regulator (Ohsumi, 2014). ATG1 interacts with ATG13 and FIP200 to serve as a scaffold for recruitment of other ATG proteins to the pre-autophagosomal structure (PAS), initiating autophagy (Cheong et al., 2008). ATG1 also regulates other functional units to control different stages of autophagosome formation (Mercer et al., 2018). In Drosophila, Atg1 overexpression is sufficient to induce autophagy, highlighting its critical role (Scott et al., 2007).
Various signaling pathways regulate autophagy. For example, cAMP-dependent protein kinase (PKA) directly phosphorylates Atg1 and Atg13 to inhibit autophagy in yeast and mammals (Budovskaya et al., 2005; Dorsey et al., 2009; Stephan et al., 2009). AMP-activated protein kinase (AMPK) directly phosphorylates ULK1 (the mammalian ortholog of Atg1) and Beclin 1 to activate autophagy in mammals (Mihaylova and Shaw, 2011). Moreover, the PI3K/AKT/TORC1 pathway links hormonal signaling and nutritional status to autophagy in response to starvation, as TORC1 blocks autophagy through direct phosphorylation of ULK1 (Kim et al., 2011). Interestingly, a recent study reported that mTOR mediates the phosphorylation of an mRNA de-capping complex, RCK/Dcp2, to degrade Atg transcripts, suppressing autophagy (Hu et al., 2015). Thus, TORC1 regulates autophagy at multiple steps.
From a genetic screen in Drosophila, we identified the Cleavage and Polyadenylation (CPA) complex as a novel regulator of autophagy. The CPA complex consists of more than 20 proteins classified into four sub-complexes and several single proteins and is required for CPA of pre-mRNAs. The sub-complexes include the evolutionarily conserved Cleavage and Polyadenylation Specificity Factor (CPSF), Cleavage stimulation Factor (CstF), Cleavage Factor I (CFI), and Cleavage Factor II (CFII) sub-complexes ((Chan et al., 2011) and table S1). Here, we characterize the role of the CPA complex in autophagy and demonstrate that it coordinates Alternative Polyadenylation (APA, a mechanism that generates distinct 3′UTRs on mRNAs) of Atg1 and Atg8a transcripts, with Alternative Splicing (AS, a mechanism that joins different exons to form mRNAs isoforms) of Atg1. CPA activity increases ATG protein levels and ATG1 kinase complex formation, enhancing autophagy during nutrient starvation. Importantly, these RNA processing events are regulated by TORC1 signaling. We show that TORC1 activity negatively regulates two downstream kinases, CDK8 and DOA, which directly phosphorylate CPSF6 (CG7185), a key component of the CPA complex, controlling its nuclear localization and RNA-binding ability. Significantly, depletion of CDK8, DOA, or CPSF6, compromises starvation-induced autophagy and also causes energy, lipid, and protein metabolic defects in both Drosophila and mammalian cells. Our studies thus identify a new role for the CPA complex in mediating TORC1-dependent functions and demonstrate that TORC1 controls alternative RNA processing through CDK8/DOA/CPA signaling.
Results
Modifiers of Atg1-mediated autophagy
Overexpression of Atg1 in the Drosophila eye induces autophagy, leading to reduced eye size and a rough eye phenotype (Figure 1A–B) (Chen et al., 2008; Scott et al., 2007). To identify new autophagic regulators, we overexpressed ATG1 in the eye and performed an RNAi screen for modifiers. The Atg1-induced eye phenotype is rescued by Atg9-RNAi, suggesting that these phenotypes are autophagy-dependent (Figure 1C). While depletion of PCF11 (a CFII sub-complex component) enhanced the Atg1-induced rough eye phenotype, multiple RNAi lines against other sub-complex components of the CPA complex, including CPSF6 (CFI complex), CstF64 (CstF complex), WDR33 and CPSF160 (CPSF complex), rescued the rough eye phenotype (Figure 1D–H). Furthermore, co-expression of the autophagosomal marker mCherry-ATG8a with Atg1 induced mcherryATG8a punctae formation in third-instar larval eye discs (compare Figure 1A′ and B′, and Figure 1I). These ATG8a punctae were abolished by depletion of Atg9, CPSF6, CstF64, WDR33, or CPSF160, while PCF11-RNAi increased their prevalence (Figure 1C′–H′ and 1I). Thus, the CFI, CPSF, and CstF sub-complexes are required for Atg1-induced autophagosome formation, and the CFII sub-complex has the opposite function.
Figure 1. The CPA complex regulates ATG1 and starvation-induced autophagosome formation.

(A–H and A′–H′) Atg1 genetically interacts with the CPA complex. Compared with controls (A), expression of Atg1 using GMR-GAL4 results in a rough and reduced eye (B). Atg1-induced eye defects are rescued by co-expressing Atg9RNAi (C), CPSF6RNAi (D), CstF64RNAi (E), WDR33RNAi (F), or CPSF160RNAi (G), but enhanced by Pcf11RNAi (H). Third-instar larval eye imaginal discs from controls expressing mCherry-ATG8a alone (A′), or flies co-expressing mCherry-Atg8a and indicated transgenes, stained with DAPI (B′–H′). (I) Quantification of mCherryATG8a puncta per cell in A′–H′ (One-Way ANOVA followed by Bonferroni’s post hoc test; data are represented as mean ± SEM; **P<0.01, ***P<0.001). (J–R) The CPA complex regulates autophagy. Clonal depletion of CPSF5 (J), CstF64 (K), or CPSF160 (L) impaired mCherry-ATG8a puncta formation during starvation, while expression of Pcf11RNAi (M) or CbcRNAi (N) in GFP-nls-labeled cells induced mCherry-ATG8a puncta under fed conditions. Mutant clones of CPSF6091 (O) or CPSF5H11 (P), circled by dotted lines, show decreased mCherry-ATG8a puncta during starvation. Cells outside of the clones are used as controls. Fat body cells were stained with DAPI. Scale bar, 20 μm. (Q–R) Quantification of the relative number of mCherry-ATG8a dots per cell. (Student’s T-test was performed to identify significant differences between dot numbers in clones and in control cells; data represent as the mean± SEM of 3 fat-body samples per genotype; *P<0.05, **P<0.01, ***P<0.001).
We next examined the effects of CPA complex components on starvation-induced autophagy in the larval fat body. Consistently, depletion of CFI, CPSF, or the CstF sub-complex suppressed starvation-induced mCherryATG8a punctae formation, whereas reducing CFII sub-complex expression increased ATG8a punctae under fed conditions (Figure 1J–N and 1Q–R). To further confirm this result, we generated null mutations in components of the CFI sub-complex, CPSF5 and CPSF6, and both strongly inhibited starvation-induced autophagosome formation (Figure 1O–P). Together, these results demonstrate that the CFI, CPSF, and CstF sub-complexes of the CPA complex positively regulate autophagy and that the CFII sub-complex acts negatively.
CPA complex-mediated APA alters ATG1 and ATG8a protein expression levels
Since the major known function of the CPA complex is 3′ end processing, we tested whether it regulates APA of Atg transcripts. We used 3′ RACE to identify polyadenylation sites in Atg1 and Atg8a transcripts (Figure 2A and Figure S1A–B) and performed qPCR with primers that detect either total or long-3′UTR specific transcripts to determine 3′UTR changes (Figure 2A). Interestingly, starvation increased 3′-UTR length of both Atg1 and Atg8a transcripts, and knockdown of CPSF6, Clp, or CstF64 reduced it (Figure 2B). In contrast, depletion of PCF11 or Cbc extended 3′UTR lengths of Atg1 and Atg8a transcripts under fed conditions (Figure S1C). Thus, the CPA complex plays an important role in regulating 3′UTR lengths of Atg genes.
Figure 2. The CPA complex controls 3′UTR length and splicing of Atg1 and Atg8a.

(A) Primers detect the Atg1 AS isoforms (with or without the retained intron), the total (CDS), or the long-specific transcripts (Distal 3′UTR) of Atg1 and Atg8a. Poly A sites are indicated by arrows. (B) The ratio between amplicons (Distal 3′UTR/CDS) represents APA isoform changes under different conditions. Compared with control under fed conditions (starvation 0 hr), the 3′UTR length of Atg1 and Atg8a transcripts increased in control larval fat bodies, but not from CPSF6RNAi, ClpRNAi, or CstF64RNAi-expressing larva under starvation for 16 hrs. (One-Way ANOVA followed by Bonferroni’s post hoc test; data are represented as mean ± SEM; *P<0.05, **P<0.01, ***P<0.001.) (C) Long 3′UTRs of Atg1 and Atg8a enhanced mRNA stability. Firefly Luciferase reporters with the indicated 3′UTRs were transfected into S2R+ cells. After 48 hrs, cells were treated with Rapamycin (20 nM) for 24 hrs, followed by Actinomycin D (10 μg/ml) for the indicated times to measure mRNA levels of Firefly Luciferase by qPCR (One-Way ANOVA followed by Bonferroni’s post hoc test; data are represented as mean ± SEM; ***P<0.001). (D) The CPA complex mediates starvation-induced AS of Atg1. RNA extracted from larval fat bodies was subjected to RT-PCR to detect the two AS isoforms, RA and RB, and the long-UTR specific transcripts (Distal 3′UTR) of Atg1. (E–G) PKA phosphorylates ATG1-PB-S297 to promote ATG1 kinase complex assembly. Yellow boxes represent the shared amino acid sequences between ATG1-PA and ATG1-PB, and the red box indicates the peptide sequence from the retained intron of Atg1-RB. Alignment of amino acid sequences encoded by the retained intron of Atg1-RB showing the PKA phosphorylation motif (E). S2R+ cells were treated with dsRNA against LacZ or PKA-C1. After 48 hrs, cells were transfected with GFP-Atg1-RB or GFP-Atg1-RBS297A, and then subjected to immunoprecipitation (IP), followed by immunoblotting (IB) with antibodies as indicated (F). S2R+ cells transfected with plasmids as indicated were subjected to immunoprecipitation (IP). Immunoprecipitated proteins and total cell lysates (TCL) were analyzed by immunoblotting (IB) with antibodies as indicated (G). (H–I) Clonal expression of PKA-C1RNAi in GFP-nls-labeled cells increased mCherry-ATG8a puncta under fed conditions, but PKA-C1 expression failed to affect it upon starvation (H). Cells outside of the clones are used as controls. Fat body cells are stained with DAPI. Scale bar, 20 μm. (I) Quantification of the relative number of mCherry-ATG8a dots. (Student’s T-test was performed to identify significant differences between dot numbers in clones and in control cells; data are represented as mean ± SEM of 3 fat-body samples per genotype; *P<0.05).
3′UTRs contain binding sites for microRNAs or RNA-binding proteins that affect mRNA stability, localization, or translational efficiency (Miura et al., 2014). We observed that knockdown of CPSF6, Clp, or CstF64, decreased Atg1 and Atg8a mRNA levels, indicating that the CPA complex-dependent 3′UTR changes of Atg1 and Atg8 may affect mRNA stability (Figure S1D). We therefore treated S2R+ cells with the transcriptional inhibitor Actinomycin D, and measured luciferase activity and RNA levels with long or short 3′UTRs. The half-life of Luciferase mRNAs with the short Atg1 or Atg8a 3′UTRs was less than those with the long 3′UTRs (Figure 2C), and a Luciferase assay also revealed that long 3′UTRs increase protein expression levels (Figure S1E). Consistently, depletion of CPSF6, CstF64, or Clp, reduced ATG1 and ATG8a protein levels in starved larval fat bodies, whereas Cbc or Pcf11 knockdown increased them under fed conditions (Figure S1F–G). Thus, the CPA complex is required for the starvation-induced 3′UTR elongation of Atg1 and Atg8a, promoting mRNA stability and protein expression.
AS of Atg1 increases ATG1 complex formation during starvation
Some CPA complex components regulate AS by interacting with splicing factors (Cardinale et al., 2007; Kyburz et al., 2006; Millevoi et al., 2006; Shi et al., 2009). To investigate whether the CPA complex modulates AS of Atg1, we examined the two major Atg1 isoforms, Atg1-RA and Atg1-RB. Atg1-RB contains a translatable retained intron, spliced-out in Atg1-RA (Figure 2A and 2E). Levels of Atg1-RB slightly decreased upon starvation, but Atg1-RA was increased (Figure 2D). This phenomenon was blocked by reduction of CPSF6, Clp, or CstF64 expression (Figure 2D). In contrast, depletion of Pcf11 or Cbc increased Atg1-RA levels under fed conditions (Figure S1H), suggesting that the CPA complex regulates AS of Atg1 upon starvation.
To identify the splicing factors influencing Atg1 intron retention, we tested those that associate with the CPA complex in mammalian cells (Cardinale et al., 2007; Kyburz et al., 2006; Millevoi et al., 2006; Shi et al., 2009). Depletion of 9G8 triggered mCherry-ATG8a puncta formation and increased Atg1-RA under fed conditions, while 9G8 overexpression reduced starvation-induced ATG8a punctae and blocked AS of Atg1 triggered by Cbc depletion (Figure S1I–K and S1P). Depletion of U2AF50 suppressed starvation-induced autophagy and Cbc-RNAi induced AS of Atg1 (Figure S1I and S1O–P). In contrast, depletion of SRm300, Rbp1-like, and Tra2, did not affect mCherry-ATG8a punctae formation (Figure S1L–N and S1P). We further noted that changes in the Atg1 3′UTR correlate with Atg1 isoform alteration (Figure 2D and S1H). Thus, APA and AS of Atg1 are coordinately regulated by the CPA complex and splicing factors, including 9G8 and U2AF50.
As the Atg1 retained intron was largely removed by splicing during starvation, we tested whether it contained sequences important for ATG1 regulation. The peptide sequence encoded by the retained intron of Atg1-RB contains a PKA consensus phosphorylation site (RRXS*) at Ser-297 (Figure 2E) (Smith et al., 2011). PKA phosphorylates and inhibits ATG1 in yeast and mammalian cells (Budovskaya et al., 2005; Dorsey et al., 2009; Stephan et al., 2009). To test whether PKA phosphorylates ATG1-PB Ser-297, we examined its phosphorylation using phospho-PKA substrate antibody in S2R+ cells. Immunoblotting of anti-GFP immunoprecipitates revealed that GFP-ATG1-PB was phosphorylated and that the phosphorylation signal was abolished after knocking down PKA expression or mutating Ser-297 (Figure 2F). We performed in vitro kinase assays to further test whether a S297A mutation in ATG1-PB affects ATG1 kinase activity using wild-type ATG1-PB or ATG1-PB-S297A isolated from S2R+ transfected cells as kinases, and Myelin Basic Protein (MBP) as a substrate. The S297A mutation had no significant effect on ATG1-PB kinase activity (not shown), consistent with a study showing that PKA regulates the association of ATG1 with the preautophagosomal structure, but not its protein kinase activity (Budovskaya et al., 2005). In contrast, co-immunoprecipitation revealed that the S297A mutation enhanced the interaction between ATG1-PB and FIP200, a component of the ATG1 complex (Figure 2G). Furthermore, wild-type ATG1 and ATG1S297A transgenic flies revealed that the mutant protein induces higher autophagic activity than wild-type (Figure S1Q–S). Thus, PKA-mediated phosphorylation of ATG1-PB blocks ATG1 complex assembly to inhibit autophagy initiation. Moreover, depletion of PKA induced mCherry-ATG8a punctae formation in larval fat body from fed larvae, but had no effect on starvation-induced autophagy, while PKA overexpression failed to inhibit autophagy upon starvation (Figure 2H–I, Data not shown). These results demonstrate that PKA inhibits ATG1 protein complex formation to repress autophagy by phosphorylating ATG1-PB Ser-297 under fed conditions. Upon starvation, removal of the retained intron in Atg1 transcripts bypasses PKA negative regulation of ATG1, enhancing autophagy.
TORC1 signaling regulates Atg mRNA processing and CPSF6 phosphorylation
TORC1 signaling pathway regulates aspects of APA and AS (Chang et al., 2015; Lee et al., 2017). Thus, we tested whether TORC1 activity regulates CPA complex-mediated mRNA processing. Tsc1/2 overexpression, which blocks TORC1 activity, increased mCherry-ATG8a punctae and reduced cell size in the larval fat body (Figure 3A and E). CPSF6-RNAi, CPSF160-RNAi, or CstF64-RNAi, inhibited the TSC1/2-induced effects (Figure 3B–D and E), suggesting that the CPA complex functions as a downstream target of TORC1 signaling in the modulation of autophagy.
Figure 3. TORC1 signaling regulates the CPA complex by inhibiting CPSF6 phosphorylation.

(A–E) The CPA complex genetically interacts with TOR. Clonal expression of TSC1 and TSC2 reduced cell size and increased mCherry-ATG8a puncta in the larval fat body under fed conditions (A). These effects were suppressed by depletion of CPSF6 (B), CPSF160 (C), or CstF64 (D). Fat body cells were stained with DAPI. Scale bar, 20 μm. Quantification of the relative number of mCherry-ATG8a dots per cell (One-Way ANOVA followed by Bonferroni’s post hoc test; data are represented as mean ± SEM; ***P<0.001) (E). (F–G) PI3K/AKT/TORC1 signaling regulates APA and AS of Atg1 and Atg8a. RNA from larval fat body of indicated transgenes was subjected to qPCR or RT-PCR to detect APA and AS isoforms. One-Way ANOVA followed by Bonferroni’s post hoc test; data are represented as mean ± SEM; *P<0.05, **P<0.01. (H) Rapamycin enhances phosphorylation of CPSF6. S2R+ cells transfected with plasmids as indicated were treated with or without Rapamycin (20 nM) for 24 hrs and then subjected to immunoprecipitation (IP), followed by immunoblotting (IB).
Next, we tested whether TORC1 signaling affects mRNA processing during nutrient deprivation. RT-PCR and qPCR data indicate that expression of PI3K92E, Pten-RNAi, myr-AKT, Tsc1-RNAi or wild-type TOR (TORwt) blocked starvation-induced 3′UTR elongation of Atg1 and Atg8a as well as Atg1 isoform conversion (Figure 3F–G). Conversely, inhibition of TORC1 activity by expressing InR-RNAi, PI3K92E-RNAi, Pten, PDK1-RNAi, Rheb-RNAi, or TOR-RNAi under fed conditions phenocopied starvation-induced RNA processing, suggesting that TORC1 controls mRNA processing of Atg1 and Atg8, possibly through the CPA complex (Figure S2A–B).
We expressed GFP-tagged components of the CPA complex in S2R+ cells and then used anti-phospho-Ser and -Thr antibody to determine their phosphorylation. Immunoblotting of anti-GFP immunoprecipitates revealed that both CstF64 and CPSF6 were phosphorylated, but only CPSF6 phosphorylation was increased by the TOR inhibitor Rapamycin (Figure 3H and S2C). Furthermore, CPSF6 phosphorylation was reduced in TSC1 or TSC2 knockout mutant cells, indicating that TORC1 activity negatively regulates CPSF6 phosphorylation (Figure S2D).
CDK8 and DOA genetically interact with TOR and CPSF6
Studies in yeast and mammalian cells show that TORC1 reduces the protein levels of CDK8 and KNS1, DOA orthologs in yeast (Feng et al., 2015; Lee et al., 2012). Moreover, high-throughput AP-MS and Y2H analyses suggest that CDK8 and CLK2 (DOA ortholog in mammals) interact with many CPA components (Guruharsha et al., 2011; Rolland et al., 2014; Varjosalo et al., 2013), suggesting that these kinases might link TORC1 and CPSF6. We therefore tested whether these components genetically interact. Both TSC1/2 overexpression-induced autophagy and reduction of cell size were suppressed by depletion of CDK8, DOA, or CycC, the binding partner of CDK8, suggesting that CDK8/CycC and DOA function downstream of TORC1 (Figure 4A–D, and 4I). Moreover, overexpression of CDK8 or DOA triggers autophagy under fed conditions, and their phenotypes were inhibited by CPSF6 depletion (Figure 4E–H, and J). Interestingly, we found that CycC-RNAi or DOA-RNAi represses DOA- or CDK8-induced autophagy, respectively, and that CDK8 and DOA associate with each other under rapamycin treatment (Figures S3A–S3C), suggesting that they form a complex. Thus, these results establish TORC1-CDK8/DOA-CPA as a new signaling pathway that regulates autophagy.
Figure 4. Genetic and biochemical interactions between TOR, CDK8/CycC, DOA, and CPSF6.

(A–J) Genetic interactions between TOR, CDK8/CycC, DOA, and CPSF6. Clonal expression of TSC1 and TSC2 reduced cell size and increased mCherry-ATG8a puncta formation in the larval fat body under fed condition (A), and these effects were suppressed by depletion of CDK8 (B), CycC (C), or DOA (D). Clonal expression of CDK8 (E) or DOA (G) induced mCherry-ATG8a puncta formation under fed conditions, and depletion of CPSF6 inhibits the CDK8 or DOA-induced effects (F and H). Fat body cells were stained with DAPI. Scale bar, 20 μm. (I–J) Quantification of the relative number of mCherry-ATG8a dots per cell. (One-Way ANOVA followed by Bonferroni’s post hoc test; data are represented as mean ± SEM; *P<0.05) (K) Schematic representation of the domain structures of CPSF6 and deletion mutants. (L–M) CDK8 and DOA directly phosphorylate CPSF6 in vitro. Flag-CDK8, CDK8-KD (L), HA-DOA, or HA-DOA-KD (kinase-dead) (M) immunoprecipitated from lysates of transfected cells was used to phosphorylate bacterially expressed recombinant CPSF6-N, CPSF6-C, and CPSF5 in an in vitro kinase assay. Lower panels demonstrate the equal input of GST-fusion proteins and CDK8 or DOA immunoprecipitates.
CPSF6 is a substrate of CDK8 and DOA kinases, that are negatively regulated by TORC1
We investigated the mechanism of TORC1-dependent regulation of CDK8 and DOA. CDK8 and DOA protein levels were increased in S2R+ cells treated with Rapamycin (Figure S3D). Importantly, ubiquitination of CDK8 and DOA was increased by MG132, a proteasome inhibitor, and Rapamycin co-treatment reduced it, indicating that TORC1 negatively regulates CDK8 and DOA via ubiquitin-mediated degradation (Figure S3E).
Next, we investigated whether CPSF6 physically interacts with CDK8 or DOA. CPSF6 is a Ser/Arg-rich (SR)-like protein that contains sequences rich in Arg-Ser and/or Arg-Asp/Glu/Gly dipeptides (referred to as the RS domain) (Figure 4K). S2R+ cells were transfected with GFP-CPSF6, together with Flag-CDK8 or HA-DOA, and then treated with or without Rapamycin. Immunoblotting of anti-GFP immunoprecipitates revealed that CPSF6 co-precipitated with CDK8 and DOA following Rapamycin treatment (Figure S3G–H). However, CPSF6 carrying a deletion in the RS domain (CPSF6ΔRS) did not interact with either CDK8 or DOA (Figure S3F–H), suggesting that CDK8 and DOA interact with CPSF6 through its RS domain when TORC1 is inactivated.
Many RNA-processing proteins possess RS-domains that are extensively phosphorylated, and it is the overall charge of the phosphorylated RS domain that determines their structure and activities (Ghosh and Adams, 2011; Xiang et al., 2013). The primary sequence of the CPSF6 RS-domain reveals a total of 37 Serines and Threonines residues, of which 12 match DOA consensus phosphorylation sites and 2 match CDK8 consensus phosphorylation sites (Figure S4A). Thus, we investigated whether CDK8 and DOA are responsible for Rapamycin-induced phosphorylation of CPSF6. Label-free quantitative mass spectrometry identified two phosphorylation sites at Ser-588 (a potential DOA phosphorylation site) and Ser-596 (a potential CDK8 phosphorylation site) in a peptide corresponding to residues 585–613 of CPSF6. Both of their phosphorylation levels increased after Rapamycin treatment (Figure S4B). Depletion of either CDK8 or DOA reduced the Rapamycin-induced phosphorylation of CPSF6, indicating that CDK8 and DOA promote CPSF6 phosphorylation as TORC1 is inhibited (Figure S4C). Finally, we performed in vitro kinase assays to test whether CPSF6 is a substrate of CDK8 and DOA using CDK8 and DOA isolated from transfected cells, and recombinant CPSF6 proteins as substrates. CDK8 and DOA both phosphorylated the C-terminal region of CPSF6, CPSF6-C (amino acids 221–652), but not its N-terminal region, CPSF6-N (amino acids 1–240), or CPSF5 (Figure 4K–M). We further determined the CPSF6 phosphorylation sites of DOA and CDK8 using in-vitro kinase assays followed by mass spectrometry. MS/MS analyses demonstrate that CPSF6-S588 is directly phosphorylated by DOA, and CPSF6-S596 is phosphorylated by CDK8 (Figure S4A). Together, these data indicate that CDK8 and DOA directly phosphorylate the RS domain of CPSF6.
RS domain phosphorylation of CPSF6 is required for its nuclear localization, RNA binding, and starvation-induced RNA processing and autophagy
The RS domain of CPSF6 largely contributes to its nuclear localization and to the RNA binding ability of the CFI sub-complex (Dettwiler et al., 2004). We thus tested whether CPSF6 with mutations of all fourteen phosphorylatable sites to Alanine (CPSF614A) or CPSF6 carrying a deletion of the RS domain (CPSF6ΔRS) affected its subcellular localization and ability to associate with RNA. CPSF614A and CPSF6ΔRS exhibited decreased phosphorylation signal even after Rapamycin treatment (Figure S4C). We found that GFP-CPSF6 was enriched in the nuclei of S2R+ cells with or without Rapamycin treatment, indicating that the basal level of CPSF6 phosphorylation is sufficient to induce its nuclear localization (Figure 5A–B). However, CPSF6 redistributed to the cytoplasm in TSC1 or TSC2 knockout cells, and this was reversible by Rapamycin (Figure S4D). Ablation of either CDK8 or DOA also resulted in cytoplasmic localization of CPSF6 (Figure 5C–F). CPSF614A or CPSF6ΔRS were also cytoplasmic (Figure 5G–H), suggesting that phosphorylation of the CPSF6 RS domain controls its nuclear localization.
Figure 5. Phosphorylation of the RS domain of CPSF6 is required for its nuclear localization, binding to RNA, and autophagy.

(A–H) Phosphorylation of the RS domain of CPSF6 is required for its nuclear localization. S2R+ Cells were treated as in Fig S4C and then subjected to immunofluorescence. GFP-CPSF6 was localized in the nuclei of control cells (LacZdsRNA) with or without Rapamycin (20 nM) (A–B), whereas it redistributed to the cytoplasm in cells treated with dsRNAs against CDK8 or DOA (C–F). GFP-CPSF614A and GFP-CPSF6ΔRS were localized in the cytoplasm (G–H). S2R+ cells are stained with DAPI (blue). Bar, 10 mm. (I) Phosphorylation of the RS domain of CPSF6 is critical for RNA binding. Cells as in Fig S4C were subjected to RNA-immunoprecipitations, followed by qRT-PCR with primers (Figure 2A) which detect total transcript (CDS) of Atg1 or Atg8a. Plotted fold change values (ratios of RNA IP/input normalized to GFP control) are the mean ± SEM of triplicates. One-Way ANOVA followed by Bonferroni’s post hoc test; data are represented as mean ± SEM; *P<0.05, **P<0.01, ***P<0.001. (J–K) CDK8 and DOA affected APA and AS of Atg1 and Atg8a transcripts. RNA extracts from larval fat body in control or mutants as indicated were subjected to qPCR (J) or RT-PCR (K) analysis to detect the APA and AS isoforms of Atg1 and Atg8a. One-Way ANOVA followed by Bonferroni’s post hoc test. Data are represented as mean ± SEM; *P<0.05, **P<0.01, ***P<0.001. (L–P) CDK8 and DOA-mediated CPSF6 phosphorylation is required for autophagosome formation. Clonal expression of indicated transgenes (L–O) in GFP-nls-labeled cells reduced mCherry-ATG8a puncta upon starvation. Cells outside the clones are used as controls. Fat body cells were stained with DAPI. Scale bar, 20 μm. (P) Quantification of the relative number of mCherry-ATG8a dots per cell. (Student’s T-test was performed to identify significant differences between dot numbers in clones and in control cells; data represent as the mean± SEM of 3 fat-body samples imaged per genotype; *P<0.05, **P<0.01, ***P<0.001).
RNA-IP revealed that CPSF6 co-precipitated with Atg1 and Atg8a transcripts and that these interactions were enhanced by Rapamycin (Figure 5I). Conversely, the interaction between CPSF6 and Atg transcripts was suppressed in TSC1 or TSC2 knockout cells and this effect was reversed by Rapamycin, suggesting that TORC1 activity regulates the RNA binding ability of CPSF6 (Figure S4E). CDK8 or DOA depletion abolished binding of CPSF6 to Atg transcripts under Rapamycin treatment, and CPSF614A or CPSF6ΔRS also exhibited reduced RNA binding (Figure 5I). Thus, CDK8 and DOA-mediated phosphorylation of the CPSF6 RS domain is required not only for its nuclear localization but also for RNA binding.
We further tested the in-vivo function of CPSF6 phosphorylation on regulating starvation-induced RNA processing and autophagosome formation. Starvation-induced Atg1 and Atg8a 3′UTR extensions and Atg1 isoform conversion were repressed in the larval fat body of CDK8 mutant, DOA mutant, CPSF614A-, or CPSF6ΔRS-expressing flies (Figure 5J–K and S4F–G). In addition, CDK8 or DOA mutations decreased ATG1 and ATG8a protein levels (Figure S4H). Likewise, overexpression of CPSF614A or CPSF6ΔRS reduced ATG1 and ATG8a protein levels (Figure S4I). Finally, starvation-induced ATG8a punctae formation was blocked by CDK8-RNAi, DOA-RNAi, CPSF614A or CPSF6ΔRS (Figure 5L–P), confirming that CDK8- and DOA-mediated CPSF6 phosphorylation is required for RNA processing of Atg transcripts and autophagy upon nutrient deprivation.
The CDK8/DOA/CPA axis regulates lipid, protein, and energy metabolism
Besides inhibiting autophagy, TORC1 promotes protein synthesis, lipogenesis, and energy metabolism (Saxton and Sabatini, 2017). To test whether CDK8/DOA/CPA signaling mediates other physiological effects of TORC1, we performed global transcriptomic analysis of wild type or CPSF5 mutant larval fat bodies under fed or starvation conditions. CFSF5 and CPSF6 are constitutive components of the CFI subcomplex, and knockdown of either CPSF5 or CPSF6 results in preferential use of proximal polyadenylation sites in mammalian cells (Martin et al., 2012). Because CPSF6 mutants mostly die during the first larval instar, we examined CPSF5 mutants as they survive to third instar and exhibit phenotypes similar to CPSF6 mutants (Figure 1O–P, 5J–K, and S4I).
Gene set enrichment analysis demonstrated that biosynthetic processes were down-regulated during starvation, while many stress responses were up-regulated (Figure 6A and S5A–E). Comparison of the transcriptome changes in the fat body of 16 hr-starved larvae versus starved CPSF5 mutant larvae revealed that 585 of 1933 differentially-expressed genes responding to starvation were CPSF5-dependent (Figure 6A and Table S2). Gene set enrichment analyses further suggest that autophagy, changes of lipid, protein and energy metabolism, but not chromosome organization or intracellular transport, are regulated by CPSF5 (Figure 6A and S5A–E). These CPSF5-dependent genes were further visualized by generating a protein-protein interaction network (Figure 6B). We also used COMPLEAT (Hu et al., 2017) to perform a protein complex-enrichment analysis and identify several protein complexes required for autophagy and nutrient and energy metabolism (Figure S5F). These analyses indicate that the CPA complex is involved in starvation-induced metabolic changes.
Figure 6. The CDK8/DOA/CPA complex controls lipid, protein and energy metabolism.

(A–B) RNA-seq analysis of transcriptome changes in the fat body of fed larvae, 16 hr-starved larvae, and 16 hr-starved CPSF5 mutant larvae. Heatmap displaying the -log10 (p-value) for each GO term. Color represents strength of significance (A). Network presentation of Gene Set Enrichment Analysis results. Genes are shown as nodes. Green node color indicates genes that are downregulated in starvation and that are CPSF5-dependent. Pink node color represents genes that are upregulated in starvation and CPSF5-dependent (B). (C) 3′-UTR changes of transcripts involved in metabolism. RNA samples as in Fig S5G were subjected to qPCR to detect CDS and the long-UTR specific transcripts (distal 3′UTR) of genes involved in metabolism. The relative distal 3′UTR/CDS results for the other six genes mentioned in Fig S5G could not be measured due to qPCR primers design issues. One-Way ANOVA followed by Bonferroni’s post hoc test. Measurements shown are mean ± SEM; *P<0.05, **P<0.01. (D) Illustration of the percentage of genes in various functional groups undergoing CPSF5-dependent alternative splicing among those responding to starvation. (E) CDK8, DOA, and CPSF5 are critical for starvation-induced metabolomic changes. ATP, proteins, and triglyceride (TAG) levels were measured using whole control or mutant larvae as indicated and normalized to their weights. One-Way ANOVA test was performed followed by Bonferroni’s post hoc test. Measurements shown are mean ± SEM; *P<0.05, **P<0.01, ***P<0.001.
qPCR confirmed that the expression of multiple genes involved in glycolysis, translation, and energy metabolism was enhanced in the fat body of Tsc2, CDK8, DOA, and CPSF5 mutant starved larvae, compared to wild-type starved larvae (Figure S5G). In contrast, the expression of genes involved in autophagy and lipid digestion was significantly reduced in Tsc2, CDK8, DOA, or CPSF5 mutants (Figure S5G). Furthermore, many metabolic genes exhibited starvation-induced APA in a Tsc2/CDK8/DOA/CPA dependent manner (Figure 6C). We also found strong correlation of CPSF5 dependency with genes that are alternatively spliced among metabolic genes (Figure 6D; Table S3), indicating that the CDK8/DOA/CPA complex regulates AS and APA of metabolic genes during starvation.
To investigate the role of the CDK8/DOA/CPA complex in the regulation of metabolism, we measured triglyceride (TAG), protein, and ATP levels in whole larvae. Compared to fed conditions, starvation reduced TAG, protein, and ATP levels, but these nutrient levels were increased in CDK8, DOA, or CPSF5 mutants (Figure 6E). Nile red staining for monitoring lipid droplets in the larval fat body confirmed that depletion of CDK8, DOA, or CPSF6 enhanced lipid storage (Figure S5H). These results demonstrate that CDK8/DOA/CPA regulate autophagy, as well as lipid, protein, and energy metabolism.
CDK8/CLK2/CPSF6 are essential for metabolic reprogramming during starvation in mammalian cells
The role of the CPA complex in regulating 3′UTR lengths was reported in mammalian cells (Martin et al., 2012). To investigate whether the CDK8/DOA/CPSF6 pathway is conserved in mammals, we tested whether CPSF6, CDK8, and CLK2, the closest human ortholog of DOA among CLK kinases, interact. Consistent with our observations in Drosophila, co-immunoprecipitations revealed that CPSF6 physically interacts with CDK8 and CLK2 during starvation (Figure S6A). In addition, in vitro kinase assays showed that CDK8 and CLK2 directly phosphorylate the RS domain of human CPSF6 (Figure 7A–C). Furthermore, treatment of cells with Senexin A (a CDK8 inhibitor) or TG003 (a CLK kinase inhibitor) abrogate starvation-induced CPSF6 phosphorylation, its nuclear localization, and RNA binding ability (Figure S6B–D). These results demonstrate the evolutionarily conserved roles of human CDK8 and CLK2 in the regulation of CPSF6.
Figure 7. CDK8/CLK2/CPSF6 regulate autophagy and metabolism in mammalian cells.

(A) Schematic representation of the domain structures of human CPSF6 and the C-terminal deletion mutant (CPSF6-N). (B–C) CDK8 and CLK2 directly phosphorylate human CPSF6 in vitro. Recombinant His-CDK8/CycC (B) or CLK2 catalytic domain (CLK2-cat) (C) was used to phosphorylate recombinant CPSF6 or CPSF6-N. The lower panels represent the amounts of recombinant proteins. (D–G) CDK8/CLK2/CPSF6 are required for autophagy and metabolomic changes during starvation. MCF7 cells, treated with 50 μM Senexin-A (CDK8 inhibitor) or 50 μM TG003 (CLK2 inhibitor) (D and F) or infected with lentivirus expressing control (shLuc) or CPSF6 shRNA (E and G), were cultured in DMEM (Fed) or EBSS (Starvation) with or without BafA1 for 2 hrs, and immunoblotted with antibodies as indicated (D–E) or subjected to ATP, protein, and TAG analysis (F–G). One-Way ANOVA followed by Bonferroni’s post hoc test. Measurements shown are mean ± SEM; *P<0.05, **P<0.01.
We further investigated the roles of CDK8/CLK2/CPSF6 in regulating starvation-induced autophagy and metabolic changes. MCF7 cells transfected with CPSF6 shRNA or treated with Senexin A or TG003 were incubated in a starvation medium (EBSS) in the presence or absence of the lysosomal inhibitor bafilomycin A1 (BafA1). Depletion of CPSF6, or inactivation of CDK8 or CLK2, reduced LC3 puncta and suppressed the conversion of cytosolic LC3 (LC3-I) to the lipidated form of LC3 (LC3-II) (Figure 7D–E and S6E–F). Moreover, depletion of CPSF6 or inhibition of CDK8 or CLK2 increased TAG, protein, and ATP levels (Figure 7F–G). Thus, the roles of CPSF6, CDK8 and CLK2 in the control of autophagy, lipid, protein, and energy metabolism, are conserved from flies to mammals.
Discussion
In this study, we found that the CPA complex acts as a downstream effector of TORC1 signaling to regulate AS and APA, affecting protein levels and functions. CDK8 and DOA are inhibited by TORC1-dependent ubiquitination and phosphorylate CPSF6. Phosphorylated CPSF6 translocates to the nucleus and induces alternative RNA processing of transcripts involved in autophagy and metabolism. Depletion of CDK8, DOA, or the CPA complex compromises autophagy and promotes altered metabolism during nutrient deprivation. Importantly, the functions of CDK8, DOA/CLK2, and CPSF6 are conserved in mammals (Figure S7).
CPA complex-regulated AS and APA control autophagy
APA and AS are prevalent RNA processing events that regulate mRNA turnover (Miura et al., 2014) and generate different protein isoforms (Chabot and Shkreta, 2016). We found that the CPA complex and two splicing factors, 9G8 and U2AF50, control AS and APA of Atg transcripts. However, we were unable to identify global 3′UTR changes by SUPPA (Alamancos et al., 2015) or using the APA analysis method (Chang et al., 2015). Using qPCR to detect long-UTR specific transcripts provides an alternative method to examine 3′UTR changes of individual genes. However, in some cases, primer designs can be problematic as it is difficult to design optimal primers to detect transcripts with very short 3′UTRs or 3′UTRs that overlap with other genes.
RegRNA, a web server for identifying functional RNA motifs and sites, predicts a stem-loop structure in the long 3′UTR, but not in the short 3′UTR of Atg1 (Chang et al., 2013a). Stem-loop structures direct RNA folding or protect mRNAs from degradation (Svoboda and Di Cara, 2006). AU-rich elements (ARE) and Cytoplasmic polyadenylation elements (CPE) were also identified in the long 3′UTR, but not in short 3′UTR of Atg8a. CPE enhance translation efficiency and ARE promote mRNA stability under starvation conditions (Miura et al., 2014; Yaman et al., 2002).
Our results show that the CPA complex selects distal poly-A sites during starvation, generating Atg1 and Atg8a mRNAs with long 3′UTRs, where putative stem-loop structures, CPE, or ARE sequence elements are located - thus promoting Atg1 and Atg8a mRNA stability and protein expression. These findings provide the first evidence that APA and AS are synchronized to regulate autophagy and reveal an additional layer of complexity in the regulation of starvation responses.
Posttranslational modifications of the CDK8/DOA/CPA complex
Previous studies suggested a relationship between 3′UTR lengths and sensitivity to CPA factor expression (Akman et al., 2015; Baier et al., 2017). However, we did not detect any changes in CPSF6 levels using a GFP-trap line in the larval fat body during starvation. Moreover, overexpression of CPSF5 or CPSF6 failed to induce any phenotypes or autophagy, indicating that starvation does not alter the protein levels of the CFI complex (unpublished data). Instead, Rapamycin treatment increased CPSF6 phosphorylation, revealing that posttranslational modifications regulate the activity of the CPA complex. We could not map all the phosphorylation sites on CPSF6, as the RS domain contains many Arginine residues and in-gel trypsin digestion generated short peptides. Chymotrypsin digestion identified two sites, but many potential phosphorylation sites of CPSF6 were still not detected. Thus, we generated CPSF6 with mutations in all potential phosphorylation sites and a CPSF6 RS domain deletion mutant to characterize the functions of phosphorylated CPSF6.
CDK8 and DOA are ubiquitinated and degraded in a TORC1-dependent manner. Interestingly, Thr31 (T-Y motif) and Thr196 (T-F motif) of CDK8 are potential TORC1 phosphorylation sites and DOA Ser337 is a potential S6K phosphorylation site (Hsu et al., 2011). Thus, TORC1 or S6K may phosphorylate CDK8 or DOA to regulate their protein stabilities. A study showed that TORC1 activation increases the levels of many E2 and E3 components of ubiquitin ligase complexes (Chang et al., 2015). It will be interesting to investigate whether and how TORC1-dependent phosphorylation of CDK8 or DOA regulates their protein stabilities.
The CDK8/DOA/CPA signaling is a conserved regulator of metabolism
An RNAi screen in S2R+ cells showed depletion of multiple CPA complex components increases ATP levels (Mohr et al., 2015). CDK8 is implicated in lipid metabolism, and KNS1 (DOA ortholog in yeast) in the regulation of protein homeostasis (Lee et al., 2012; Zhao et al., 2012). Here, we found the CDK8/DOA/CPA complex regulates autophagy and metabolism in both Drosophila and mammalian cells. As TORC1 hyperactivation and metabolic reprogramming contribute to malignancy, our study provides insight into new cancer therapeutic strategies targeting RNA metabolism downstream of the TORC1 signaling pathway.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Norbert Perrimon (perrimon@receptor.med.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Experimental Animals
Species: Drosophila melanogaster
Flies were raised in a humidified incubator at 25°C with 12/12 hrs dark/light cycles (lights on at 7 am) on standard lab food containing per liter: 15 g yeast, 8.6 g soy flour, 63 g cornmeal, 5g agar, 5g malt, 74 mL corn syrup. Mixed sex second instar larvae were used in experiments. The following Drosophila strains from previous studies, Bloomington Drosophila stock center (BDSC), or Vienna Drosophila Resource center (VDRC), were used: UAS-Atg1 (BDSC51655), UAS-mcherry-Atg8a (BDSC37750), r4-mCherry-Atg8a Act>CD2>GAL4 UAS-GFP-nls (Arsham and Neufeld, 2009), pmCherry-Atg8a (Chang et al., 2013), UAS-Luc-RNAi (BDSC31603), UAS-Atg9-RNAi (VDRC10045), UAS-Pcf11-RNAi (BDSC32411), UAS-Cbc-RNAi (VDRC20998 and VDRC10686), UAS-CPSF6-RNAi (BDSC34804 and VDRC107147), UAS-CPSF5-RNAi (BDSC32883), UAS-CstF64-RNAi (VDRC21045), UAS-Su(f)-RNAi (BDSC43273), UAS-CstF50-RNAi (VDRC43716), UAS-CPSF160-RNAi (BDSC42478 and BDSC55698), UAS-Clp-RNAi (BDSC36816), UAS-CG1109-RNAi (BDSC55249), UAS-Fip1-RNAi (VDRC27317), UAS-CPSF73-RNAi (BDSC55696 and VDRC39557), UAS-Sym-RNAi (BDSC43227), UAS-CPSF100-RNAi (BDSC50893), UAS-Pka-C1-RNAi (BDSC31277 and BDSC58355), UAS-Pka-C1-Flag (BDSC35555), UAS-TSC1, UAS-TSC2 (Potter et al., 2001), UAS-PI3K92E (BDSC8286), UAS-Pten-RNAi (BDSC25841 and BDSC33643), UAS-myr-AKT (BDSC50758), UAS-TSC1-RNAi (BDSC54034 and BDSC52931), RhebAV4 (BDSC9690), UAS-TORwt (BDSC7012), UAS-DOA-RNAi (VDRC19066 and VDRC102520), UAS-CycC-RNAi (BDSC33753 and VDRC48834), UAS-CDK8-HA (FlyORF F001713), Tsc2109 (BDSC4739), UAS-3XFlag-DOA (Zhao et al., 2013), CDK8K185 (Zhao et al., 2012), CycCy5 (Zhao et al., 2012), DOAHD and DOADEM (Yun et al., 1994), UAS-CDK8-RNAi (VDRC45371), Df(3L)AC1 (BDSC997), UAS-GFP-Atg8a (Scott et al., 2007), UAS-9G8-RNAi (VDRC100226), UAS-GFP-9G8 (Gabut et al., 2007), UAS-SRm300-RNAi (BDSC21815 and BDSC52936), UAS-tra2-RNAiG1X; UAS-tra2-RNAi82A3 (a gift of Dr. W. Mattox.) (Fortier and Belote, 2000), UAS-Rbp1-like-RNAi (VDRC105883), UAS-U2AF50-RNAi (BDSC27542), UAS-InR-RNAi (BDSC31594 and BDSC51518), UAS-PI3K92E-RNAi (BDSC61182), UAS-Pten (BDSC9689 and (Potter et al., 2001)), UAS-PDK1-RNAi (BDSC34936 and BDSC27925), UAS-AKT-RNAi (BDSC31701), UAS-Rheb-RNAi (BDSC33966), and UAS-TOR-RNAi (BDSC33578 and BDSC34639). The UAS-HA-Atg1, UAS-HA-Atg1S297A, UAS-HA-CPSF614A, and UAS-HA-CPSF6ΔRS transgenes were generated from plasmids described below and cloned into the Drosophila Gateway vector pTWH. Mutant alleles for CPSF5 and CPSF6 were generated using CRISPR technology. Two independent guide RNAs (gRNAs) per gene were designed using the gRNA design tool: http://www.flyrnai.org/crispr/index.html. Oligonucleotides were annealed and cloned into the pl100 vector. Vectors were injected into w;nos-cas9/CyO embryos. Wings of single flies were clipped to isolate DNA and high-resolution melt analysis (HRMA) analysis was used to detect indel mutations (Housden and Perrimon, 2016). Candidate flies were crossed with y w; TM3/TM6b to establish stocks. The CPSF5H11 allele was obtained using the gRNA sequence GTCTCAAACAAATCGGGCTC and found to carry to a 13 bp deletion leading to a frame shift and a potential 36 a.a peptide (Table S4). The CFSF6091 allele was obtained using the gRNA sequence GTACAGATCCAAGACCACGT and found to correspond to an indel with a premature stop codon and a potential truncated protein of 31a.a (Table S4). CPSF5H11 allele is homozygous lethal and failed to complement deficiencies uncovering CPSF5 (BDSC8970 and BDSC997). CFSF6091 is homozygous lethal and failed to complement deficiencies uncovering CPSF6 (BDSC7591, BDSC24400, BDSC1541, and BDSC27576).
Cell lines
Drosophila S2R+ cells (sex: male) were cultured in Schneider’s medium supplemented with 10% fetal bovine serum (FBS) at 25°C. HEK293T (sex: female) and MCF7 (sex: female) cells were cultured at 37°C in DMEM (Invitrogen, 10-017-C V) medium supplemented with 10% FBS.
METHOD DETAILS
Plasmid
The full-length cDNAs of Atg1 (LD18893), CPSF5 (SD03330), CPSF6 (LD25239), CstF64 (RE27227), CPSF160 (LD38533), Cbc (LD15072) and Pcf11 (MIP05908), were cloned into the Drosophila Gateway vector pAWG. GFP was cloned into pAWM as a control. GFP-CPSF6ΔRS (amino acids 1–400) which contains the RRM domain was created by PCR amplification and verified by DNA sequencing. Flag-CDK8 and HA-DOA were constructed following PCR from cDNA clones (RE13344 and RE04477) and cloned into pcDNA3.1-Flag and pWALIUM10-moe, respectively. Using PCR mutagenesis, we generated CDK8-KD (kinase-dead) and DOA-KD mutants by replacing the conserved ATP-binding Lysine 52 and Lysine 193 in the kinase domain with Alanine, respectively. The Atg1-PB-S279A mutant was generated by replacing Serine 297 with Alanine. CPSF614A was generated by custom gene synthesis (IDT) and replacing Serines 459, 468, 525, 527, 531, 541, 543, 549, 571, 573, 588, 596, 622, and Threonine 404 with Alanines and cloned into Drosophila Gateway vector pAWG. For the generation of the GST fusion protein, DNA sequences corresponding to amino acids 1–240 and 221–652 of CPSF6 and full length of CPSF5 were PCR-amplified and subcloned into the pGEX-2T vector. GFP-human CPSF6 was obtained from GeneCopoeia (EX-Mm18988-M29). UAS-Flag-Fip200 was a gift of Dr. Jun Hee Lee (Kim et al., 2013)
Larval starvation procedure
Second larvae were collected 72–96 hrs after egg laying and then cultured in fresh fly media supplemented with yeast paste (Fed), or in vials containing 20% sucrose (Starved) for 4hrs or 16 hrs.
3′ Rapid Amplification of cDNA Ends (3′ RACE)
Total RNA was extracted from larval fat body using TRIzol® reagent (Invitrogen, 15596-018). 4 μg of total RNA was used for cDNA synthesis and 3′RACE by using SMARTer RACE 5′/3′ Kit (Clontech, 634858). Gene-specific forward primers and a universal reverse primer were used for the first-round and nested PCR amplification as indicated. The PCR products were cloned using TOPO® TA Cloning vector (Invitrogen, K4500-01) and sequenced. Gene specific primers are listed in Table S5.
3′ UTR luciferase reporter assay
The firefly luciferase reporter plasmids containing long or short 3′ UTRs of Atg1 and Atg8a were constructed using the primers listed in Table S5. Experiments were performed in 96-well plates excluding the outer wells. Cells were transfected with firefly luciferase reporter plasmids and Renilla luciferase reporter plasmid for transfection control. After 72 hrs, luciferase activities were measured using DualGlo (Promega, E2980).
Cell culture, reagents, RNA interference, and transfection
Drosophila wild-type, TSC1KO and TSC2KO S2R+ cells (Housden et al., 2015) were cultured in Schneider’s medium supplemented with 10% fetal bovine serum (FBS) at 25°C. For Rapamycin (LC Laboratories, R-5000), MG132 (Calbiochem, 474791), or Actinomycin D (Calbiochem, 114666) treatment, S2R+ cells were treated with 20nM Rapamycin or 20 μM MG132 for 24hrs, or 10μg/ml Actinomycin D for the indicated time points.
For RNAi experiments, PCR templates for dsRNA against PKA (DRSC03399 and DRSC31381) CDK8 (DRSC28684 and DRSC41558) and DOA (DRSC16650 and DRSC40713) were prepared using the MEGAscript® T7 Transcription Kit (Invitrogen, AMB13345). DsRNA against the bacterial β-galactosidase gene (lacZ) was used as a control. S2R+ cells were dispensed into assay plates containing dsRNAs at a standard concentration for the ‘bathing’ method (https://fgr.hms.harvard.edu/drsc-cell-rnai).
HEK293T and MCF7 cells were cultured at 37°C in DME M (Invitrogen, 10-017-CV) medium supplemented with 10% FBS (complete medium). For nutrient starvation, cells were starved for 2 hrs in serum-free Earle’s balanced salt solution (EBSS) medium (Sigma, 14155063) plus 100 nM Bafilomycin A1 (Sigma, B1793), 50 μM Senexin A (Tocris, 4875), or 50 μM TG003 (Sigma, T5575) where indicated. The production and infection of lentivirus carrying CPSF6 shRNA clones were performed as described previously (Tang et al., 2013; Tang et al., 2011). The target sequences of these clones are CPSF6 shRNA #1: (Clone ID: TRCN0000237833) 5′ GTTGTAACTCCATGCAATAAA 3′ and CPSF6 shRNA #2: (Clone ID: TRCN0000244314) 5′ GGTGATTATGGGAGTGCTATT 3′. Luciferase shRNA was used as a control.
Antibodies
Antibodies used for the study were: anti-Atg8 (LSBio, LS-B4021), anti-GFP (Molecular Probes, A6455), anti-phospho-Ser (Santa Cruz, sc-81514), anti-phospho-Threnonine (Cell Signaling, 9381), anti-Phospho-PKA substrate (Cell Signaling, 9624), anti-Flag (Sigma, F3165), anti-HA (Covance/BioLegend, MMS-101P), anti-CDK8 (Santa Cruz, sc-13155 ), anti-LC3B (Cell Signaling, 2775), anti-human CPSF6 (Santa Cruz, sc-100692), anti-ubiquitin FK2 (Enzo life Science, BML-PW8810-0100), and anti-Tubulin (Sigma, T5168). Anti-Atg1 was a gift of Dr. Jun Hee Lee (Kim et al., 2013). Anti-CLK2 was produced by cloning via PCR a 1.3 kb fragment encoding the catalytic domain of human CLK2 from a full-length cDNA in frame into the BamHI site in the pMAL-C2 vector (New England Biolabs). It was expressed in E. coli as a fusion protein with the Maltose Binding Protein. Following purification on an amylose column, the recombinant protein was injected into two rats for antibody production.
Immunofluorescence
Eye imaginal discs from wandering third instar larvae were dissected, fixed with 4% paraformaldehyde, and mounted. GFP-marked flip clones in the larval fat body were generated through heat shock-independent induction as previously described (Tang et al., 2013). For lipid droplet staining, larvae were dissected in PBS and fixed in 4% paraformaldehyde for 30 min at room temperature. Larval fat body was then washed with PBS, incubated for 15 min in 2μg/ml Nile red/PBS, and then mounted. S2R+ cells were fixed with 4% paraformaldehyde and MCF7 cells were fixed with ice-cold 100% methanol. Cells were then permeabilized with 0.1% triton and processed for immunostaining. DAPI (1 μg/ml) was used to stain nuclei. Samples were examined using a confocal laser scanning microscope (LSM780; Carl Zeiss Inc.) equipped with a 63× Plan-Apochromat (NA1.4) objective lens.
Immunoblotting
Larvae were collected 72–96 hrs after egg laying, cultured in fresh fly media supplemented with yeast paste (Fed) and 10 mg/ml CQ (Fed+CQ), or in vials containing 20% sucrose (Starved) and 10 mg/ml CQ (Starved+CQ) for 4hrs or 16 hrs prior to dissection. After dissection, the samples were then boiled in SDS sample buffer, run on a 4–20% polyacrylamide gel (Bio-Rad, 4561096), and transferred to an Immobilon-P polyvinylidene fluoride (PVDF) membrane (Millipore). The membrane was blocked by 5%BAS in TBST (TBS with 0.1% Tween-20) in room temperature for 1 hr and then probed with primary antibody in 1X TBST with 5% BSA overnight, followed by HRP-conjugated secondary antibody, and signal was detected by enhanced chemiluminescence (ECL; Amersham, RPN2209; Pierce, 34095).
Quantification of mRNA expression
Total RNA was extracted from larval fat body using TRIzol® reagent (Invitrogen 15596-018). We synthesized first strand cDNA with 1 μg of total RNA using iScriptTM Reverse Transcription Supermix (BIO-RAD, 1708896) and then performed RT-PCR using GoTaq Green Master mix (Promega, M7122) or quantitative PCR with CFX96 Real-Time System (BIO-RAD) using iQTM SYBR Green Supermix (BIO-RAD, 1708880). All expression values were normalized to RpL32 (also known as rp49). All assays were performed in triplicate. The primer sequences used for PCR are listed in Table S5.
ATP, Triglyceride, and protein measurements
MCF7 cells or six larvae from each group were homogenized in PBS supplemented with 0.1% Triton X-100 and Proteinase inhibitor cocktail (Pierce, 78440), heated at 70C for 5 min, and the supernatant collected after centrifugation at 14,000 rpm for 10 min. Supernatants were then subjected to TAG and Protein measurement using a Serum Triglyceride Determination kit (Sigma, T2449) and a BCA protein assay (Pierce, 23227) following the manufacturer’s protocol. For ATP assay, larvae or cells were lysed in CellTiter-Glo buffers and lysates were subjected to ATP measurement using the CellTiter-Glo luminescent cell viability assay kit (Promega, G7573). For the Drosophila studies, ATP, triglyceride, and protein values were normalized to larval weight.
Immunoprecipitation
DNA was transfected into S2R+ or HEK293T cells in a 10cm plate with Effectene transfection reagent (Qiagen, 301427) following the manufacturer’s protocol. After 3 days of incubation, cells were lysed with lysis buffer (Pierce 89901 and 87788) with 2X protease and phosphatase inhibitor cocktail (Pierce, 78440) or RNasin Plus RNase inhibitor (Promega, N2611). Lysate was incubated with Chromotek-GFP-Trap (Bulldog Biotechnology, gta-20), anti-HA agarose (Sigma, A2095) or anti-Flag agarose (Sigma, A2220) for 1–2hrs at 4°C to precipitate the protein complexes. Beads were washed 3–4 times with 1 ml lysis buffer. Phosphorylated protein-RNA complexes were subjected to label-free quantitative mass spectrometry with in-gel digestion using Chymotrypsin or detected by Western blotting or qPCR. The primer sequences used for RNA-Immunoprecipitation are listed in Table S5.
In vitro kinase assay
Recombinant human proteins used for the study were: His-tagged CDK8/CycC (Thermo Fisher Scientific, PV4402), CLK2-cat (amino acids 137–499) (Thermo Fisher Scientific, PV4201), GST-tagged CPSF6-N (NovoPro Bioscience, 506834), and GST-tagged CPSF6 (Abnova, H00011052-P01). To generate recombinant Drosophila proteins, various segments of CPSF6 cDNA were cloned into pGEX-2T and recombinant CPSF6 proteins were purified from bacteria and eluted from Glutathione Sepharose (Clontech, 635607) as suggested by the manufacturer. S2R+ cells expressing HA-DOA, GFP-ATG1-PB, or ATG1-PB-S297A or HEK 293T cells expressing FLAG-CDK8 were lysed in lysis buffer (Pierce, 87788) with protease and phosphatase inhibitor cocktail (Pierce, 78440) and the lysates were immunoprecipitated. Kinase reactions were carried out in a kinase reaction buffer containing the immune complex, recombinant proteins, and [r-32P] ATP as described previously (Tang et al., 2011).
RNA Sequencing Analysis
Total RNA was extracted from larval fat body using TRIzol® reagent (Invitrogen, 15596-018). After assessing RNA quality with an Agilent Bioanalyzer, libraries constructed with an Illumina TruSeq Stranded Total RNA Library Prep Kit with Ribo-Zero Gold were sequenced using an Illumina HiSeq 4000 at the Columbia Genome Center (http://systemsbiology.columbia.edu/genome-center). We multiplexed samples in each lane, which yields a targeted number of paired-end 100-bp reads for each sample. The raw data files of sequencing reads were processed at Harvard Chan Bioinformatics Core with the bcbio-nextgen pipeline, version 1.0.0a0-4708de9 (http://bcbio-nextgen.readthedocs.io/). For quality control purposes, the reads were aligned to Drosophila genome version BDGP6 with STAR version 2.5.3 (https://github.com/alexdobin/STAR) and the alignments were evaluated based on the mappability to transcripts and the complexity of the transcriptome as well as the quality among other custom metrics. The expression of transcripts was quantified using Sailfish 0.10.1 (https://github.com/kingsfordgroup/sailfish) taking 30 bootstrap samples for each sample. Gene level expression was calculated by collapsing the transcript-level quantification with tximport (https://github.com/mikelove/tximport). Differentially expressed genes were analyzed with DESeq2 version 1.14.1 (https://bioconductor.statistik.tu-dortmund.de/packages/3.4/bioc/html/DESeq2.html) while differentially expressed transcripts were analyzed with sleuth version 0.28.1 (https://github.com/pachterlab/sleuth), leveraging the bootstrap samples to account for technical variability assigning reads to individual transcripts. The final hits of differentially expressed genes as well as transcripts were selected based on both fold changes comparing to the control larval fat body as well as adjusted p value calculated by DESeq or sleuth. TPM values were used to calculate the fold changes. Hits were selected if 2 or more-fold changes were consistently observed among the replicates and the adjusted P value is less than 0.05. The genes or transcripts that do not meet the 2-fold change cutoff with any of the replicates but with adjusted p-value of 0.01 or less are included as low confidence hits in Table S2 but were not selected for enrichment analysis. Gene set enrichment analysis was performed using an in-house java program based on hyper-geometric distribution. For Drosophila genes, gene sets were assembled using gene ontology annotation, pathway annotation from GLAD, and protein complex annotation from COMPLEAT (Hu et al., 2017). Human pathway annotation of Reactome and KEGG were mapped to Drosophila gene sets using DIOPT (Hu et al., 2017). The heatmap of the selected gene sets were obtained using TM4 software suite (http://mev.tm4.org/). We selected the starvation responsive genes that are CPSF5 dependent and also part of selected gene sets (energy metabolism, ribosome, lipid metabolism, glycolysis and autophagy) and built a protein-protein interaction network based on integrated network from BioGRID, InAct, MINT, DIP, DroID, DPiM and FlyBase. Solid edges are protein-protein interactions identified in Drosophila while the dotted lines are for interologs, protein-protein interactions derived from data in other species. Network visualization was done using Cytoscape vs 3.1.0 (http://www.cytoscape.org/) (https://github.com/cytoscape).
QUANTIFICATION AND STATISTICAL ANALYSES
Statistical analyses were performed in GraphPad Prism. Student’s t-test was used for pairwise comparisons, whereas multiple comparisons were analyzed with one-way analysis of variance (ANOVA) and Bonferroni’s post hoc test. No particular methods were used to determine whether the data met assumptions of the statistical approach. Statistical parameters can be found in the figure legends.
DATA AND SOFTWARE AVAILABILITY
The accession number for the RNA-seq data reported in this paper is GEO: GSE99509.
Supplementary Material
Research Highlights.
TORC1 negatively inhibits CDK8 and DOA kinases.
CDK8 and DOA phosphorylate CPSF6 to induce alternative RNA processing.
Depletion of CPSF6 impairs autophagy and metabolic changes during starvation.
The CDK8-CLK2/DOA-CPSF6 axis is conserved in mammals.
Acknowledgments
We thank: Harvard Catalyst and Dr. Liming Liang (Harvard T.H. Chan School of Public Health) for statistical consulting, R. Kirchner (Harvard Chan Bioinformatics Core) for assistance with RNA-seq analysis, Dr. J.-Y. Ji, Dr. William Mattox, the Vienna Drosophila Resource Center and Bloomington Drosophila Stock Center for fly stocks. We also thank G. Lee, J. Blenis, and members of the Perrimon laboratory for critical comments on the manuscript. This work was supported by the NIH (R01 AR057352 and 5P01CA120964) and the STARR Consortium (N.P.). H.-W. Tang is supported by the Human Frontier Science Program and the Postdoctoral Research Abroad Program, sponsored by Ministry of Science and Technology, Taiwan. N.P. is a Howard Hughes Medical Institute investigator.
Footnotes
Author Contributions
H.T. conceived the study and designed and performed experiments. Y.H. performed bioinformatics analyses. C.C. generated CRISPR mutants. B.X. performed 3′RACE experiments. J.Z. generated transgenic flies. M.Y. and J.A. performed mass spectrometry experiments. L.R. provided the CLK2 antibody. H.T., L.R., and N.P., discussed results. H.T. and N.P. wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akman HB, Oyken M, Tuncer T, Can T, Erson-Bensan AE. 3′UTR shortening and EGF signaling: implications for breast cancer. Hum Mol Genet. 2015;24:6910–6920. doi: 10.1093/hmg/ddv391. [DOI] [PubMed] [Google Scholar]
- Alamancos GP, Pages A, Trincado JL, Bellora N, Eyras E. Leveraging transcript quantification for fast computation of alternative splicing profiles. RNA. 2015;21:1521–1531. doi: 10.1261/rna.051557.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsham AM, Neufeld TP. A genetic screen in Drosophila reveals novel cytoprotective functions of the autophagy-lysosome pathway. PLoS One. 2009;4:e6068. doi: 10.1371/journal.pone.0006068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baier RV, Picao-Osorio J, Alonso CR. Molecular regulation of alternative polyadenylation (APA) within the Drosophila nervous system. J Mol Biol. 2017 doi: 10.1016/j.jmb.2017.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budovskaya YV, Stephan JS, Deminoff SJ, Herman PK. An evolutionary proteomics approach identifies substrates of the cAMP-dependent protein kinase. Proc Natl Acad Sci U S A. 2005;102:13933–13938. doi: 10.1073/pnas.0501046102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardinale S, Cisterna B, Bonetti P, Aringhieri C, Biggiogera M, Barabino SM. Subnuclear localization and dynamics of the Pre-mRNA 3′ end processing factor mammalian cleavage factor I 68-kDa subunit. Mol Biol Cell. 2007;18:1282–1292. doi: 10.1091/mbc.E06-09-0846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabot B, Shkreta L. Defective control of pre-messenger RNA splicing in human disease. J Cell Biol. 2016;212:13–27. doi: 10.1083/jcb.201510032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan S, Choi EA, Shi Y. Pre-mRNA 3′-end processing complex assembly and function. Wiley Interdiscip Rev RNA. 2011;2:321–335. doi: 10.1002/wrna.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JW, Zhang W, Yeh HS, de Jong EP, Jun S, Kim KH, Bae SS, Beckman K, Hwang TH, Kim KS, et al. mRNA 3′-UTR shortening is a molecular signature of mTORC1 activation. Nat Commun. 2015;6:7218. doi: 10.1038/ncomms8218. [DOI] [PubMed] [Google Scholar]
- Chang TH, Huang HY, Hsu JB, Weng SL, Horng JT, Huang HD. An enhanced computational platform for investigating the roles of regulatory RNA and for identifying functional RNA motifs. BMC Bioinformatics. 2013a;14(Suppl 2):S4. doi: 10.1186/1471-2105-14-S2-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TK, Shravage BV, Hayes SD, Powers CM, Simin RT, Wade Harper J, Baehrecke EH. Uba1 functions in Atg7- and Atg3-independent autophagy. Nat Cell Biol. 2013b;15:1067–1078. doi: 10.1038/ncb2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen GC, Lee JY, Tang HW, Debnath J, Thomas SM, Settleman J. Genetic interactions between Drosophila melanogaster Atg1 and paxillin reveal a role for paxillin in autophagosome formation. Autophagy. 2008;4:37–45. doi: 10.4161/auto.5141. [DOI] [PubMed] [Google Scholar]
- Cheong H, Nair U, Geng J, Klionsky DJ. The Atg1 kinase complex is involved in the regulation of protein recruitment to initiate sequestering vesicle formation for nonspecific autophagy in Saccharomyces cerevisiae. Mol Biol Cell. 2008;19:668–681. doi: 10.1091/mbc.E07-08-0826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dettwiler S, Aringhieri C, Cardinale S, Keller W, Barabino SM. Distinct sequence motifs within the 68-kDa subunit of cleavage factor Im mediate RNA binding, protein-protein interactions, and subcellular localization. J Biol Chem. 2004;279:35788–35797. doi: 10.1074/jbc.M403927200. [DOI] [PubMed] [Google Scholar]
- Dorsey FC, Rose KL, Coenen S, Prater SM, Cavett V, Cleveland JL, Caldwell-Busby J. Mapping the phosphorylation sites of Ulk1. J Proteome Res. 2009;8:5253–5263. doi: 10.1021/pr900583m. [DOI] [PubMed] [Google Scholar]
- Feng D, Youn DY, Zhao X, Gao Y, Quinn WJ, 3rd, Xiaoli AM, Sun Y, Birnbaum MJ, Pessin JE, Yang F. mTORC1 Down-Regulates Cyclin-Dependent Kinase 8 (CDK8) and Cyclin C (CycC) PLoS One. 2015;10:e0126240. doi: 10.1371/journal.pone.0126240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortier E, Belote JM. Temperature-dependent gene silencing by an expressed inverted repeat in Drosophila. Genesis. 2000;26:240–244. doi: 10.1002/(sici)1526-968x(200004)26:4<240::aid-gene40>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Gabut M, Dejardin J, Tazi J, Soret J. The SR family proteins B52 and dASF/SF2 modulate development of the Drosophila visual system by regulating specific RNA targets. Mol Cell Biol. 2007;27:3087–3097. doi: 10.1128/MCB.01876-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh G, Adams JA. Phosphorylation mechanism and structure of serine-arginine protein kinases. FEBS J. 2011;278:587–597. doi: 10.1111/j.1742-4658.2010.07992.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guruharsha KG, Rual JF, Zhai B, Mintseris J, Vaidya P, Vaidya N, Beekman C, Wong C, Rhee DY, Cenaj O, et al. A protein complex network of Drosophila melanogaster. Cell. 2011;147:690–703. doi: 10.1016/j.cell.2011.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Housden BE, Perrimon N. Detection of Indel Mutations in Drosophila by High-Resolution Melt Analysis (HRMA) Cold Spring Harb Protoc. 2016;2016 doi: 10.1101/pdb.prot090795. pdb prot090795. [DOI] [PubMed] [Google Scholar]
- Housden BE, Valvezan AJ, Kelley C, Sopko R, Hu Y, Roesel C, Lin S, Buckner M, Tao R, Yilmazel B, et al. Identification of potential drug targets for tuberous sclerosis complex by synthetic screens combining CRISPR-based knockouts with RNAi. Sci Signal. 2015;8:rs9. doi: 10.1126/scisignal.aab3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PP, Kang SA, Rameseder J, Zhang Y, Ottina KA, Lim D, Peterson TR, Choi Y, Gray NS, Yaffe MB, et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science. 2011;332:1317–1322. doi: 10.1126/science.1199498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G, McQuiston T, Bernard A, Park YD, Qiu J, Vural A, Zhang N, Waterman SR, Blewett NH, Myers TG, et al. A conserved mechanism of TOR-dependent RCK-mediated mRNA degradation regulates autophagy. Nat Cell Biol. 2015;17:930–942. doi: 10.1038/ncb3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Comjean A, Roesel C, Vinayagam A, Flockhart I, Zirin J, Perkins L, Perrimon N, Mohr SE. FlyRNAi.org-the database of the Drosophila RNAi screening center and transgenic RNAi project: 2017 update. Nucleic Acids Res. 2017;45:D672–D678. doi: 10.1093/nar/gkw977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P, Mizushima N. Autophagy and human diseases. Cell Res. 2014;24:69–79. doi: 10.1038/cr.2013.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Park HL, Park HW, Ro SH, Nam SG, Reed JM, Guan JL, Lee JH. Drosophila Fip200 is an essential regulator of autophagy that attenuates both growth and aging. Autophagy. 2013;9:1201–1213. doi: 10.4161/auto.24811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyburz A, Friedlein A, Langen H, Keller W. Direct interactions between subunits of CPSF and the U2 snRNP contribute to the coupling of pre-mRNA 3′ end processing and splicing. Mol Cell. 2006;23:195–205. doi: 10.1016/j.molcel.2006.05.037. [DOI] [PubMed] [Google Scholar]
- Lee G, Zheng Y, Cho S, Jang C, England C, Dempsey JM, Yu Y, Liu X, He L, Cavaliere PM, et al. Post-transcriptional Regulation of De Novo Lipogenesis by mTORC1-S6K1-SRPK2 Signaling. Cell. 2017;171:1545–1558. e1518. doi: 10.1016/j.cell.2017.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Moir RD, McIntosh KB, Willis IM. TOR signaling regulates ribosome and tRNA synthesis via LAMMER/Clk and GSK-3 family kinases. Mol Cell. 2012;45:836–843. doi: 10.1016/j.molcel.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G, Gruber AR, Keller W, Zavolan M. Genome-wide analysis of pre-mRNA 3′ end processing reveals a decisive role of human cleavage factor I in the regulation of 3′ UTR length. Cell Rep. 2012;1:753–763. doi: 10.1016/j.celrep.2012.05.003. [DOI] [PubMed] [Google Scholar]
- Mercer TJ, Gubas A, Tooze SA. A Molecular Perspective of Mammalian Autophagosome Biogenesis. J Biol Chem. 2018 doi: 10.1074/jbc.R117.810366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millevoi S, Loulergue C, Dettwiler S, Karaa SZ, Keller W, Antoniou M, Vagner S. An interaction between U2AF 65 and CF I(m) links the splicing and 3′ end processing machineries. EMBO J. 2006;25:4854–4864. doi: 10.1038/sj.emboj.7601331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura P, Sanfilippo P, Shenker S, Lai EC. Alternative polyadenylation in the nervous system: to what lengths will 3′ UTR extensions take us? Bioessays. 2014;36:766–777. doi: 10.1002/bies.201300174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr SE, Hu Y, Rudd K, Buckner M, Gilly Q, Foster B, Sierzputowska K, Comjean A, Ye B, Perrimon N. Reagent and Data Resources for Investigation of RNA Binding Protein Functions in Drosophila melanogaster Cultured Cells. G3 (Bethesda) 2015;5:1919–1924. doi: 10.1534/g3.115.019364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsumi Y. Historical landmarks of autophagy research. Cell Res. 2014;24:9–23. doi: 10.1038/cr.2013.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter CJ, Huang H, Xu T. Drosophila Tsc1 functions with Tsc2 to antagonize insulin signaling in regulating cell growth, cell proliferation, and organ size. Cell. 2001;105:357–368. doi: 10.1016/s0092-8674(01)00333-6. [DOI] [PubMed] [Google Scholar]
- Rolland T, Tasan M, Charloteaux B, Pevzner SJ, Zhong Q, Sahni N, Yi S, Lemmens I, Fontanillo C, Mosca R, et al. A proteome-scale map of the human interactome network. Cell. 2014;159:1212–1226. doi: 10.1016/j.cell.2014.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017;169:361–371. doi: 10.1016/j.cell.2017.03.035. [DOI] [PubMed] [Google Scholar]
- Scott RC, Juhasz G, Neufeld TP. Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr Biol. 2007;17:1–11. doi: 10.1016/j.cub.2006.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Di Giammartino DC, Taylor D, Sarkeshik A, Rice WJ, Yates JR, 3rd, Frank J, Manley JL. Molecular architecture of the human pre-mRNA 3′ processing complex. Mol Cell. 2009;33:365–376. doi: 10.1016/j.molcel.2008.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith FD, Samelson BK, Scott JD. Discovery of cellular substrates for protein kinase A using a peptide array screening protocol. Biochem J. 2011;438:103–110. doi: 10.1042/BJ20110720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan JS, Yeh YY, Ramachandran V, Deminoff SJ, Herman PK. The Tor and PKA signaling pathways independently target the Atg1/Atg13 protein kinase complex to control autophagy. Proc Natl Acad Sci U S A. 2009;106:17049–17054. doi: 10.1073/pnas.0903316106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svoboda P, Di Cara A. Hairpin RNA: a secondary structure of primary importance. Cell Mol Life Sci. 2006;63:901–908. doi: 10.1007/s00018-005-5558-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang HW, Liao HM, Peng WH, Lin HR, Chen CH, Chen GC. Atg9 interacts with dTRAF2/TRAF6 to regulate oxidative stress-induced JNK activation and autophagy induction. Dev Cell. 2013;27:489–503. doi: 10.1016/j.devcel.2013.10.017. [DOI] [PubMed] [Google Scholar]
- Tang HW, Wang YB, Wang SL, Wu MH, Lin SY, Chen GC. Atg1-mediated myosin II activation regulates autophagosome formation during starvation-induced autophagy. EMBO J. 2011;30:636–651. doi: 10.1038/emboj.2010.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varjosalo M, Keskitalo S, Van Drogen A, Nurkkala H, Vichalkovski A, Aebersold R, Gstaiger M. The protein interaction landscape of the human CMGC kinase group. Cell Rep. 2013;3:1306–1320. doi: 10.1016/j.celrep.2013.03.027. [DOI] [PubMed] [Google Scholar]
- Xiang S, Gapsys V, Kim HY, Bessonov S, Hsiao HH, Mohlmann S, Klaukien V, Ficner R, Becker S, Urlaub H, et al. Phosphorylation drives a dynamic switch in serine/arginine-rich proteins. Structure. 2013;21:2162–2174. doi: 10.1016/j.str.2013.09.014. [DOI] [PubMed] [Google Scholar]
- Yaman I, Fernandez J, Sarkar B, Schneider RJ, Snider MD, Nagy LE, Hatzoglou M. Nutritional control of mRNA stability is mediated by a conserved AU-rich element that binds the cytoplasmic shuttling protein HuR. J Biol Chem. 2002;277:41539–41546. doi: 10.1074/jbc.M204850200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun B, Farkas R, Lee K, Rabinow L. The Doa locus encodes a member of a new protein kinase family and is essential for eye and embryonic development in Drosophila melanogaster. Genes Dev. 1994;8:1160–1173. doi: 10.1101/gad.8.10.1160. [DOI] [PubMed] [Google Scholar]
- Zhao S, Chen D, Geng Q, Wang Z. The highly conserved LAMMER/CLK2 protein kinases prevent germ cell overproliferation in Drosophila. Dev Biol. 2013;376:163–170. doi: 10.1016/j.ydbio.2013.01.023. [DOI] [PubMed] [Google Scholar]
- Zhao X, Feng D, Wang Q, Abdulla A, Xie XJ, Zhou J, Sun Y, Yang ES, Liu LP, Vaitheesvaran B, et al. Regulation of lipogenesis by cyclin-dependent kinase 8-mediated control of SREBP-1. J Clin Invest. 2012;122:2417–2427. doi: 10.1172/JCI61462. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.