

SUMMARY
Cancer cell survival is dependent on oxidative-stress defenses against reactive oxygen species (ROS) that accumulate during tumorigenesis. Here, we show a non-canonical oxidative-stress defense mechanism through TRPA1, a neuronal redox-sensing Ca2+-influx channel. In TRPA1-enriched breast and lung cancer spheroids, TRPA1 is critical for survival of inner cells that exhibit ROS accumulation. Moreover, TRPA1 promotes resistance to ROS-producing chemotherapies, and TRPA1 inhibition suppresses xenograft tumor growth and enhances chemosensitivity. TRPA1 does not affect redox status but upregulates Ca2+-dependent anti-apoptotic pathways. NRF2, an oxidant-defense transcription factor, directly controls TRPA1 expression, thus providing an orthogonal mechanism for protection against oxidative stress together with canonical ROS-neutralizing mechanisms. These findings reveal an oxidative-stress defense program involving TRPA1 that could be exploited for targeted cancer therapies.
Graphical Abstract
In Brief
Takahashi et al. show that TRPA1, a neuronal redox-sensing Ca2+-influx channel overexpressed in human cancer, upregulates Ca2+-dependent anti-apoptotic pathways to promote ROS resistance. NRF2 directly controls TRPA1 expression and TRPA1 inhibition suppresses xenograft tumor growth and enhances chemosensitivity.
INTRODUCTION
Tumor suppressor and oncogenic pathways frequently mutated in cancer commonly cause increased accumulation of reactive oxygen species (ROS) (Gorrini et al., 2013). Moreover, conditions associated with tumorigenesis, such as detachment from extracellular matrix (ECM), hypoxia, and inflammation, can all lead to generation of ROS and impose further oxidative stress on tumor cells (Gorrini et al., 2013; Schafer et al., 2009; Tennant et al., 2010). These highly reactive metabolites can damage cellular components and induce apoptosis (Gorrini et al., 2013; Schieber and Chandel, 2014). Mounting evidence suggests that during tumor progression, there is a selection for cancer cells that have induced oxidative-stress defense programs to adapt to oxidative stress (Gorrini et al., 2013; Schieber and Chandel, 2014; Tennant et al., 2010).
Oxidative-stress defense is of particular importance for cancer cells in the acquisition of anchorage independence. Epithelial cells are dependent on interactions with specific ECM components for survival, proliferation, and differentiation functions (Debnath and Brugge, 2005). When normal cells are displaced from their ECM niches, they undergo anoikis, a form of apoptotic cell death. The ability to avoid anoikis is an important characteristic of most epithelial tumors. Using three-dimensional (3D) culture models, we previously found that the centrally localized, ECM-deprived cells within mammary epithelial MCF-10A acini accumulate ROS, which contribute to cell death and the development of a hollow lumen (Schafer et al., 2009). Treatment with ROS scavengers can prevent inner cell death in MCF-10A acini, suggesting that oxidative-stress defense is required for cancer cells to fill the luminal space, a hallmark of epithelial tumors.
Oxidative-stress defense mechanisms that reduce ROS have been investigated in multiple steps of tumorigenesis. Through disruption of Gclm encoding a rate-limiting enzyme for the synthesis of glutathione (GSH) that can neutralize ROS, GSH was shown to be required for cancer initiation in the MMTV-PyMT mouse breast cancer model (Harris et al., 2015). Moreover, metastasizing melanoma cells undergo metabolic changes involving upregulation of NADPH-generating enzymes that can increase GSH/oxidized GSH (GSSG) ratio, and treatment with N-acetyl-L-cysteine (NAC), an ROS-scavenging agent, enhances metastasis (Le Gal et al., 2015; Piskounova et al., 2015). Reductive glutamine metabolism was shown to mitigate mitochondrial ROS and promote growth of lung cancer spheroids (Jiang et al., 2016). Notably, the KEAP1-NRF2 pathway, which plays a central role in protecting cells against oxidative stress through induction of ROS-neutralizing gene expression (Suzuki et al., 2013), was shown to stimulate cancer initiation (DeNicola et al., 2011) and support tumor maintenance in pancreatic cancer (Chio et al., 2016). However, it is not straight-forward to target canonical ROS-neutralizing programs because of the increased oxidative stress in normal tissues.
In addition to the KEAP1-NRF2 system, a subset of the mammalian transient receptor potential (TRP) family proteins, which comprise 28 subtypes of ion channels, detects oxidants/electrophiles, including ROS, and induces Ca2+/cation influx (Clapham, 2003; Shimizu et al., 2014). TRPA1, TRPC5, and TRPV1–TRPV4 channels are directly activated by oxidants/electrophiles through cysteine modifications (Hinman et al., 2006; Macpherson et al., 2007; Shimizu et al., 2014; Takahashi et al., 2011), whereas TRPM2 and TRPM7 are indirectly activated by ROS (Shimizu et al., 2014). Each redox-sensitive TRP channel senses a specific range of redox potential (Takahashi et al., 2011). TRPA1, which was originally found as the receptor of mustard oil in sensory neurons (Jordt et al., 2004), exhibits by far the highest sensitivity to oxidants due to the presence of hyper-reactive cysteines in its cytoplasmic region and plays a pivotal role in detecting cysteine-reactive irritants and augmenting sensory or vagal nerve discharges to evoke pain and cough (Takahashi et al., 2011). TRPA1 is also activated by cancer therapies in sensory neurons, which is associated with therapy-induced pain (Fusi et al., 2014; Nassini et al., 2011). Recently, a subset of TRP channels has been found overexpressed in cancer (Déliot and Constantin, 2015; Park et al., 2016). However, their importance in cancer initiation or progression remains largely unknown. Given the fundamental role of Ca2+ signaling in a wide range of cellular responses, including cell proliferation and survival (Clapham, 2007), it is important to understand if and how upregulated redox-sensitive TRP channels affect oxidative-stress defense programs in cancer cells.
RESULTS
TRPA1 Is Functionally Overexpressed in Diverse Cancer Types
Analysis of The Cancer Genome Atlas (TCGA) datasets showed that some redox-sensitive TRP channels exhibit altered mRNA expression in tumors relative to corresponding normal tissues (Figure 1A and Table S1). Among these, TRPA1 was the most highly upregulated in diverse cancer types. Interestingly, among all TRP channels, TRPA1 was the most highly upregulated in invasive ductal breast carcinoma (BRCA, enriched in all breast cancer subtypes except for normal-like subtype), kidney renal clear cell carcinoma (KIRC), and lung squamous cell carcinoma (LUSC) and was the second-most highly upregulated TRP channel in lung adenocarcinoma (LUAD) (Figures S1A and S1B and Table S1). TRPA1 mRNA was also highly upregulated in malignant peripheral nerve sheath tumor (MPNST) (Figures S1C and S1D) (Miller et al., 2009).
Figure 1. TRPA1 Is Functionally Overexpressed in Diverse Tumor Types.
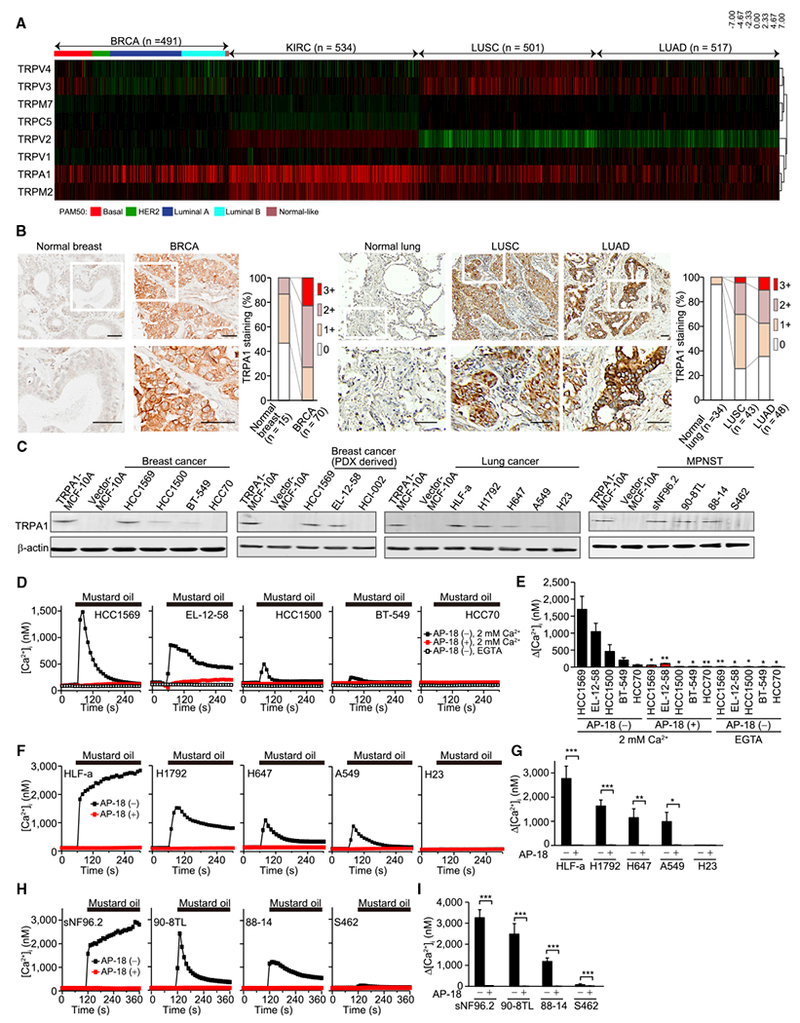
(A) Heatmap of redox-sensitive TRP channel mRNA levels in TCGA BRCA, KIRC, LUSC, and LUAD relative to corresponding normal tissues (n = 121 for BRCA, n = 72 for KIRC, n = 51 for LUSC, and n = 59 for LUAD). The samples, whose PAM50 has not been identified, are not included for BRCA.
(B) Representative images of TRPA1 IHC in normal breast tissue and breast tumor sections and in normal lung tissue and lung tumor sections, and the score of staining level. An enlarged view of the boxed region in the top images is shown in the corresponding lower image. Scale bar, 50 μm.
(C) Immunoblot analysis of TRPA1 in the indicated breast cancer and lung cancer or MPNST cells.
(D and E) Averaged time courses of [Ca2+]i changes (D) and the maximal [Ca2+]i rises (Δ[Ca2+]i) (E) evoked by 30 μM mustard oil in Ca2+-free, 0.5 mM EGTA- or 2 mM Ca2+-containing solution are shown for the indicated breast cancer cells treated with or without 10 μM AP-18. Data are shown as means ± SEM (n = 13–117). *p < 0.05 and **p < 0.01 compared with AP-18 (–) in 2 mM Ca2+-containing solution (Student’s t test).
(F–I) Averaged time courses of [Ca2+]i changes (F and H) and Δ[Ca2+]i (G and I) evoked by 30 μM mustard oil in 2 mM Ca2+-containing solution in the indicated lung cancer (F and G) and MPNST cells (H and I) treated with or without 10 μM AP-18. Data are shown as means ± SEM (n = 21–60 for F and G, n = 12–100 for H and I). *p < 0.05, **p < 0.01, and ***p < 0.001 (Student’s t test).
See also Figure S1 and Tables S1 and S2.
TRPA1 is frequently amplified in BRCA (10%) and MPNST (27%), but rarely so in LUSC (1.7%) and LUAD (2.2%) and KIRC (0.9%). In BRCA, TRPA1 is located at the peak of the chromosome 8 amplicon, but TRPA1 mRNA weakly correlates with increased copy number (Figure S1E). TRPA1 mutations were found in some cancers; however, none (among 61 mutants tested) led to a constitutive TRPA1 activation (Figure S1F and Table S2).
Consistent with TRPA1 mRNA upregulation, immunohistochemistry (IHC) staining of a panel of breast or lung cancer and corresponding normal tissue samples revealed that TRPA1 protein expression is significantly higher in breast (χ2 test: p = 0.0001) and lung tumors (χ2 test: p = 0.0004 in LUSC, p < 0.0001 in LUAD) (Figures 1B, S1G, and S1H). High TRPA1 mRNA expression was also observed in 2 of 12 patient-derived xenografts (PDXs) from BRCA (HCI-003 and HCI-011) (DeRose et al., 2011) (Figure S1I), which was validated in HCI-011 by IHC analysis (Figure S1J).
Quantitative PCR and immunoblot analysis showed that TRPA1 is expressed in a subset of breast, lung, and MPNST cancer cell lines (Figures 1C, S1K, and S1L). A lack of available kidney cancer cell lines with high TRPA1 expression (based on CCLE; http://www.broadinstitute.org/software/cprg) precluded our ability to assess TRPA1 expression in this cancer type. We also examined TRPA1 expression in two basal-like breast cancer cell lines derived from breast cancer PDXs (DeRose et al., 2011; Johnson et al., 2016). TRPA1 expression was enriched in EL-12-58 cells but not in HCI-002 cells (Figure 1C). To address whether TRPA1 is functionally active in these cancer cells, we examined the effect of mustard oil, the TRPA1 agonist (Jordt et al., 2004), on intracellular Ca2+ mobilization. Mustard oil elicited increases in intracellular Ca2+ concentration ([Ca2+]i), which were abolished by either the TRPA1 specific inhibitor AP-18, TRPA1 knockdown by short hairpin RNA (shRNA), or removal of extracellular Ca2+ (Figures 1D–1I and S1M).
TRPA1 Promotes Oxidative-Stress Defense in Response to ROS
We next investigated the impact of TRPA1 on cell survival in response to H2O2, one of the most important ROS in pathophysiology of cancer (Schieber and Chandel, 2014).
H2O2 induced an increase in [Ca2+]i and also oscillatory Ca2+ responses that were suppressed by TRPA1 inhibition in TRPA1-enriched cancer cell lines (Figures 2A–2C and S2A–S2C), indicating that TRPA1 is the major channel responsible for Ca2+ entry in response to H2O2 in these cancer cells. This is consistent with evidence that knockdown of TRPM2, another redox-sensitive channel (Shimizu et al., 2014) whose expression partially overlaps that of TRPA1 in human tumors (Figure 1A), does not affect Ca2+ responses to H2O2 in HCC1569 and H1792 cells (Figures S2D and S2E). Notably, AP-18 or TRPA1 knockdown, which did not affect normal cell growth in monolayer culture (Figure S2F), decreased live cell number and increased cell death in response to H2O2 in TRPA1-enriched cancer cells, but not in those with low TRPA1 expression (Figures 2D–2I, S2G, and S2H). Ectopic TRPA1 expression increased the survival of H2O2-treated cells with low basal TRPA1 expression (Figures 2J and S2I), further supporting that TRPA1 promotes oxidative-stress defense. The intracellular Ca2+ chelator BAPTA-AM suppressed cell viability in shGFP-transduced control HCC1569 cells but not in shTRPA1-transduced cells upon treatment with H2O2 (Figure 2K), indicating the importance of TRPA1-mediated Ca2+ influx in the phenotype.
Figure 2. TRPA1 Increases [Ca2+]i and Cell Survival in Response to H2O2 in Cancer Cells.
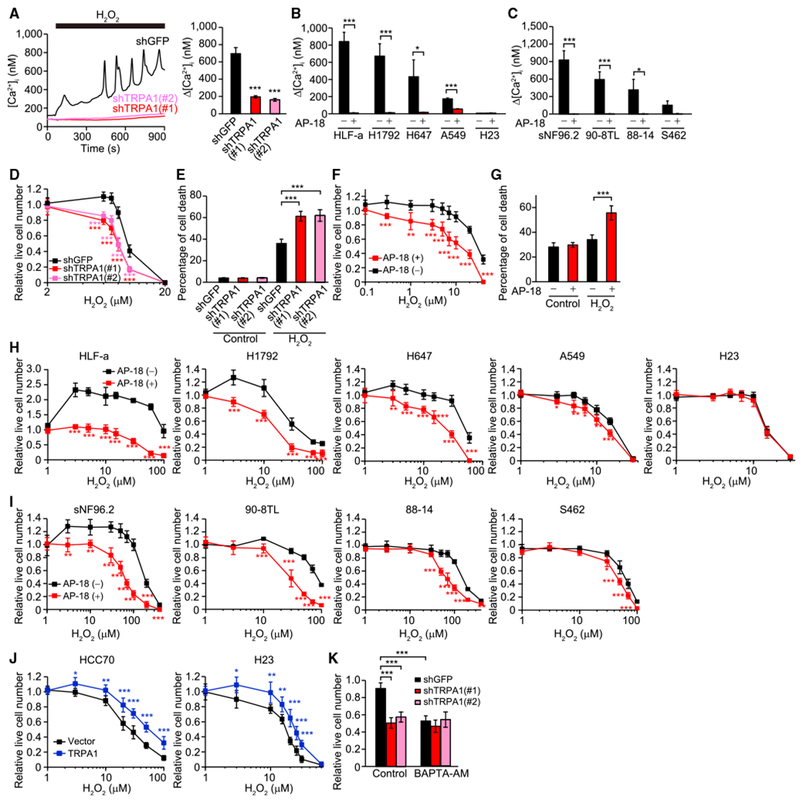
(A) Representative time courses and Δ[Ca2+]i evoked by 10 μM H2O2 in the indicated HCC1569 cells. Data are shown as means ± SEM (n = 35–58). ***p < 0.001 compared with shGFP (Student’s t test).
(B and C) Δ[Ca2+]i evoked by 10 μM H2O2(B) or 30 μM H2O2 (C) in the indicated lung cancer (B) and MPNST cells (C) treated with or without 10 μM AP-18. Data are shown as means ± SEM (n = 21–120 for B, n = 28–107 for C). *p < 0.05 and ***p < 0.001 (Student’s t test).
(D) Live cell numbers relative to H2O2-untreated cells in the indicated HCC1569 cells upon treatment with H2O2 for 96 hr. ***p < 0.001 compared with shGFP (Student’s t test).
(E) Percentage of cell death in the indicated HCC1569 cells treated with or without 10 μM H2O2 for 96 hr. ***p < 0.001 (Student’s t test).
(F) Live cell numbers relative to H2O2-untreated cells in EL-12-58 cells treated with H2O2 for 72 hr in the presence or absence of 10 μM AP-18. **p < 0.01 and ***p < 0.001 compared with vehicle control for AP-18 (AP-18 [–]) (Student’s t test).
(G) Percentage of cell death in EL-12-58 cells treated with or without 20 μM H2O2 for 72 hr in the presence or absence of 10 μM AP-18. ***p < 0.001 (Student’s t test).
(H–J) Live cell numbers relative to H2O2-untreated cells in the indicated cancer cells upon treatment with H2O2 for 72 hr. In (H and I), cells were treated with or without 10 μM AP-18. In (J), cells were transduced with an empty lentiviral vector or TRPA1. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with vehicle control for AP-18 (AP-18 [–]) in (H) and (I) and vector in (J) (Student’s t test).
(K) Live cell numbers relative to H2O2-untreated cells in the indicated HCC1569 cells treated with 8 μM H2O2 in the presence or absence of 10 μM BAPTA-AM for 96 hr. ***p < 0.001 (one-way ANOVA).
In (D–K), data are shown as means ± SD from the sum of three independent experiments performed in duplicate. See also Figure S2.
We next examined whether ectopic TRPA1 expression induces oxidative-stress defense in MCF-10A cells, a well-established immortalized mammary epithelial cell line used for the analysis of phenotypic changes induced by oncogene transduction (Debnath and Brugge, 2005). We used MCF-10A culture medium without EGF, as EGF receptor is largely downregulated in ECM-deprived MCF-10A cells (Reginato et al., 2003). Ectopic TRPA1 expression increased [Ca2+]i and live cell number and suppressed cell death and apoptosis in response to H2O2 (Figures S2J–S2M). BAPTA-AM suppressed viability of TRPA1-expressing cells treated with H2O2 (Figure S2N), indicating the importance of [Ca2+]i rises in cell survival. Together, these results indicate that TRPA1-mediated Ca2+ influx can induce oxidative-stress defense in cancer cells.
TRPA1 Induces Ca2+ Influx in Response to ROS Generated in the Inner Cells of Tumor Spheroids and Protects Them from Apoptotic Death
To examine the role of TRPA1 in a more functional context, we cultured TRPA1-enriched HCC1569 and H1792 cells with reconstituted basement membrane (Matrigel) to form 3D spheroids and examined the effect of TRPA1 on ROS, [Ca2+]i, and cell survival. Both HCC1569 and H1792 formed solid spheroids. Interestingly, analysis of the ROS probe Hyper-2 revealed that the intracellular ROS level was substantially higher in the inner cells of spheroids (Figures 3A and 3B), indicating that survival of the inner cells cannot be simply explained by a lack of oxidative stress. The higher ROS levels in the inner cells were unaffected by TRPA1 inhibition, but were abolished by NAC (Figures 3A, 3B, and S3A), suggesting that TRPA1 does not contribute to reduction of ROS. The observed increase in ROS in the inner cells cannot be attributed to cell death, as the majority of the cells were alive at the time point examined (day 5). Notably, the inner cells showed elevated [Ca2+]i that was suppressed by TRPA1 inhibition or NAC (Figures 3A, 3B, and S3A), indicating that TRPA1 induces Ca2+ influx in response to ROS generated in the inner spheroid cells.
Figure 3. TRPA1 Induces Ca2+ Influx in Response to ROS Generated in Inner Cells of Tumor Spheroids and Prevents Clearance of the Inner Cells.
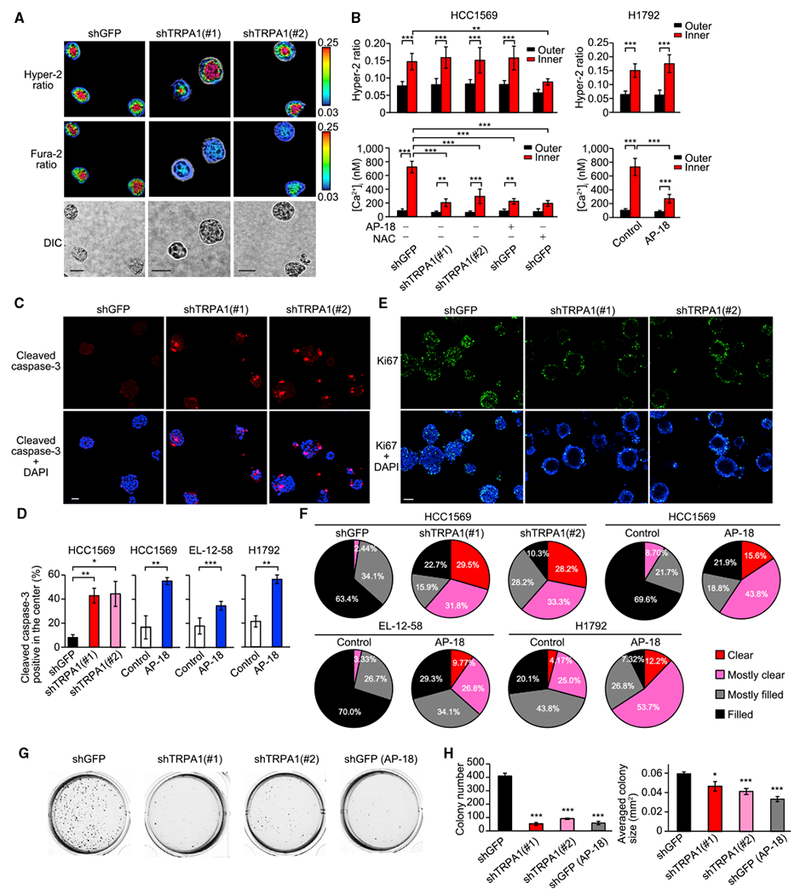
(A) Representative Hyper-2 and fura-2 ratio images of day-5 HCC1569 spheroids transduced with shTRPA1 or shGFP. Scale bar, 50 μm. For quantifying signal, a 10-μm width of the peripheral region was defined as the outer region.
(B) Averaged Hyper-2 ratio and [Ca2+]i in the inner and outer region of the indicated day-5 HCC1569 and H1792 spheroids treated with or without 10 μM AP-18 or 5 mM NAC (pH was adjusted to 7.4). Data are shown as means ± SD (n = 3–11 for HCC1569, n = 12 for H1792: sum of two independent experiments). **p < 0.01 and ***p < 0.001 (one-way ANOVA).
(C) Representative images of day-10 HCC1569 spheroids stained with cleaved caspase-3 and DAPI. Scale bar, 50 μm.
(D) Percentage of day-10 HCC1569, day-10 EL-12-58, or day-6 H1792 spheroids with cleaved caspase-3 staining in the center. The spheroids were treated with or without 10 μM AP-18. Data are shown as means ± SEM from three independent experiments. *p < 0.05, **p < 0.01, and ***p < 0.001 (Student’s t test).
(E) Representative images of day-15 HCC1569 spheroids stained with Ki67 and DAPI. Scale bar, 100 μm.
(F) Pie chart of clearance levels of day-15 HCC1569 spheroids transduced with shTRPA1 or shGFP (n = 39–44: sum of three independent experiments), day-15 HCC1569 spheroids treated with or without 10 μM AP-18 (n = 23 for control and n = 32 for AP-18: sum of two independent experiments), day-15 EL-12-58 spheroids treated with or without 10 μM AP-18 (n = 90 for control and n = 41 for AP-18: sum of three independent experiments), day-10 H1792 spheroids treated with or without 10 μM AP-18 (n = 48 for control and n = 41 for AP-18: sum of two independent experiments).
(G and H) The indicated HCC1569 cells were grown in soft agar with or without 10 μM AP-18 and after 20 days, images were taken after iodonitrotetrazolium chloride staining (G). Colony number and averaged colony size are also shown (H). Data represent means ± SEM of three independent experiments. *p < 0.05 and ***p < 0.001 compared with shGFP (Student’s t test). See also Figure S3.
TRPA1 inhibition increased apoptosis of cells localized in the inner region of spheroids (Figures 3C, 3D, and S3B). Furthermore, when cells were cultured with Matrigel for 10–15 days, TRPA1 inhibition induced clearance of cells from the inner space (Figures 3E, 3F, and S3C). Collectively, these results suggest that TRPA1-mediated Ca2+ influx is required for the survival of the ROShigh inner spheroid tumor cells.
TRPA1 Promotes Anchorage Independence
Since inner cells of tumor spheroids are deprived of the strong ECM signals from Matrigel, it is possible that improved survival by TRPA1 is at least in part due to its ability to promote anchorage independence through oxidative-stress defense mechanisms. Indeed, TRPA1 inhibition impaired the anchorage-independent growth of HCC1569 cells, reducing both the number and average size of colonies (Figures 3G and 3H). Furthermore, TRPA1 expression increased live cell number and suppressed anoikis in suspended MCF-10A cells (Figure S3D), which do not possess anchorage-independent ability. We next investigated ROS and [Ca2+]i levels following detachment. The H2O2-specific probe peroxy green-1 (Miller et al., 2007) showed that detachment induced a significant increase in intracellular H2O2 level that was unaffected by TRPA1 expression (Figure S3E). Consistently, detachment, as well as H2O2 treatment, induced expression of ROS-neutralizing genes, including NQO1 and GCLM, through NRF2 (Figures S3F and S3G). TRPA1 did not affect NRF2-mediated induction of NQO1 and GCLM mRNA. Methylglyoxal adducts, a major marker of dicarbonyl stress that weakly activates TRPA1 (Figures S3H and S3I) (Cao et al., 2012; Eberhardt et al., 2012; Koivisto et al., 2012), were not increased by detachment (Figures S3J and S3K). Notably, detachment induced a TRPA1-dependent increase in [Ca2+]i that was abolished by NAC (Figure S3E). Thus, these results suggest that TRPA1 mediates Ca2+ entry in response to ROS induction, and not dicarbonyl stress, during detachment.
In 3D culture, the wild-type but not the C199S Hyper-2 mutant, which is unable to react with ROS, showed increased signal in the inner cells of MCF-10A acini (Figures S3L–S3N), which was abolished by NAC but not by TRPA1 expression or AP-18. The ECM-deprived MCF-10A cells also exhibited marginally increased [Ca2+]i that was augmented by TRPA1 expression (Figures S3L and S3M), which was abolished by AP-18 or NAC. Moreover, TRPA1 expression suppressed apoptosis of the cells localized in the inner region of the MCF-10A acini (Figure S3O). Collectively, these results suggest that ROS produced as a result of ECM deprivation induce TRPA1-mediated Ca2+ influx that in turn suppresses anoikis without altering cellular ROS levels.
TRPA1-Mediated Ca2+ Influx Decreases Chemosensitivity
Oxidative-stress defense programs are known to mediate therapy resistance, as many anticancer treatments cause cellular redox perturbation that induces cell death (Galluzzi et al., 2012; Gorrini et al., 2013; Liu et al., 2016). In particular, platinum-based drugs strongly generate reactive chemicals, including ROS, that either directly trigger apoptosis or exacerbate drug-induced DNA damage (Galluzzi et al., 2012). Given that platinum-based drugs induce neuronal TRPA1 activation through electrophilic reactions (Nassini et al., 2011), we next investigated the role of TRPA1 in carboplatin resistance.
Carboplatin induced an increase in [Ca2+]i and also oscillatory Ca2+ responses that were suppressed by TRPA1 inhibition in TRPA1-enriched cancer cell lines (Figures 4A–4D and S4A–S4C). TRPM2 knockdown did not affect Ca2+ responses to carboplatin (Figure S4D). Notably, TRPA1 inhibition decreased live cell number and increased cell death in response to carboplatin in TRPA1-enriched cancer cells, but not in those with low TRPA1 expression (Figures 4E–4K, S4E, and S4F). BAPTA-AM suppressed cell viability in shGFP-transduced HCC1569 cells but not in shTRPA1-transduced cells upon treatment with carboplatin (Figure 4L), indicating the importance of [Ca2+]i rises in carboplatin resistance. Carboplatin-induced ROS generation (Galluzzi et al., 2012) was unaffected by TRPA1 knockdown (Figure S4G). TRPA1-mediated carboplatin resistance was eliminated by NAC (Figures 4F and 4G), suggesting that TRPA1 activation by carboplatin through electrophilic reaction of carboplatin (Galluzzi et al., 2012; Nassini et al., 2011) induces therapy resistance programs.
Figure 4. TRPA1 Increases [Ca2+]i and Cancer Cell Survival in Response to Carboplatin.
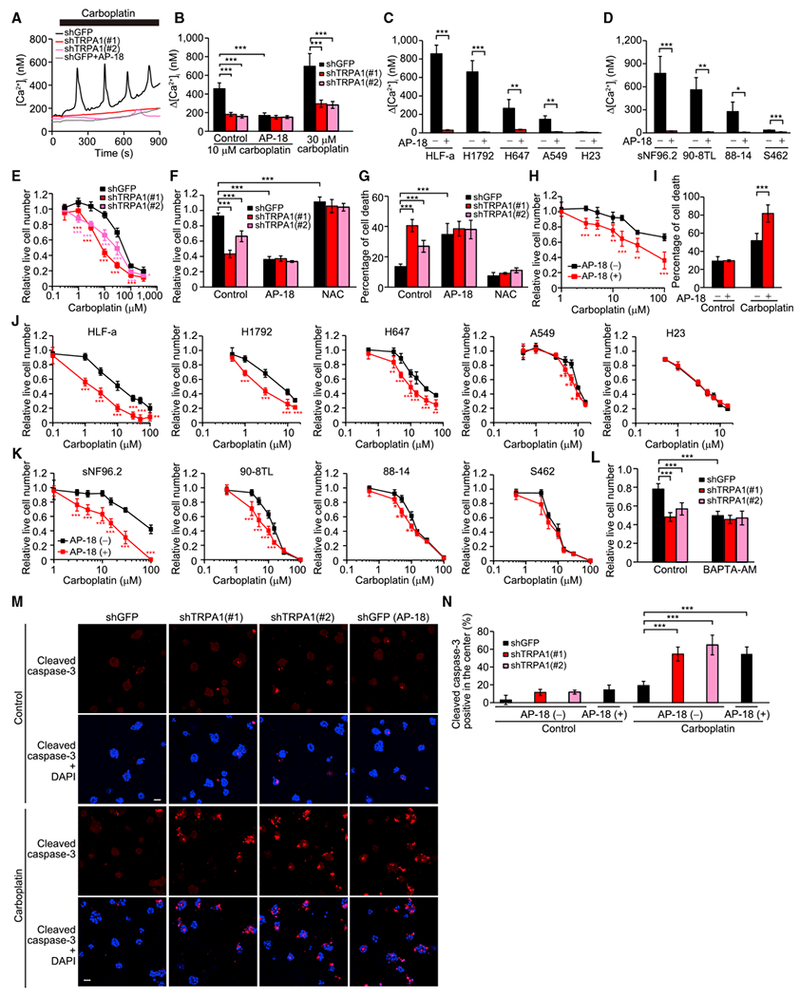
(A) Representative time courses of [Ca2+]i changes in the indicated HCC1569 cells upon treatment with 10 μM carboplatin in the presence or absence of 10 μM AP-18.
(B) Δ[Ca2+]i evoked by carboplatin in the indicated HCC1569 cells treated with or without 10 μM AP-18. Data are shown as means ± SEM (n = 22–61). ***p < 0.001 (one-way ANOVA).
(C and D) Δ[Ca2+]i evoked by 10 μM carboplatin in the indicated lung cancer (C) and MPNST cells (D) treated with or without 10 μM AP-18. Data are shown as means ± SEM (n = 12–129 for lung cancer, n = 30–72 for MPNST). *p < 0.05, **p < 0.01, and ***p < 0.001 (Student’s t test).
(E) Live cell numbers relative to carboplatin-untreated cells in the indicated HCC1569 cells treated with carboplatin for 72 hr. *p < 0.05 and ***p < 0.001 compared with shGFP (Student’s t test).
(F) Live cell numbers relative to carboplatin-untreated cells in the indicated HCC1569 cells treated with 10 μM carboplatin in the presence or absence of 10 μM AP-18 or 5 mM NAC (pH was adjusted to 7.4) for 72 hr. ***p < 0.001 (one-way ANOVA).
(G) Percentage of cell death in the indicated HCC1569 cells treated with 30 μM carboplatin in the presence or absence of 10 μM AP-18 or 5 mM NAC (pH was adjusted to 7.4) for 72 hr. ***p < 0.001 (one-way ANOVA).
(H) Live cell numbers relative to carboplatin-untreated cells in EL-12-58 cells treated with carboplatin in the presence or absence of 10 μM AP-18 for 72 hr. **p < 0.01 and ***p < 0.001 compared with vehicle control for AP-18 (AP-18 [–]) (Student’s t test).
(I) Percentage of cell death in EL-12-58 cells treated with or without 100 μM carboplatin for 72 hr in the presence or absence of 10 μM AP-18. ***p < 0.001 (Student’s t test).
(J and K) Live cell numbers relative to carboplatin-untreated cells in the indicated lung cancer (J) and MPNST cells (K) treated with carboplatin in the presence or absence of 10 μM AP-18 for 72 hr. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with vehicle control for AP-18 (AP-18 [–]) (Student’s t test).
(L) Live cell numbers relative to carboplatin-untreated cells in the indicated HCC1569 cells treated with 10 μM carboplatin in the presence or absence of 10 μM BAPTA-AM for 72 hr. ***p < 0.001 (one-way ANOVA).
(M) Representative images of day-7 HCC1569 spheroids treated with or without 10 μM AP-18, 10 μM carboplatin, or both for 48 hr. The spheroids were stained with cleaved caspase-3 and DAPI. Scale bar, 50 μm.
(N) Percentage of spheroids with cleaved caspase-3 staining in the center. Data are shown as means ± SD from three independent experiments. ***p < 0.001 (one-way ANOVA).
In (E–L), data are shown as means ± SD from the sum of two or three independent experiments performed in duplicate. See also Figure S4.
In 3D culture, TRPA1 inhibition strongly enhanced apoptosis of HCC1569 cells localized in the inner region of day 7 spheroids treated with carboplatin (Figures 4M and 4N). The more prominent effect of TRPA1 inhibition on cell death observed in 3D versus monolayer culture is probably due to the higher ROS levels in the inner region of spheroids than in monolayer culture upon carboplatin treatment.
TRPA1 inhibition also sensitized HCC1569 and H1792 cells to doxorubicin, gemcitabine, and paclitaxel, all of which can cause cellular redox perturbation (Arora et al., 2013; Gorrini et al., 2013; Liu et al., 2016), and this effect was eliminated by NAC (Figures S4H and S4I). Thus, these results suggest that TRPA1 allows cancer cells to adapt to standard-of-care therapies.
TRPA1 Inhibition Suppresses Tumor Growth and Enhances Chemosensitivity In Vivo
To investigate whether TRPA1 contributes to tumor growth and chemoresistance in vivo, HCC1569 cells, which are pronounced resistance to cisplatin (http://www.cancerrxgene.org), stably expressing TRPA1 shRNAs were transplanted into cleared mammary fat pads of immunocompromised mice and tumor growth was measured. TRPA1 knockdown not only suppressed tumor growth (Figure 5A, vehicle controls) but also sensitized tumors to carboplatin treatment, which was associated with increased apoptosis (Figures 5A–5C).
Figure 5. TRPA1 Inhibition Suppresses Tumor Growth and Enhances Chemosensitivity in Xenograft Models.
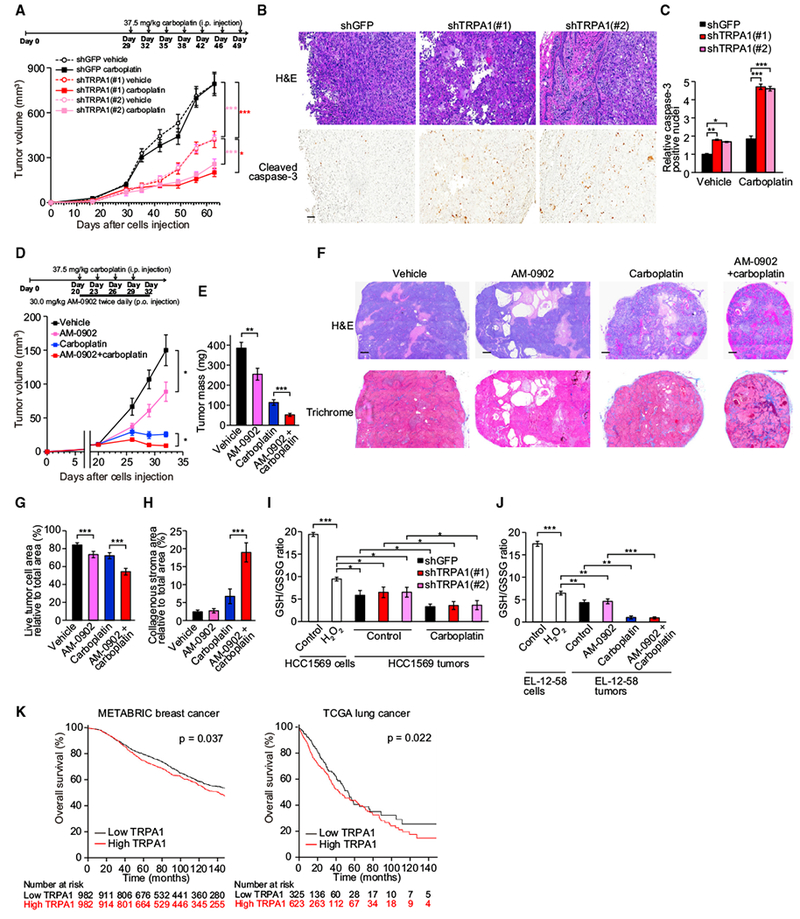
(A) shGFP- or shTRPA1-transduced HCC1569 cells were injected into cleared mammary fat pads of NOD-Rag1null Il2rgnull (NRG) mice and tumor growth was monitored over time (n = 7–9). Carboplatin was administrated as indicated. *p < 0.05 and ***p < 0.001 (two-way ANOVA).
(B) Representative field from H&E- and cleaved caspase-3-stained slides of tumors treated with carboplatin. Scale bar, 100 μm.
(C) Quantification of cleaved caspase-3-positive nuclei. Data normalized to shGFP-expressing tumors treated with vehicle and shown as mean ± SEM (n = 3–9). *p < 0.05, **p < 0.01, and ***p < 0.001 (one-way ANOVA).
(D) EL-12-58 cells were injected into cleared mammary fat pads of NRG mice and tumor growth was monitored over time (n = 10 for each arm). Drugs were administered as indicated. *p < 0.05 (two-way ANOVA).
(E) Tumor mass. **p < 0.01 and ***p < 0.001 (Student’s t test).
(F) Representative field from H&E- or trichrome-stained slides of the indicated EL-12-58 tumors. Scale bar, 500 μm.
(G and H) Quantification of live tumor cell area (G) or collagenous stroma area relative to total area (H) in the indicated EL-12-58 tumors. Data are shown as means ± SD (n = 5 for each arm). ***p < 0.001 (Student’s t test).
(I and J) GSH/GSSG ratio in the indicated HCC1569 cells or tumors (I) and the indicated EL-12-58 cells or tumors (J). Cells were cultured in monolayer in the presence or absence of 10 μM H2O2 for 24 hr. Data are shown as means ± SD of three independent experiments. *p < 0.05, **p < 0.01, and ***p < 0.001 (one-way ANOVA).
(K) Kaplan-Meier survival curves for overall survival of METABRIC breast or TCGA lung (LUSC and LUAD) cancers with low (below median) or high TRPA1 (above/including median). In TCGA lung cancers, TRPA1 expression was based on FKPM values.
In (B), (C), and (E–J), tumors were resected from mice in (A) or (D) at the endpoint. In (A), (D), and (E), data are shown as means ± SEM. See also Figure S5.
We next examined the effect of pharmacological TRPA1 inhibition on tumor growth in a PDX model. Although several TRPA1 inhibitors have been developed as pain and respiratory therapies, their pharmacokinetics are not optimal. We used AM-0902, which is an oral bioactive TRPA1 inhibitor and has the best pharmacokinetics reported to date but still exhibits a short plasma half-life (T1/2 = 2.8 hr) (Schenkel et al., 2016). Despite this limitation, oral administration of AM-0902 twice daily reduced both tumor volume and mass in EL-12-58 xenograft models without significant side toxicities (0.188% ± 2.42% weight loss at day 32) (Figures 5D and 5E). Moreover, combined administration of AM-0902 and carboplatin substantially reduced tumor growth compared with each single treatment. Although 3 of 10 animals treated with AM-0902 + carboplatin exhibited significant weight loss during the last 3 days (from day 29 to day 32), most likely due to diarrhea, animals treated with either AM-0902 + carboplatin or carboplatin alone showed comparable weight loss until day 29 and there was no correlation between weight loss and tumor growth at any time points (Figure S5A). H&E staining of tumor sections revealed that AM-0902 treatment reduced live tumor cell area relative to total area, compared with vehicle or single carboplatin treatment (Figures 5F and 5G). Furthermore, Masson trichrome staining showed that the population of collagenous stroma was enhanced by AM-0902 + carboplatin treatment (Figures 5F and 5H), probably due to a desmoplastic response to tumor cell death.
To address whether oxidative stress is induced in our tumor models, we evaluated redox status expressed as GSH/GSSG ratio. Both HCC1569 and EL-12-58 tumors exhibited reduced GSH/GSSG ratio, which was further decreased by carboplatin treatment, compared with HCC1569 and EL-12-58 cells treated with vehicle or 10 μM H2O2, which can activate TRPA1 (Figure 2A), in monolayer cultures (Figures 5I and 5J). This indicates that these tumor cells are exposed to high levels of oxidative stress. Notably, TRPA1 inhibition did not affect the GSH/GSSG ratio (Figures 5I and 5J) or the expression of NQO1 and GCLM (Figures S5B and S5C) in these tumors, indicating that TRPA1 does not contribute to reduction of ROS in tumor cells. The induction of oxidative stress was supported by the findings that significant levels of 4-hydroxynonenal (4-HNE) and 8-hydroxyguanosine (8-OHdG), which are commonly used as readouts for oxidative stress, were detected in a subset of tumor cells and were strongly enhanced by carboplatin treatment (Figures S5D and S5E). Methylglyoxal adducts were not accumulated in these tumors (Figures S5F and S5G).
We next investigated the correlation between TRPA1 expression and prognosis in human patients. In univariate analyses, high TRPA1 expression was associated with statistically significantly worse overall survival (Figure 5K). TRPA1 remained statistically significant in multivariate analyses including parameters known to be associated with breast cancer survival (hazard ratio [HR] 1.17, 95% confidence interval [CI] 1.02–1.34, p = 0.021) and lung cancer survival (HR 1.34, 95% CI 1.02–1.77, p = 0.039) (Table 1). Collectively, these results support the importance of TRPA1 in tumor progression and chemoresistance and highlight this channel as a potential target for therapeutic intervention.
Table 1.
Results of Multivariable Cox Proportional Hazards Model on Overall Survival
| 95% Confidence Interval |
||||
|---|---|---|---|---|
| Variables | Hazard Ratio | Lower | Upper | p |
| Breast Cancer (METABRIC) | ||||
| TRPA1 expression | ||||
| Low (below median) | reference | reference | reference | reference |
| High (above/including median) | 1.17 | 1.02 | 1.34 | 0.021 |
| Primary receptor status | ||||
| ER+/HER2− | reference | reference | reference | reference |
| HER2+ | 1.37 | 1.16 | 1.61 | 1.75 × 10−4 |
| Triple-negative | 1.07 | 0.89 | 1.29 | 0.497 |
| Primary stage at diagnosis | ||||
| Stage I | reference | reference | reference | reference |
| Stage II | 1.25 | 1.06 | 1.48 | 0.008 |
| Stage III | 2.82 | 2.12 | 3.76 | 1.22 × 10−12 |
| Stage IV | 6.43 | 3.17 | 13.04 | 2.48 × 10−7 |
| Grade at diagnosis | ||||
| Grade 1 | reference | reference | reference | reference |
| Grade 2 | 1.26 | 0.94 | 1.69 | 0.124 |
| Grade 3 | 1.61 | 1.20 | 2.15 | 1.45 × 10−3 |
| Lung Cancer (TCGA) | ||||
| TRPA1 expression | ||||
| Low (below median) | reference | reference | reference | reference |
| High (above/including median) | 1.34 | 1.02 | 1.77 | 0.039 |
| Gender | ||||
| Female | reference | reference | reference | reference |
| Male | 1.10 | 0.85 | 1.41 | 0.47 |
| Tobacco use | ||||
| Never smoker | reference | reference | reference | reference |
| Former smoker, quit >15 years | 0.77 | 0.47 | 1.27 | 0.31 |
| Former smoker, quit <15 years | 0.72 | 0.45 | 1.14 | 0.16 |
| Current smoker | 0.79 | 0.49 | 1.29 | 0.35 |
| Age at diagnosis | ||||
| Under 50 years | Ref | Ref | Ref | Ref |
| 50 to 59 years | 1.09 | 0.59 | 2.04 | 0.78 |
| 60 to 69 years | 1.07 | 0.59 | 1.92 | 0.83 |
| Over/including 70 years | 1.47 | 0.81 | 2.64 | 0.20 |
| Stage at diagnosis | ||||
| Stage I | reference | reference | reference | reference |
| Stage II | 1.59 | 1.20 | 2.10 | 1.14 × 10−3 |
| Stage III | 2.46 | 1.84 | 3.27 | 8.19 × 10−10 |
| Stage IV | 2.65 | 1.53 | 4.59 | 5.10 × 10−4 |
| Histology | ||||
| Adenocarcinoma | reference | reference | reference | reference |
| Squamous | 0.97 | 0.74 | 1.28 | 0.84 |
TRPA1 Mediates Oxidative-Stress Defense through Upregulation of Ca2+-Dependent Anti-apoptotic Signaling Pathways
To identify the relevant TRPA1-regulated signaling pathways that mediate oxidative-stress defense, we performed an unbiased analysis of a large panel (>300) of signaling proteins/pathways using reverse phase protein array (RPPA). We first examined TRPA1-expressing MCF-10A cells in order to identify pathways that TRPA1 is able to activate in a minimally altered immortalized cell line. In MCF-10A cells treated with H2O2 or deprived of matrix attachment, ectopic TRPA1 expression increased the phosphorylation of multiple pro-survival proteins, including components of the RAS-ERK (p-MAPK, p-B-RAF, p-p90RSK, p-MEK1, or p-ELK), PI3K/AKT (p-AKT, p-GSK-3α-β, or p-PRAS40), and mammalian target of rapamycin (mTOR) signaling pathways (p-S6, p-eIF4E, p-4E-BP1, or p-mTOR), as well as upstream components of these pathways (p-SRC, p-SHP-2, or p-SHC) (Figure 6A). Moreover, among the BCL-2 family of apoptosis regulators, the anti-apoptotic protein MCL-1 was the most highly upregulated by TRPA1 under these conditions (Figures 6B and S6A). These pro-survival/anti-apoptotic proteins were unaffected by TRPA1 in vehicle-treated or attached cells (Figures S6B–S6D). RPPA results were validated by immunoblot analysis (Figures 6C and S6E), and RAS activation was confirmed by RAS-GTP pull-down assay (Figures 6D and S6F).
Figure 6. TRPA1 Mediates Oxidative-Stress Defense through Upregulation of Ca2+-Dependent Anti-Apoptotic Signaling Pathways.
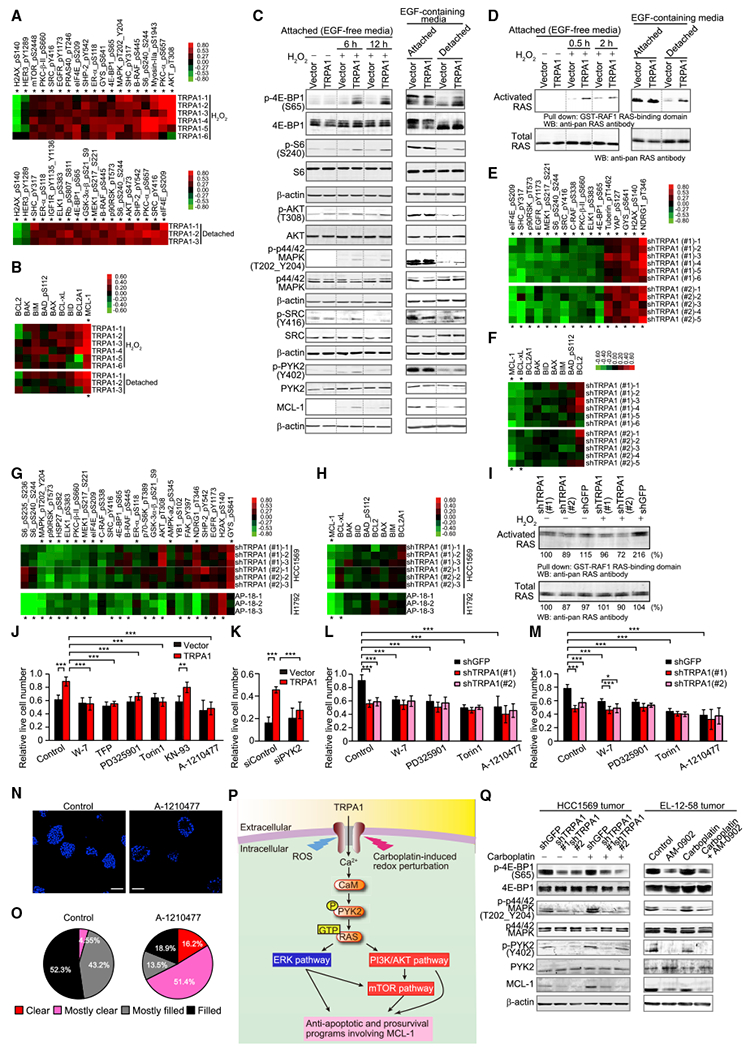
(A) Heatmap of significantly changed phosphorylated proteins (*p < 0.05) between TRPA1- and vector-transduced MCF-10A cells in response to H2O2 or matrix deprivation (detached). Lysates of cells treated with 100 μM H2O2 for 24 hr or cultured in suspension for 24 hr were analyzed by RPPA and all signals from all samples were normalized to vector control.
(B) Heatmap of BCL-2 family proteins in TRPA1-transduced MCF-10A cells treated as in (A). *p < 0.05 comparing TRPA1 and vector.
(C) Immunoblot of lysates from the indicated MCF-10A cells treated with 100 μM H2O2 for 6 hr or 12 hr or cultured in suspension for 24 hr. Dotted lines indicate the cropping lines, and uncropped blots shown in Figure S5E.
(D) Ras activity in the indicated MCF-10A cells treated with 100 μM H2O2 for 0.5 hr or 2 hr or cultured in suspension for 24 hr, as determined by Ras-GTP pull-down assay. Uncropped blots shown in Figure S5F.
(E) Heatmap of significantly changed phosphorylated proteins (*p < 0.05) between shTRPA1 (#1 and #2)- and shGFP-transduced HCC1569 cells. Cells were treated with 10 μM H2O2 for 24 hr. Samples normalized to shGFP.
(F) Heatmap of BCL-2 family proteins in shTRPA1-transduced HCC1569 cells treated as in (E).
(G) Heatmap of significantly changed phosphorylated proteins (*p < 0.05), either between shTRPA1 (#1 and #2)- and shGFP-transduced HCC1569 cells or between AP-18- and its vehicle-treated H1792 cells. Cells were treated with 10 μM carboplatin in the presence or absence of 10 μM AP-18 for 24 hr. Samples normalized to shGFP for HCC1569 cells and AP-18 (–) for H1792 cells.
(H) Heatmap of BCL-2 family proteins in HCC1569 and H1792 cells treated as in (G).
(I) Ras activity in the indicated HCC1569 cells treated with 10 μM H2O2 for 2 hr. Normalized densitometric values are shown below each band.
(J and K) Live cell numbers relative to H2O2-untreated cells in the indicated MCF-10A cells treated with 70 μM H2O2 (J) or 100 μM H2O2 (K) in the presence or absence of 1 μM W-7, 10 μM TFP, 1 nM PD325901, 100 nM Torin1, 10 μM KN-93, or 3 μM A-1210477 for 72 hr. **p < 0.01 and ***p < 0.001 (one-way ANOVA).
(L and M) Live cell numbers relative to H2O2-untreated (L) or carboplatin-untreated cells (M) in the indicated HCC1569 cells treated with 8 μM H2O2 (L) or 10 μM carboplatin (M) in the presence or absence of 1 μM W-7, 10 nM PD325901, 100 nM Torin1, or 3 μM A-1210477 for 96 hr (L) or 72 hr (M). Data for control in (M) are from Figure 4L, as these samples were analyzed concurrently. *p < 0.05 and ***p < 0.001 (one-way ANOVA).
(N) Representative images of day-13 HCC1569 (shGFP) spheroids treated with or without 3 μM A-1210477. Spheroids were stained with DAPI. Scale bar, 100 μm.
(O) Pie chart of clearance levels of Day-13 HCC1569 (shGFP) spheroids treated with or without 3 μM A-1210477 (n = 44 for control and n = 37 for A-1210477: sum of two independent experiments).
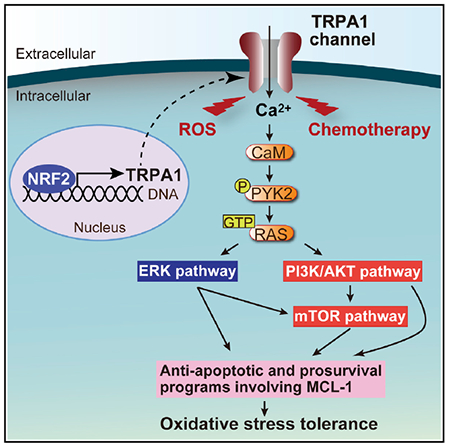
(P) Model of the relevant TRPA1-regulated signaling pathways that contribute to oxidative-stress defense in cancer cells.
(Q) Immunoblot of lysates from the indicated HCC1569 or EL-12-58 tumors that were resected from mice in Figure 5A for HCC1569 tumors or Figure 5D for EL-12-58 tumors at the end point.
In (J–M), data are shown as means ± SD from the sum of three independent experiments performed in duplicate. See also Figures S6 and S7.
Consistent with these observations, TRPA1 inhibition decreased the phosphorylation of RAS-ERK/AKT/mTOR signaling proteins and expression of MCL-1 in HCC1569 and H1792 cells (Figures 6E–6I, S6G, and S6H). These pro-survival/anti-apoptotic proteins were unaffected by TRPA1 inhibition in vehicle-treated cells (Figures S6I–S6M). Interestingly, phosphorylation of PI3K/AKT signaling proteins was unaffected by TRPA1 knockdown in HCC1569 cells treated with H2O2 or carboplatin (Figures 6E and 6G), raising the possibility that this pathway is already maximally activated due to PTEN deletion and HER2 overexpression present in these cells. Indeed, p-AKT and p-HER2 were strongly upregulated in HCC1569 cells compared with TRPA1-expressing MCF-10A cells, whereas phosphorylation levels of RAS-ERK and mTOR signaling proteins were comparable (Figure S6N). In support of these findings, the phosphorylation of RAS-ERK and mTOR signaling proteins and MCL-1 mRNA were increased in BRCA with high TRPA1 expression (Figures S7A and S7B).
TRPA1 also increased the phosphorylation of PYK2, which can be activated by [Ca2+]i rises through calmodulin (CaM) and activate the RAS-ERK/AKT/mTOR signaling pathways (Cullen and Lockyer, 2002) (Figures 6C and S6E). Phosphorylation of RAS-ERK/AKT/mTOR signaling proteins and expression of MCL-1 were suppressed by BAPTA-AM, W-7 (CaM inhibitor), AP-18, or PYK2 knockdown in TRPA1-expressing cells treated with H2O2 (Figure S7C), suggesting that TRPA1-mediated Ca2+ influx amplifies pro-survival/anti-apoptotic signaling pathways through Ca2+-CaM-mediated PYK2 activation in oxidative conditions.
W-7, trifluoperazine (TFP; another CaM inhibitor structurally distinct from W-7), PD325901 (MEK inhibitor), Torin1 (mTOR inhibitor), A-1210477 (MCL-1 inhibitor), or PYK2 knockdown, but not KN-93 (CaM kinase II inhibitor), suppressed the enhanced viability of TRPA1-expressing MCF-10A cells treated with H2O2 or HCC1569 cells treated with H2O2 or carboplatin (Figures 6J–6M). Notably, A-1210477 abolished TRPA1-mediated inhibition of apoptosis in MCF-10A or HCC1569 cells localized in the inner region of spheroids (Figures S7D–S7G) and increased clearance of cells from the inner space in shGFP-transduced control HCC1569 spheroids but not in shTRPA1-transduced spheroids (Figures 6N, 6O, S7H, and S7I). Moreover, overexpression of MCL-1 or TRPA1 rescued colony growth in TRPA1-knockdown cells (Figures S7J–S7M). These results, together with the known importance of RAS-ERK, AKT, mTOR signaling pathways in the regulation of MCL-1 protein (Yamaguchi et al., 2012), suggest that enhanced anti-apoptotic programs involving MCL-1 through the activation of Ca2+-CaM/PYK2-dependent survival pathways play an important role in TRPA1-induced oxidative-stress tolerance (Figure 6P).
In HCC1569 and EL-12-58 tumors treated with vehicle or carboplatin, TRPA1 knockdown or AM-0902 suppressed phosphorylation of PYK2, MAPK, and 4E-BP1 and expression of MCL-1 (Figure 6Q), suggesting that TRPA1 is activated and upregulates Ca2+-dependent anti-apoptotic programs in tumors.
All above results together strongly indicate that TRPA1 contributes to a non-canonical oxidative-defense program that is distinct from canonical ROS-neutralizing programs in cancer cells.
TRPA1 Expression in Tumor Cells Is Regulated by NRF2
We next asked whether cancer cells upregulate TRPA1 expression as an oxidative-stress defense program. In cancer cells with low basal TRPA1 expression, H2O2 increased TRPA1 expression substantially in serum-starved conditions (Figures S1K, S1L, and 7A) and marginally in complete media (Figure S8A). By contrast, H2O2 did not enhance TRPA1 expression in cancer cells with high basal TRPA1 expression (Figures S1K, S1L, and S8B). Interestingly, TRPA1 expression was statistically enriched in human breast and lung tumors that exhibit high activity of NRF2 (Figure 7B and Table S3), as determined by the expression level of well-characterized NRF2 target genes (DeNicola et al., 2015), suggesting not only that TRPA1-enriched tumor cells are exposed to oxidative stress but also that oxidative stress induces TRPA1 expression through NRF2 activation. Indeed, H2O2-induced TRPA1 expression was abolished by NRF2 knockdown (Figure 7C). Furthermore, H2O2 increased not only TRPA1 but also MCL-1 expression, which was suppressed by either TRPA1 inhibition or NRF2 knockdown (Figure 7D). Thus, these results suggest that oxidative stress could at least partly contribute to enhanced TRPA1 expression in human tumor cells through ROS-induced NRF2 activation.
Figure 7. TRPA1 Expression Is Induced by NRF2.
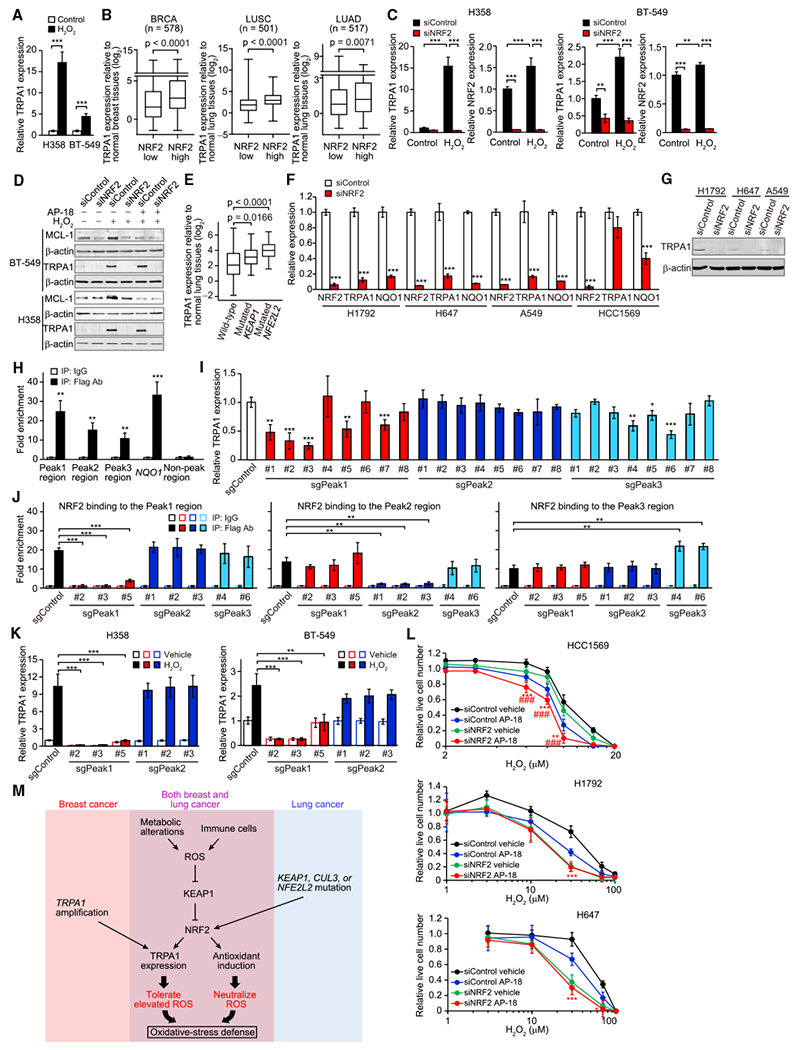
(A) mRNA expression of TRPA1 in H358 or BT-549 cells treated with or without 10 μM H2O2 for 48 hr in serum-starved conditions (0.1% fetal bovine serum). Data were normalized to vehicle control. ***p < 0.001 (Student’s t test).
(B) Boxplots displaying TRPA1 mRNA levels relative to corresponding normal tissues in low or high NRF2 tumors from TCGA BRCA, LUSC, or LUAD. Statistical significance determined by the Student’s t test.
(C) mRNA expression of TRPA1 in the indicated H358 or BT-549 cells upon treatment with or without 10 μM H2O2 for 48 hr in serum-starved conditions. Data were normalized to small interfering RNA control (siControl) cells treated with vehicle control. **p < 0.01 and ***p < 0.001 (one-way ANOVA).
(D) Immunoblot of lysates from the indicated BT-549 or H358 cells treated with or without 10 μM H2O2 in the presence or absence of 10 μM AP-18 for 48 hr in serum-starved conditions.
(E) Boxplots displaying TRPA1 mRNA levels relative to normal lung tissues in wild-type (n = 114) and KEAP1- (n = 22) or NFE2L2-mutated (n = 26) LUSC based on TCGA dataset. Statistical significance determined by the Student’s t test.
(F) mRNA expression of NRF2, TRPA1, and NQO1 in the indicated cancer cells. Data were normalized to siControl. ***p < 0.001 compared with siControl (Student’s t test).
(G) Immunoblot analysis of TRPA1 in the indicated cancer cells.
(H) ChIP with Flag antibody of the fragmented chromatin containing the Peak1, Peak2, Peak3, or non-Peak region (Chr8: 72086158–72086422 [GRCh38]) or the NQO1 promoter (Chr16: 69727000–69727111 [GRCh38]) in Flag-NRF2-overexpressing H1792 cells. Binding was normalized to that in immunoprecipitation with immunoglobulin (Ig)G control. **p < 0.01 and ***p < 0.001 compared with IgG (Student’s t test).
(I) mRNA expression of TRPA1 in the indicated H1792 cells. Data were normalized to single guide RNA control (sgControl). *p < 0.05, **p < 0.01, and ***p < 0.001 compared with sgControl (Student’s t test).
(J) ChIP with Flag antibody of the fragmented chromatin containing the Peak1, Peak2, or Peak3 region in the indicated Flag-NRF2-overexpressing H1792 cells. Binding was normalized to that in immunoprecipitation with IgG control. **p < 0.01 and ***p < 0.001 (Student’s t test).
(K) mRNA expression of TRPA1 in the indicated H358 or BT-549 cells upon treatment with or without 10 μM H2O2 for 48 hr in serum-starved conditions. Data were normalized to sgControl cells treated with vehicle control. **p < 0.01 and ***p < 0.001 (Student’s t test).
(L) Live cell numbers relative to H2O2-untreated cells in the indicated cancer cells upon treatment with H2O2 for 72 hr in the presence or absence of 10 μM AP-18. Data are shown as means ± SD from the sum of three independent experiments performed in duplicate. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with siControl cells treated with AP-18 (Student’s t test). ###p < 0.001 compared with siNRF2 cells treated with vehicle.
(M) Model of the induction of TRPA1 in breast and lung cancer.
In (A), (C), (F), and (H–K), data are shown as means ± SD of three independent experiments. In (B) and (E), box and whiskers graphs indicate the median and the 25th and 75th percentiles, with minimum and maximum values at the extremes of the whiskers. See also Figure S8 and Table S3.
Mutations or deletions in NFE2L2 (encoding NRF2), KEAP1, or CUL3 (both encoding negative regulators of NRF2) that lead to a constitutive activation of NRF2 (Suzuki et al., 2013), are frequently observed in LUSC (32.8%), LUAD (22.6%), and head-neck squamous carcinoma (HNSC, 14.3%), but rarely occur in BRCA (1.8%). Notably, in LUSC and HNSC, TRPA1 expression was highly upregulated in those harboring NFE2L2 or KEAP1 mutations (Figures 7E and S8C). Moreover, based on data from the COSMIC – Cell Lines Project (http://cancer.sanger.ac.uk/cell_lines), all TRPA1-enriched lung cancer cell lines (Figure 1C), except for HLF-a cells, whose genetic information is not available in the database, harbor KEAP1 mutations that cause constitutive activation of NRF2 (DeNicola et al., 2015). This evidence raises the possibility that constitutive activation of NRF2 leads to the induction of TRPA1 expression in lung cancer cells.
NRF2 knockdown strongly suppressed the expression of TRPA1 as well as NQO1 in lung cancer H1792, H647, and A549 cells but not in breast cancer HCC1569 cells harboring five copies of TRPA1 (Figures 7F, 7G, and S8D), indicating that constitutive NRF2 activation supports TRPA1 expression in these lung cancer cell lines. NRF2 knockdown did not suppress expression of other TRP channels, including TRPC3 and TRPV1 (Figures S8E and S8F). These results indicate that in addition to oxidative stress, genetic alterations also trigger TRPA1 expression through constitutive activation of NRF2 in lung tumors.
Based on the chromatin immunoprecipitation (ChIP)-coupled deep sequencing (ChIP-Seq) dataset (http://cistrome.org), there are three putative NRF2 binding sites, which overlap with the binding sites of H3K27Ac, H3K4me1, and H3K4me2, markers of promoter and enhancer regions (Heintzman et al., 2009; Wang et al., 2014), around the TRPA1 locus (Figure S8G). Indeed, ChIP of NRF2-associated DNA sequences showed NRF2 binding to these peak regions as well as the promoter region of NQO1 in H1792 cells (Figure 7H). We disrupted these regions by CRISPR-Cas9 technology and found that TRPA1 expression was suppressed substantially by sgPeak1-targeted constructs and marginally by sgPeak3-targeted constructs, but not by sgPeak2-targeted constructs (Figures 7I and S8H). Interestingly, NRF2 binding to the Peak1 and Peak2 was abolished by sgPeak1- and sgPeak2-targeted constructs, respectively; however, sgPeak3-targeted constructs did not reduce NRF2 binding to the Peak3 region (Figures 7J and S8I). Given that the Peak3 is located in intron 21 of TRPA1 (Figure S8G), the marginal suppression of TRPA1 expression by sgPeak3 targeting could be at least in part due to defects in splicing or other intron-mediated enhancement mechanisms, independent of NRF2 binding. Furthermore, H2O2-induced TRPA1 expression was abolished by sgPeak1-targeted constructs in BT-549 and H358 cells (Figure 7K). Collectively, these results suggest that NRF2 can directly induce TRPA1 expression through its binding to the Peak1 region.
Finally, to explore the relative contributions of TRPA1-dependent and TRPA1-independent programs in the adaptation to oxidative stress, we investigated the effect of inhibition of TRPA1, NRF2, or both on the survival of cells whose TRPA1 expression was not affected by H2O2 upon H2O2 treatment. Given that only a handful of early-stage NRF2 inhibitors have been developed to date (Singh et al., 2016) and that the selectivity of these compounds for NRF2 has not been evaluated, we performed NRF2 knockdown. In TRPA1-amplified HCC1569 cells, inhibition of either TRPA1 or NRF2 suppressed cell survival, and inhibition of both reduced cell survival more than inhibition of each one (Figure 7L), suggesting that TRPA1-dependent and TRPA1-independent NRF2-mediated programs separately contribute to oxidative-stress defense in this cell line. By contrast, in KEAP1-mutated lung cancer cells, NRF2 inhibition suppressed cell survival more than TRPA1 inhibition, and TRPA1 inhibition failed to affect cell survival in NRF2-knockdown cells (Figure 7L), suggesting that NRF2 induces both TRPA1-dependent and TRPA1-independent programs in these cell lines.
DISCUSSION
In this study, we reveal that TRPA1 is upregulated by NRF2 and promotes oxidative-stress tolerance in cancer cells. Our findings, together with the known importance of NRF2 in the induction of ROS-neutralizing gene expression, indicate that cancer cells mobilize a set of adaptive mechanisms, involving TRPA1-mediated non-canonical oxidative-stress defense as well as canonical ROS-neutralizing mechanisms, to survive harsh oxidative challenges (Figure 7M).
In glandular cancers, such as breast cancer, tumor cells are able to survive and proliferate in the lumen of hollow glandular structures, whereas normal breast epithelial cells are cleared from the lumen by apoptosis and oxidative stress (Debnath and Brugge, 2005; Mailleux et al., 2007; Schafer et al., 2009). This suggests that overcoming both apoptotic and oxidative stress is required for tumorigenesis. Our results show that even in cancer spheroids, the inner cells exhibit enhanced ROS levels. This could be in part due to altered glucose and glutamine metabolisms and mitochondrial dysfunction caused by suppressed signaling to PI3K/AKT (Jiang et al., 2016; Schafer et al., 2009). The increased ROS level in inner spheroid cells, together with our finding that inhibition of TRPA1 or MCL-1 induces clearance of cells from the inner space, suggests not only that TRPA1 and MCL-1 are essential for the survival of these cells but also that there are detrimental levels of oxidative stress on these inner cells. Indeed, increase in oxidative damage products, such as 4-HNE and 8-OHdG, correlates with the aggressiveness of tumors and poor prognosis (Patel et al., 2007; Soini et al., 2011). Thus, these observations strongly suggest that survival of the inner spheroid tumor cells cannot be simply explained by a lack of oxidative stress and that oxidative-stress tolerance programs are required for the survival of these inner cells.
MCL-1 possesses many phosphorylation sites, and differential phosphorylation of MCL-1 by ERK and GSK-3β, which is inactivated by AKT, results in different fates (Akgul, 2009). Given this, together with the fact that AKT and ERK induce MCL-1 transcription (Yamaguchi et al., 2012), TRPA1-mediated activation of RAS-ERK and AKT may upregulate MCL-1 protein expression. Whereas our results indicate a critical role of MCL-1 in TRPA1-induced oxidative-stress tolerance, we cannot exclude the possibility that other factors also contribute to TRPA1-mediated effects, because TRPA1 upregulated RAS-ERK/AKT/mTOR signaling pathways, which can affect a broad range of cellular processes.
Our results reveal that in addition to ROS-neutralizing programs, NRF2 promotes an oxidative-stress tolerance program through TRPA1. Recent studies have also shown additional roles of NRF2, such as the promotion of EGFR signaling pathways (Chio et al., 2016) and the regulation of metabolic programs (DeNicola et al., 2015; Mitsuishi et al., 2012). Interestingly, mutations or deletions in NFE2L2, KEAP1, or CUL3 are frequently observed in LUSC, LUAD, HNSC, and esophageal carcinoma, all of which are directly in contact with external environment. Given that these tissues can be exposed to strong oxidative stress due to cigarette smoke or higher partial pressure of O2 compared with other tissues, it is possible that the cells possessing genetic alterations of these genes are selected to constitutively upregulate TRPA1 and ROS-neutralizing factors in these tumors. Indeed, cigarette smoke extract induces TRPA1 expression in lung cancer A549 cells and tracheal epithelial cells (Nie et al., 2016; Zhong et al., 2015). Furthermore, our analysis based on TCGA shows that TRPA1 expression weakly but significantly correlated with cigarette smoking history pack-year values in LUAD (r = 0.029, p = 0.0015) and HNSC patients (r = 0.14, p = 0.020). TRPA1 expression is also induced by tumor necrosis factor α and hypoxia-inducible factor-1α in human synoviocytes and odontoblasts (Hatano et al., 2012; El Karim et al., 2016), suggesting that TRPA1 expression in tumor cells is mediated by tumor microenvironmental factors in addition to genetic alterations. This may, in part, explain the reason why the number of TRPA1-enriched cancer cell lines is limited in contrast to marked TRPA1 upregulation in human tumors.
TRPA1 functions as a major stress sensor in nociceptive neurons (Shimizu et al., 2014; Takahashi et al., 2011). From this perspective, it is possible that TRPA1 activation by other types of stress, such as hypoxia, acid, and inflammatory mediators, also contribute to tumor development and progression. Interestingly, TRPA1 was shown to induce NRF2 activation to detoxify dicarbonyls under dicarbonyl stress in Caenorhabditis elegans (Chaudhuri et al., 2016). Based on our data, dicarbonyl stress is not induced in matrix-detached cells and in our tumor models, and TRPA1 does not affect NRF2 activities in cancer cells. However, given that dicarbonyls, by-products of glycolysis, are increased in several cancer types (Chiavarina et al., 2017), dicarbonyl stress can be a potential source of TRPA1 activation in tumor cells. In mammals, TRPA1 activation by dicarbonyls is associated with metabolic neuropathy and insulin release in sensory neurons and pancreatic β cells, respectively (Cao et al., 2012; Eberhardt et al., 2012; Koivisto et al., 2012). Given that cellular responses induced by Ca2+ influx are context specific and dependent on both the amplitude and pattern of Ca2+ responses and the presence of certain regulatory signals and/or partner proteins (Clapham, 2007; Cullen and Lockyer, 2002), a better understanding of the effect of other TRPA1 agonists in tumor cells is likely to provide an overall picture of TRPA1-mediated phenotypes in cancer.
Recently, TRPA1 interaction with fibroblast growth factor receptor 2 (FGFR2) was shown to enhance FGFR2 signaling pathways and regulate metastasis in LUAD (Berrout et al., 2017). However, our analysis based on TCGA and CCLE indicates that TRPA1 expression exhibits a strong negative correlation with FGFR2 expression in lung (r = −0.20, p = 0.0067) and other cancer cell lines (r = −0.14, p < 0.0001) as well as LUAD (r = −0.227, p < 0.0001). Importantly, all TRPA1-enriched lung cancer cell lines used in this study exhibit undetectable FGFR2 expression. These lines of evidence indicate that the TRPA1-FGFR2 interaction is a very rare event in human cancer and that the TRPA1 function described here is more relevant to the biology of cancer.
There is now compelling evidence that oxidative-stress defense programs represent a vulnerability of tumor cells. Therefore, the development of therapies targeting oxidative-stress defense represents an attractive strategy to enhance the sensitivity of tumor cells to existing therapies. However, it is not straightforward to target canonical antioxidant programs due to the side effects resulting from increased oxidative stress in normal tissues. In contrast, TRPA1 inhibitors are currently under clinical evaluation as pain and respiratory therapies and have not shown any evidence of central nervous system or other drug-related side effects to date. Thus, although improvement of TRPA1 drug pharmacokinetics and optimization of administration protocols are necessary, our results highlight the potential clinical utility of targeting oxidative-stress defense in diverse cancer types.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal antibody anti-TRPA1 | Novus | Cat# NB110-40763; RRID: AB_715124 |
| Rabbit polyclonal antibody anti-TRPA1 | Sigma-Aldrich | Cat# HPA026630; RRID: AB_1858360 |
| Rabbit polyclonal antibody anti-Cleaved Caspase-3 (Asp175) | Cell Signaling Technology | Cat# 9661; RRID: AB_2341188 |
| Mouse monoclonal antibody anti-Ki-67 | Dako | Cat# M7240; RRID: AB_2142367 |
| Mouse monoclonal anti-Flag M2 antibody | Sigma-Aldrich | Cat# F1804; RRID: AB_259529 |
| Mouse monoclonal antibody anti-beta-Actin | Sigma-Aldrich | Cat# A1978; RRID: AB_476692 |
| Rabbit polyclonal antibody anti-Phospho-4E-BP1 (Ser65) | Cell Signaling Technology | Cat# 9451; RRID: AB_330947 |
| Rabbit monoclonal antibody anti-4E-BP1 | Cell Signaling Technology | Cat# 9644S; RRID: AB_10691384 |
| Mouse monoclonal antibody anti-Phospho-S6 Ribosomal Protein (Ser240) | Dako | Cat# M7300; RRID: AB_2182542 |
| Rabbit monoclonal antibody anti-S6 Ribosomal Protein | Cell Signaling Technology | Cat# 2217; RRID: AB_331355 |
| Rabbit monoclonal antibody anti-Phospho-AKT (Thr308) | Cell Signaling Technology | Cat# 4056; RRID: AB_331163 |
| Mouse monoclonal antibody anti-AKT | Cell Signaling Technology | Cat# 2920; RRID: AB_1147620 |
| Rabbit monoclonal antibody anti-Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) | Cell Signaling Technology | Cat# 4370; RRID: AB_2315112 |
| Rabbit polyclonal antibody anti-p44/42 MAPK (Erk1/2) | Cell Signaling Technology | Cat# 9102; RRID: AB_330744 |
| Rabbit polyclonal antibody anti-Phospho-PYK2 (Tyr402) | Cell Signaling Technology | Cat# 3291S; RRID: AB_2300530 |
| Mouse monoclonal antibody anti-PYK2 | Cell Signaling Technology | Cat# 3480S; RRID: AB_2174093 |
| Rabbit monoclonal antibody anti-MCL-1 | Cell Signaling Technology | Cat# 5453S; RRID: AB_10694494 |
| Rabbit polyclonal antibody anti-Phospho-SRC (Tyr416) | Cell Signaling Technology | Cat# 2101; RRID: AB_331697 |
| Rabbit polyclonal antibody anti-SRC | Cell Signaling Technology | Cat# 2108; RRID: AB_331137 |
| Mouse monoclonal antibody anti-Methylglyoxal | Cell Biolabs, Inc | Cat# STA-011 |
| Rabbit polyclonal antibody anti-4 Hydroxynonenal | Abcam | Cat# ab46545; RRID: AB_722490 |
| Mouse monoclonal antibody anti-8 Hydroxyguanosine | Abcam | Cat# ab48508; RRID: AB_867461 |
| Biological Samples | ||
| Unstained breast cancer tissue arrays | US Biomax | BR1504a |
| Unstained normal breast tissue arrays | US Biomax | BN08111 |
| Unstained lung cancer tissue arrays | US Biomax | BC041115d |
| Unstained normal lung tissue arrays | US Biomax | LCN241 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| AP-18 | Sigma-Aldrich | Cat# A7232, CAS: 55224-94-7 |
| AM-0902 | Schenkel et al., 2016 | N/A |
| Fura-2 | Thermo Fisher Scientific | Cat# F1201 |
| N-Acetyl-L-cysteine | Sigma-Aldrich | Cat# A7250, CAS: 616-91-1 |
| Hydrogen peroxide solution | Sigma-Aldrich | Cat# H1009, CAS: 7722-84-1 |
| Allyl isothiocyanate (Mustard oil) | Sigma-Aldrich | Cat# W203408, CAS: 57-06-7 |
| BAPTA, AM, cell permeant chelator | Thermo Fisher Scientific | Cat# B1205 |
| Peroxy green 1 | Miller et al., 2007 | N/A |
| Carboplatin | Sigma-Aldrich | Cat# C2538, CAS: 41575-94-4 |
| Gemcitabine hydrochloride | Sigma-Aldrich | Cat# G6423, CAS: 122111-03-9 |
| Doxorubicin hydrochloride | Sigma-Aldrich | Cat# 44583, CAS: 25316-40-9 |
| Paclitaxel | Sigma-Aldrich | Cat# T7402, CAS: 33069-62-4 |
| CellEvent™ Caspase-3/7 Green Detection Reagent | Thermo Fisher Scientific | Cat# C10423 |
| N-(6-Aminohexyl)-5-chloro-1-naphthalenesulfonamide hydrochloride (W-7) | Sigma-Aldrich | Cat# A3281 CAS: 61714-27-0 |
| Trifluoperazine dihydrochloride (TFP) | Sigma-Aldrich | Cat# T8516 CAS: 440-17-5 |
| PD325901 | Selleckchem | Cat# S1036 |
| Torin1 | Tocris | Cat# 4247 |
| KN-93 | Santa Cruz Biotechnology | Cat# sc-202199 CAS: 139298-40-1 |
| A-1210477 | Selleckchem | Cat# S7790 |
| Critical Commercial Assays | ||
| Active Ras Pull-Down and Detection Kit | Thermo Fisher Scientific | Cat# 16117 |
| Mem-PER™ Plus Membrane Protein Extraction Kit | Thermo Fisher Scientific | Cat# 89842 |
| Fluo-4 NW Calcium Assay Kit | Thermo Fisher Scientific | Cat# F36206 |
| Trichrome Stain | Abcam | Cat# ab150686 |
| GSH/GSSG-Glo™ Assay | PROMEGA | Cat# V6611 |
| Deposited Data | ||
| RNA-seq, gene alteration, paired clinical feature, and RPPA data for tumors and corresponding normal tissues | TCGA | https://gdac.broadinstitute.org or http://www.cbioportal.org |
| Normalized gene expression data and paired clinical feature data including survival | METABRIC | European Genome-phenome Archive: EGAD00010000210, EGAD0001000021 |
| Expression dataset for MPNST | Miller et al., 2009 | GEO: GSE14038 |
| RNA-seq data for HCI PDX models | This paper | GEO: GSE113476 |
| Experimental Models: Cell Lines | ||
| MCF-10A | Laboratory of Dennis Slamon | N/A |
| HCC1569 | Laboratory of Dennis Slamon | N/A |
| HCC38 | Laboratory of Dennis Slamon | N/A |
| HCC202 | Laboratory of Dennis Slamon | N/A |
| HCC70 | Laboratory of Dennis Slamon | N/A |
| HCC1500 | Laboratory of Dennis Slamon | N/A |
| MDAMB-175 | Laboratory of Dennis Slamon | N/A |
| T47D | Laboratory of Dennis Slamon | N/A |
| BT-549 | Laboratory of Dennis Slamon | N/A |
| H1792 | ATCC | Cat# CRL-5895; RRID: CVCL_1495 |
| H647 | ATCC | Cat# CRL-5834; RRID: CVCL_1574 |
| A549 | ATCC | Cat# CCL-185; RRID: CVCL_0023 |
| H358 | ATCC | Cat# CRL-5807; RRID: CVCL_1559 |
| H23 | ATCC | Cat# CRL-5800; RRID: CVCL_1547 |
| sNF96.2 | ATCC | Cat# CRL-2884; RRID: CVCL_K281 |
| 90-8TL | Laboratory of Eric Legius | N/A |
| 88-14 | Laboratory of Jonathan Fletcher | N/A |
| S462 | Laboratory of Eric Legius | N/A |
| EL-12-58 | Johnson et al., 2016 | N/A |
| HCI-002 | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: NOD-Rag1null Il22rgnull (NRG) | Jackson Laboratory | Stock#: 007799 |
| Oligonucleotides | ||
| Primers for the construction of TRPA1 mutants: listed in Table S2 | This paper | N/A |
| Primers for the construction of LentiCRISPR plasmids: listed in Table S4 | This paper | N/A |
| Primers for quantitative PCR: listed in Table S5 | This paper | N/A |
| Recombinant DNA | ||
| pENTR223.1-TRPA1 | GE Healthcare | Cat# OHS5893-202502591 |
| pC1-HyPer-2 | Addgene | Cat# 42211 |
| pC1-HyPer-C199S | Addgene | Cat# 42213 |
| pLX304 | Addgene | Cat# 25890 |
| pCDH (Puromycin resistance) | System Bioscience | Cat# CD510B-1 |
| pCDH (Hygromycin B resistance) | System Bioscience | Cat# CD515B-1 |
| pCI-neo-TRPA1 | Takahashi et al., 2011 | N/A |
| pCI-neo-TRPA1 mutant | This paper | N/A |
| pCDNA3.1-Flag-NRF2 | Addgene | Cat# 36971 |
| LentiCRISPR plasmids | This paper | N/A |
| pLX304-TRPA1 | This paper | N/A |
| pLX304-MCL-1 | GE Healthcare | Cat# OHS6085-213573654 |
| pCDH-Hyper-2 (Puro and Hygromycin B) | This paper | N/A |
| pCDH-HyPer-C199S (Puro and Hygromycin B) | This paper | N/A |
| shRNA: TRPA1 #1 | The RNAi Consortium | TRCN0000044801 |
| shRNA: TRPA1 #2 | The RNAi Consortium | TRCN0000434290 |
| shRNA: GFP | Addgene | 30323 |
| Inducible shRNA: NFE2L2 #1 | GE Healthcare | V3THS_306092 |
| Inducible shRNA: NFE2L2 #2 | GE Healthcare | V3THS_306096 |
| Inducible shRNA: non-silencing control | GE Healthcare | RHS4743 |
| SMARTpool siRNA for PYK2 | GE Healthcare | L-003165-00 |
| SMARTpool siRNA for NFE2L2 | GE Healthcare | L-003755-00 |
| SMARTpool siRNA for TRPM2 | GE Healthcare | L-004193-00 |
| siGENOME Non-Targeting siRNA Pool #2 | GE Healthcare | D-001206-14 |
| Software and Algorithms | ||
| GraphPad Prism 7.0 | GraphPad Prism Software | N/A |
| Cluster 3.0 | N/A | http://bonsai.hgc.jp/~mdehoon/software/cluster/software.htm |
| Java TreeView | N/A | http://jtreeview.sourceforge.net/ |
| Metafluor | Molecular Devices | N/A |
| Other | ||
| RPPA assay | RPPA Core Facility - MD Anderson Cancer Center | N/A |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to and will be fulfilled by the corresponding author Joan S. Brugge (joan_brugge@hms.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals and In Vivo Procedures
All animal studies were performed according to protocols approved by the Institutional Animal Care and Use Committee, the Standing Committee on Animals at Harvard University. One million HCC1569 cells stably expressing either TRPA1 or GFP (control) shRNAs or two million EL-12-58 cells were injected into cleared mammary fatpads of 8-week-old female NOD-Rag1null Il2rgnull (NRG) mice. Tumor volume (mm3) was monitored by caliper measurements in two perpendicular dimensions (the formula: (d2 × D) / 2, where d and D represent the shortest and longest diameters, respectively) at the indicated time points. After tumors reached 3 to 5 mm in diameter, animals were randomized into the following treatment arms: (i) 37.5 mg/kg carboplatin or (ii) its vehicle (H2O) administrated via intraperitoneal injection for HCC1569 cell-transplanted animals; (i) oral administration of 30.0 mg/kg AM-0902, (ii) intraperitoneal administration of 37.5 mg/kg carboplatin, (iii) oral administration of 30.0 mg/kg AM-0902 plus intraperitoneal administration of 37.5 mg/kg carboplatin, or (iv) oral administration of 1% Tween 80/2% HPMC/97% water/methanesulfonic acid pH 2.2 (vehicle for AM-0902) plus intraperitoneal administration of H2O (vehicle for carboplatin) for EL-12-58 cell-transplanted animals. Power calculations are based on the standard deviation and effect size from pilot experiments in HCC1569 cells. The sample size of seven mice per group provides greater than 95% power to detect a ~80% difference in change in tumor volume between control and shTRPA1-treated cells with a two-sided type I error rate of 5%. Averaged tumor volume represents mean ± SEM.
Human Samples
Unstained breast or lung cancer and normal breast or lung tissue arrays were purchased from US Biomax (BR1504a for breast tumors, BN08111 for normal breast tissues, BC041115d for lung tumors, LCN241 for normal lung tissues), which collects all tissues under HIPPA approved protocols and the highest ethical standards with the donor being informed completely and with their consent (https://www.biomax.us/). Patient consent for and generation of the breast cancer EL-12-58-PDX model was obtained under protocols approved by the Dana-Farber/Harvard Cancer Center Institutional Review Board and HCI PDX models under protocols approved by the University of Utah Institutional Review Board, as previously described (DeRose et al., 2011; Johnson et al., 2016).
Development of Patient Derived HCI-002 Cell Line
Patient-derived xenograft HCI-002 was expanded in mammary fat pads of NOD-SCID mice to 1cm diameter (DeRose et al., 2011). Tumors were mechanically dissociated then digested with collagenase/hyaluoronidase (Stem Cell Technologies) followed by TrypLE Express (Thermo Fisher Scientific) with DNase (Roche Diagnostics). The resultant organoid suspension was plated in conditioned media from irradiated J2 3T3 fibroblasts with Rho kinase inhibitor. Contaminating fibroblasts were minimized by a combined approach of differential trypsinization and differential adhesion over passages 1–3 with a highly pure human epithelial population (>95% EPCAM-positive by FACS). The cell culture remained phenotypically stable over multiple passages and recapitulated the histologic appearance of the parent patient-derived xenograft upon orthotopic implantation into mammary fat pads of immunocompromised mice. Early-passage cultures (passage 4–8) were used for all experiments.
Cell Culture
H1792, H647, A549, H23, H358, and sNF96.2 were obtained from, and authenticated by, ATCC. MCF-10A, HCC1569, HCC38, HCC202, HCC70, HCC1500, MDAMB-175, T47D, and BT-549 were obtained from, and authenticated by, Dennis Slamon’s laboratory. 90-8TL and S462 were generated in laboratory of Eric Legius. 88-14 was generated in laboratory of Jonathan Fletcher. PDX EL-12-58 was derived from a liver metastasis of a heavily pretreated basal-like triple-negative breast cancer in our laboratory (Johnson et al., 2016). The HCI-002 cell line was established as part of this study from the corresponding PDX model described previously (DeRose et al., 2011). Cells were negative for mycoplasma contamination. HCC1569, HCC38, HCC202, HCC70, HCC1500, MDAMB-175, T47D, BT-549, H1792, H647, A549, H358, and H23 cells were cultured in RPMI (Life Technologies) + 10% fetal bovine serum (FBS) (Life Technologies) with penicillin and streptomycin (P/S) (Life Technologies). HLF-a cells were cultured in Eagle’s Minimum Essential Medium (ATCC) + 10% FBS with P/S. sNF96.2, 90-8TL, 88-14, and S462 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Life Technology) + 10% FBS with P/S, 1 mM sodium pyruvate (Thermo Fisher Scientific), and 2 mM L-glutamine (Thermo Fisher Scientific). EL-12-58 and HCI-002 cells were cultured in F-12 (Lonza)/DMEM (3:1 mixture) + 5% FBS with P/S, hydrocortisone (0.4 μg/mL, Sigma), cholera toxin (8.4 ng/mL, Sigma), insulin (5 μg/mL, Sigma), EGF (10 ng/mL, PeproTech), and Y-27632 (5 μM, ENZO life sciences). MCF-10A cells were maintained as described (http://brugge.hms.harvard.edu). All cells were routinely cultured in 37 °C with 5% CO2. For 3D culture, HCC1569, EL-12-58, H1792 cells were plated on Matrigel-coated glass bottom 24-well plates in the regular growth media supplemented with 2% Growth Factor Reduced Matrigel (BD Biosciences). MCF-10A cells were plated on Matrigel-coated glass bottom 24-well plates in DMEM/F12 (Life Technologies), 2% horse serum (Life Technologies), EGF (5 ng/mL), hydrocortisone (0.5 μg/mL), cholera toxin (100 ng/mL), insulin (10 μg/mL), and P/S supplemented with 2% Growth Factor Reduced Matrigel (BD Biosciences). The cells were re-fed with fresh media every three days. For suspension culture, MCF-10A cells were plated on poly HEMA (poly-2-hydroxyethyl methacrylate, Sigma)-coated plates in the regular MCF-10A growth media. In 3D and suspension culture, cells were maintained in 37°C with 5% CO2 and 6% O2 (physiological partial pressure of O2).
METHOD DETAILS
Plasmids, siRNAs, shRNAs, CRISPR/Cas9, and Virus Production
TRPA1 shRNAs were acquired from the RNAi Consortium, and have 100% sequence homology for TRPA1 (shRNA #1 is TRCN0000044801 and shRNA #2 is TRCN0000434290). shRNA against GFP was purchased from Addgene (plasmid #: 30323). Inducible NRF2 shRNAs were obtained from GE Healthcare (Clone ID: V3THS_306092 and V3THS_306096). Inducible lentiviral non-silencing shRNA control was obtained from GE Healthcare (RHS4743). TRPA1 mutants were constructed using pCI-neo-TRPA1 (Takahashi et al., 2011) according to standard Quickchange protocol. Primers used for the construction are shown in Table S2. The nucleotide sequences of the mutants were verified by sequencing the corresponding cDNA. The pC1-HyPer-2 (Cat#: 42211) and pC1-HyPer-C199S (Cat#: 42213) plasmids were obtained by Addgene. The lentiviral constructs of pCDH (Puro or Hygromycin B)-HyPer-2 and pCDH (Puro or Hygromycin B)-Hyper-C199S were constructed by transferring the NheI and BamHI digested fragments of HyPer-2 and HyPer-C199S from the pC1 constructs into the NheI and BamHI digested pCDH (Puro or Hygromycin B) vector. pENTR223.1-TRPA1 was purchased from GE Healthcare and cloned into a Gateway compatible pLX304 vector. The lentiviral construct of MCL-1 was obtained by GE Healthcare. LentiCRISPR plasmids were constructed using lentiCRISPRv2 containing two expression cassettes, hSpCas9 and the chimeric guide RNA, according to the standard protocol (Sanjana et al., 2014). Design of gRNAs and off-target sites prediction were performed by using the Zhang laboratory CRISPR design tool (http://crispr.mit.edu). All constructs were sequence verified and primer sequence information is shown in Table S4. LentiCRISPRv2-sgControl (the target sequence: 5’-GAGGCTAAGCGTCGCAA-3’) was obtained from Dr. Vidyasagar Koduri (Harvard Medical School). Lentiviruses for shRNAs, CRISPR/Cas9, pLX304-TRPA1, pLX304-MCL-1, pCDH-HyPer2, and pCDH-HyPer2 (C199S) were made in 293T cells according to standard protocol and selected with Blasticidin, Puromycin, or Hygromycin B for at least 1 week. pCI-neo-TRPA1 construct plus pEGFP-F (Clontech) as a transfection marker were transfected using SuperFect Transfection Reagent (Qiagen). pCDNA3.1-Flag-NRF2 was purchased from Addgene (Cat#: 36971) and transfected using polyethylenimine. SMARTpool siRNA for Pyk2 (L-003165-00), NRF2 (L-003755-00), TRPM2 (L-004193-00), and siGENOME Non-Targeting siRNA Pool #2 (D-001206-14) were purchased from GE Healthcare and was transfected according to standard protocol and experiments were performed within 48–96 hr of transfection.
Ca2+ Measurement
For the measurement of [Ca2+]i levels in cells cultured in 2D or 3D in Matrigel, cells were stained with 3.5 μM fura-2-AM (Life Technology) in the presence of 2.5 mM probenecid (Sigma) and 0.02% Pluronic F-127 (Sigma) for 45 min at 37°C. The fura-2 fluorescence was measured in HEPES-buffered saline [HBS; 150 mM NaCl, 6 mM KCl, 1.2 mM MgSO4, 2 mM CaCl2, 11.5 mM glucose, and 25 mM HEPES (pH adjusted to 7.4 with NaOH)] with 1.25 mM probenecid at 37°C. Fluorescence images of the cells were acquired through the Eclipse Ti-E microscope with ORCA-R2 cooled CCD camera and analyzed with Metafluor. The 340:380-nm ratio images were obtained on a pixel-by-pixel basis and were converted to Ca2+ concentrations by in vivo calibration using 5 μM ionomycin. 3D images are shown as one section from midstructure. For the measurement of [Ca2+]i levels in cells cultured in suspension, cell clumps were dissociated with trypsin and [Ca2+]i levels were determined by using Fluo-4 NW Calcium Assay Kit (Molecular probe). Cells were stained with Fluo-4 according to the manufacturer’s instructions and subjected to flow cytometric analysis (FACSCalibur) using HBS as sheath fluid. Time courses of [Ca2+]i rises in 2D culture are representative of two or three independent experiments and averaged Δ[Ca2+]i represents mean ± SEM from the sum of two or three independent experiments. Fura-2 ratio images in 3D culture are representative of two independent experiments and averaged [Ca2+]i in 3D culture represents mean ± SEM from the sum of two independent experiments.
Immunohistochemistry Staining
Formalin-fixed tumor samples derived from mouse xenografts were processed and embedded in paraffin. Tissue sections were deparaffinized and antigen retrieval was achieved by use of heat-induced epitope retrieval with pH 6.0 citrate buffer (Sigma). Tissue sections were stained with hematoxylin and eosin (H&E), anti-TRPA1 antibody (Sigma), or anti-Cleaved Caspase-3 (Asp175) antibody (Cell Signaling). TRPA1 antibody was validated using sections from tumors that expressed TRPA1 shRNA vectors (Figure S1G). Immunostained slides were counterstained with hematoxylin (Sigma). The score of TRPA1 staining level was based on blinded analysis of epithelial staining by veterinary pathologist R.T.B.. The score of 2+ or greater was used for χ2 test. Images of tissue sections were captured at 4× magnification to create a merged image of the entire section and at 20× magnification for quantification of cleaved caspase-3-positive nuclei, using a Olympus VS120 Slide Scanner. Images of tumor sections stained with H&E, cleaved caspase-3, 4-HNE, and 8-OHdG are representative of at least two independent experiments. Relative apoptosis was quantified by determining the percentage of cleaved caspase-3-positive cells from more than five random fields, per section, from each tumor in the group, and normalized to the shGFP-expressing tumors treated with vehicle.
Quantitative PCR
mRNA prepared from cell or tumor extracts using the RNeasy Mini Kit (QIAGEN) was reverse-transcribed into cDNA using the qScript cDNA synthesis kit (Quanta Biosciences). Real-time PCR was performed on an ABI PRISM 7900HT or QuantStudio 7 Flex Real-Time PCR System (Life Technologies) with Power SYBR Green PCR Mix (Life Technologies). Primers used for the analysis are shown in Table S5.
Cell Viability and Apoptosis
For the measurements of live cell number and cell death under treatment with H2O2 or chemotherapy, cells were cultured with EGF-free medium for MCF-10A cells and standard culture medium for all other cell lines in 96-well plates in the presence or absence of these drugs for indicated times and then stained with 5 μM bisBenzimide H 33342 trihydrochloride (Hoechst; Sigma) and 5 μM propidium iodide (PI; Sigma) for 1 hr at 37°C. Hoechst- and/or PI-positive cells were counted using a laser scanning cytometer, Acumen Cellista (TTPLabTech). For the measurement of apoptosis induced by H2O2, MCF-10A cells were cultured with EGF-free medium for the indicated time and then stained with Annexin V-Alexa Fluor® 488 conjugate (Life Technologies) and PI in Ca2+-containing solution (10 mM HEPES pH 7.4, 140 mM NaCl, and 2.5 mM CaCl2), followed by flow cytometric measurement of fluorescence using a FACSCalibur. Flow cytometry plots are representative of three independent experiments. For the counting of viable cells cultured in suspension, MCF-10A cells were cultured in standard MCF-10A medium for indicated time and then cell clumps were dissociated with trypsin and stained with trypan blue. Viable cells were counted using the Countless Automated Cell Counters (Thermo Fisher Scientific). For the measurement of percentage of dead cells cultured in suspension, MCF-10A cells were cultured in standard MCF-10A medium and then stained with 5 μM Hoechst and 5 μM PI for 1 hr at 37°C without enzymatic dissociation of cell clumps. Hoechst- and/or PI-positive areas were measured using Acumen Cellista. For the measurement of apoptosis induced by detachment, MCF-10A cells were cultured in standard MCF-10A medium for indicated time and then stained with 5 μM Hoechst and 5 μM of CellEvent™ Caspase-3/7 Green Detection Reagent (Thermo Fisher Scientific) for 1 hr at 37°C without enzymatic dissociation of cell clumps. Hoechst- and/or CellEvent™ Caspase-3/7 Green Detection Reagent-positive areas were measured using Acumen Cellista.
ROS and GSH/GSSG Measurement
For the measurement of ROS levels in HCC1569 and H1792 spheroids and MCF-10A acini, cells were transduced with a lentiviral vector encoding Hyper-2 or Hyper-2 (C199S). The Hyper-2 fluorescence was measured at 37°C in HBS solution. Fluorescence images of the cells were acquired through the Eclipse Ti-E microscope (Nikon) with ORCA-R2 cooled CCD camera (Hamamatsu Photonics) and analyzed with Metafluor image acquisition software (Molecular Devices). The 488:405-nm ratio images were obtained on a pixel-by-pixel basis. 3D images are shown as one section from midstructure. For the measurement of [H2O2]i levels in MCF-10A cells cultured in suspension, cell clumps were dissociated with trypsin and stained with 10 μM peroxy green-1 for 30 min at 37°C. Cells were then subjected to flow cytometric analysis (FACSCalibur; BD Biosciences). ROS levels determined by flow cytometry are reported as mean fluorescence intensity expressed in arbitrary units (Harris et al., 2015). GSH/GSSG ratio was assessed using the GSH/GSSG-Glo™ Assay kit (PROMEGA) according to the manufacturer’s instructions, and the luciferase signal was measured by SpectraMax M5 (Molecular Devices).
Immunofluorescence Staining
HCC1569, EL-12-58, and H1792 spheroids and MCF-10A acini were fixed with 4% paraformaldehyde and stained as previously described (http://brugge.hms.harvard.edu). Antibodies used were Ki67 (DAKO) and Cleaved Caspase-3 (Asp175) antibody (Cell Signaling). Fluorescent images were acquired through the Eclipse Ti-E microscopes with A1R point scanning confocal (Nikon). Data are processed with NIS Elements software (Nikon). 3D images are shown as one section from midstructure. Images shown are representative of at least three independent experiments. For examination of luminal filling, spheroids were scored as clear (~90–100% clear), mostly clear (~50–90% clear), mostly filled (~10–50% clear), or clear (~0–10% clear), as previously reported (Schafer et al., 2009).
Soft Agar Assay
Cells were added to the regular growth media for HCC1569 or H1792 cells (± AP-18) plus 0.4% low-melt agarose (Sigma) and layered onto a bed of growth media plus 0.5% low-melt agarose. Cells were fed every 4 days with growth media (± AP-18) plus 0.4% low-melt agarose. At the indicated times, viable colonies were stained with iodonitrotetrazolium chloride (Sigma). Colony number and colony size were determined using ImageJ. Images shown are representative of at least three independent experiments.
Masson Trichrome Staining
Formalin-fixed tumor samples derived from mouse xenografts were processed and embedded in paraffin. Tissue sections were deparaffinized and stained with trichrome using trichrome stain kit (Abcam) according to the manufacturer’s instructions. Images of tissue sections were captured at 4× magnification to create a merged image of the entire section and at 10× magnification, using a Olympus VS120 Slide Scanner. Collagenous stroma area relative to total area was quantified from the whole field of each tumor section, using ImageJ. Masson trichrome selectively stains collagenous components blue and non-collagenous components red.
RPPA and Immunoblot Assay
For RPPA assay, sample preparation and probing with antibodies to the indicated proteins, normalization of data points, and analysis were performed as previously described (Hennessy et al., 2010). For immunoblot analysis for all the indicated proteins except for TRPA1, samples were lysed in cell lysis buffer (1% Triton X-100, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 100 mM NaF, 10 mM Na pyrophosphate, 10% glycerol, 50 mM HEPES pH 7.4) with protease and phosphatase inhibitor cocktails (Roche) and 1 mM Na3VO4 (Sigma). Protein concentrations were determined by BCA protein assay (Life Technologies). Equal protein amounts were denatured in SDS sample buffer (10% Glycerol, 2% SDS, 62.5 mM Tris-HCL, pH 6.8) with 1% β-mercaptoethanol for 10 min at 95°C. For immunoblot analysis for TRPA1 protein, membrane fraction was isolated by using Mem-PER™ Plus Membrane Protein Extraction Kit (Life Technologies) according to the manufacturer’s instructions. Equal protein amounts were denatured in SDS sample buffer with 50 mM dithiothreitol using a vortex mixer for at least 30 min at room temperature. Proteins were analyzed by SDS-PAGE and immunoblot using the indicated antibodies. Blots were imaged with the Odyssey CLx infrared imaging system (LI-COR) and are representative of at least two independent experiments where indicated. Ras activities were assessed using the Active Ras Pull-Down and Detection Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions.
Chromatin Immunoprecipitation
H1792 cells overexpressing Flag-NRF2 were cross-linked in the culture medium with 1% formaldehyde for 10 min at room temperature. The reaction was stopped by the addition of 100 mM glycine, followed by 5 mg/mL BSA in PBS and then two washing in cold PBS. Cells were harvested and resuspended in lysis buffer [50 mM Tris·HCl, pH 8.0, 10 mM EDTA, 1% SDS, 1×Protease Inhibitor Cocktail (Selleck Chemicals)], and then sonicated with a Sonic Dismembrator (Fisher Scientific, Model FB120) to obtain a fragment size of 300–500 bp. Fragmented chromatin was diluted in IP buffer (20 mM Tris·HCl, pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 1×Protease Inhibitor Cocktail) and incubated overnight at 4°C with Protein G magnetic beads (Dynabeads; Thermo Fisher Scientific) that had been pre-incubated with monoclonal anti-Flag M2 antibody (Sigma, F1804) or mouse IgG (Thermo Fisher Scientific). Immunoprecipitates were washed three times with wash buffer (50 mM Hepes, pH 7.6, 0.5 M LiCl, 1 mM EDTA, 0.7% Na deoxycholate, and 1% Nonidet P-40) and twice with TE buffer. Immunoprecipitated (or no IP input) DNA was recovered in Elution Buffer (1% SDS and 0.1 M NaHCO3) over 6 hr at 65°C, and then column-purified with QiaQuick columns (Qiagen). Quantitative PCR was performed with Fast SYBR Green Master Mix (Life Technologies) on a QuantStudio 7 Flex Real-Time PCR System (Life Technologies). Primer sequences are listed in Table S5.
QUANTIFICATION AND STATISTICAL ANALYSIS
Bioinformatics and Statistical Analysis
Analysis of the TCGA data was conducted on processed RNA-seq, gene alteration data, paired clinical feature data, and RPPA data downloaded through the Broad GDAC Firehose (https://gdac.broadinstitute.org) or cBioPortal (http://www.cbioportal.org). Normalized gene expression data and paired clinical feature data including survival from METABRIC were obtained from the publicly available European Genome-phenome Archive: EGAD00010000210, EGAD0001000021. Published MPNST datasets were downloaded from Miller et al. (Miller et al., 2009) (GEO: GSE14038). Microarray data processing and statistical analyses were performed in R version 3.1.3. mRNA and protein heatmaps were generated in Cluster 3.0 as a hierarchical cluster using Pearson Correlation and a center metric. The resulting heatmap was visualized in Java Treeview. Kaplan-Meier curves and log likelihood ratio statistic were calculated using the ‘packHV’ package. Error bars represent either the SD or SEM, as described in the figure legends. The sample size for each experiment, n, is included in the associated figure legend. Box and whiskers graphs indicate the median and the 25th and 75th percentiles, with minimum and maximum values at the extremes of the whiskers. Statistical significance was determined by the Student’s t test and the one-way or two-way ANOVA followed by Tukey’s test using GraphPad Prism 7.0. p values < 0.05 were considered significant. p values for each experiment are also included in the associated figure legends.
DATA AND SOFTWARE AVAILABILITY
RNA-seq data for HCI PDX models were deposited in GEO: GSE113476.
Supplementary Material
Highlights.
TRPA1 is overexpressed in cancer and mediates a non-canonical ROS defense program
TRPA1 promotes ROS tolerance via induction of Ca2+-dependent anti-apoptotic pathways
NRF2, an oxidant-defense transcription factor, directly controls TRPA1 expression
Targeting TRPA1 reduces xenograft tumor growth and enhances chemosensitivity
Significance.
Accumulating evidence has shown that oxidative-stress defense programs are critically involved in tumor progression and therapy resistance and represent an “Achilles heel” or vulnerability of tumor cells. However, molecular mechanisms by which tumor cells adapt to oxidative stress are poorly understood. Our study, focusing on a neuronal redox-sensing channel TRPA1, which is overexpressed in human cancer, reveals a non-canonical oxidative-stress defense mechanism distinct from well-characterized ROS-neutralizing mechanisms. TRPA1 does not affect canonical ROS-neutralizing programs but senses redox stress and upregulates Ca2+-dependent anti-apoptotic programs that promotes oxidative-stress tolerance. TRPA1 inhibition reduces tumor growth and enhances chemotherapy sensitivities in breast cancer xenograft models. Our study reveals the existence of oxidative-stress tolerance programs involving TRPA1 and highlights its potential as a therapeutic target.
ACKNOWLEDGMENTS
We thank Patricia Cho, Xiangkun Han, Jonathan Coloff, Hendrik Johannes Kuiken, Carman Li, Grace Gao, Tony Yeung, Satoshi Hamano, and Astrid Ruefli-Brasse for reagents and/or helpful discussions and Angie Martinez Gakidis for scientific editing. Image analysis and cell viability assay were performed at The Nikon Imaging Center and ICCB-Longwood Screening Facility at Harvard Medical School. This research was supported by the Susan G. Komen for the Cure Foundation (SAC170002, J.S.B.), the Breast Cancer Research Foundation (J.S.B.), CCSG grant CA016672 (G.B.M.), SAC110052 (G.B.M.), BCRF-16-109 (G.B.M.), and by funding from the ROADS Program funded by F. Hoffmann-La Roche Ltd to J.S.B.
Footnotes
STAR★METHODS
- KEY RESOURCES TABLE
- CONTACT FOR REAGENT AND RESOURCE SHARING
- EXPERIMENTAL MODEL AND SUBJECT DETAILS
-
○Animals and In Vivo Procedures
-
○Human Samples
-
○Development of Patient Derived HCI-002 Cell Line
-
○Cell Culture
-
○
- METHOD DETAILS
-
○Plasmids, siRNAs, shRNAs, CRISPR/Cas9, and Virus Production
-
○Ca2+ Measurement
-
○Immunohistochemistry Staining
-
○Quantitative PCR
-
○Cell Viability and Apoptosis
-
○ROS and GSH/GSSG Measurement
-
○Immunofluorescence Staining
-
○Soft Agar Assay
-
○Masson Trichrome Staining
-
○RPPA and Immunoblot Assay
-
○Chromatin Immunoprecipitation
-
○
- QUANTIFICATION AND STATISTICAL ANALYSIS
-
○Bioinformatics and Statistical Analysis
-
○
- DATA AND SOFTWARE AVAILABILITY
SUPPLEMENTAL INFORMATION
Supplemental Information includes eight figures and five tables and can be found with this article online at https://doi.org/10.1016/j.ccell.2018.05.001.
DECLARATION OF INTERESTS
J.S.B. receives funding from F. Hoffmann-La Roche Ltd. G.B.M. holds advisory positions on SAB for AstraZeneca and Immunomet and has received funding support from Karus, Pfizer, Tesaro, AstraZeneca, and Immunomet. This work is related to a patent application: PCT/US18/20937 titled “Methods of Use for TRP Channel Antagonist-Based Combination Cancer Therapies”.
REFERENCES
- Akgul C (2009). Mcl-1 is a potential therapeutic target in multiple types of cancer. Cell. Mol. Life Sci 66, 1326–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora S, Bhardwaj A, Singh S, Srivastava SK, McClellan S, Nirodi CS, Piazza GA, Grizzle WE, Owen LB, and Singh AP (2013). An undesired effect of chemotherapy: gemcitabine promotes pancreatic cancer cell invasiveness through reactive oxygen species-dependent, nuclear factor κB- and hypoxia-inducible factor 1α-mediated up-regulation of CXCR4. J.Biol. Chem 288, 21197–21207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrout J, Kyriakopoulou E, Moparthi L, Hogea AS, Berrout L, Ivan C, Lorger M, Boyle J, Peers C, Muench S, et al. (2017). TRPA1-FGFR2 binding event is a regulatory oncogenic driver modulated by miRNA-142-3p. Nat. Commun 8, 947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao DS, Zhong L, Hsieh T, Abooj M, Bishnoi M, Hughes L, and Premkumar LS (2012). Expression of transient receptor potential ankyrin 1 (TRPA1) and its role in insulin release from rat pancreatic beta cells. PLoS One 7, e38005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri J, Bose N, Gong J, Hall D, Rifkind A, Bhaumik D, Peiris TH, Chamoli M, Le CH, Liu J, et al. (2016). A Caenorhabditis eiegans model elucidates a conserved role for TRPA1-Nrf signaling in reactive α-dicarbonyl detoxification. Curr. Biol 26, 3014–3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiavarina B, Nokin MJ, Bellier J, Durieux F, Bletard N, Sherer F, Lovinfosse P, Peulen O, Verset L, Dehon R, et al. (2017). Methylglyoxal-mediated stress correlates with high metabolic activity and promotes tumor growth in colorectal cancer. Int. J. Mol. Sci 18, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chio IIC, Jafarnejad SM, Ponz-Sarvise M, Park Y, Rivera K, Palm W, Wilson J, Sangar V, Hao Y, Öhlund D, et al. (2016). NRF2 promotes tumor maintenance by modulating mRNA translation in pancreatic cancer. Cell 166, 963–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham DE (2003). TRP channels as cellular sensors. Nature 426,517–524. [DOI] [PubMed] [Google Scholar]
- Clapham DE (2007). Calcium signaling. Cell 131, 1047–1058. [DOI] [PubMed] [Google Scholar]
- Cullen PJ, and Lockyer PJ (2002). Integration of calcium and Ras signalling. Nat. Rev. Mol. Cell Biol 3, 339–348. [DOI] [PubMed] [Google Scholar]
- Debnath J, and Brugge JS (2005). Modelling glandular epithelial cancers in three-dimensional cultures. Nat. Rev. Cancer 5, 675–688. [DOI] [PubMed] [Google Scholar]
- Déliot N, and Constantin B (2015). Plasma membrane calcium channels in cancer: alterations and consequences for cell proliferation and migration. Biochim. Biophys. Acta 1848, 2512–2522. [DOI] [PubMed] [Google Scholar]
- DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, et al. (2011). Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475, 106–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNicola GM, Chen P, Mullarky E, Sudderth JA, Hu Z, Wu D,Tang H, Xie Y, Asara JM, Huffman KE, et al. (2015). NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat. Genet 47, 1475–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRose YS, Wang G, Lin YC, Bernard PS, Buys SS, Ebbert MTW, Factor R, Matsen C, Milash BA, Nelson E, et al. (2011). Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat. Med 17, 1514–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhardt MJ, Filipovic MR, Leffler A, De La Roche J, Kistner K, Fischer MJ, Fleming T, Zimmermann K, Ivanovic-Burmazovic I, Nawroth PP, et al. (2012). Methylglyoxal activates nociceptors through transient receptor potential channel A1 (TRPA1): a possible mechanism of metabolic neuropathies. J. Biol. Chem 287, 28291–28306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Karim IA, McCrudden MTC, McGahon MK, Curtis TM, Jeanneau C, Giraud T, Irwin CR, Linden GJ, Lundy FT, and About I (2016). Biodentine reduces tumor necrosis factor alpha-induced TRPA1 expression in odontoblastlike cells. J. Endod 42, 589–595. [DOI] [PubMed] [Google Scholar]
- Fusi C, Materazzi S, Benemei S, Coppi E, Trevisan G, Marone IM, Minocci D, De Logu F, Tuccinardi T, Di Tommaso MR, et al. (2014). Steroidal and non-steroidal third-generation aromatase inhibitors induce pain-like symptoms via TRPA1. Nat. Commun 5, 5736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Gal K, Ibrahim MX, Wiel C, Sayin VI, Akula MK, Karlsson C, Dalin MG, Akyurek LM, Lindahl P, Nilsson J, et al. (2015). Antioxidants can increase melanoma metastasis in mice. Sci. Transl. Med 7, 308re8. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, Castedo M, and Kroemer G (2012). Molecular mechanisms of cisplatin resistance. Oncogene 31, 1869–1883. [DOI] [PubMed] [Google Scholar]
- Gorrini C, Harris IS, and Mak TW (2013). Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov 12, 931–947. [DOI] [PubMed] [Google Scholar]
- Harris IS, Treloar AE, Inoue S, Sasaki M, Gorrini C, Lee K, Yung K, Brenner D, Knobbe-Thomsen CB, Cox MA, et al. (2015). Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 27, 211–222. [DOI] [PubMed] [Google Scholar]
- Hatano N, Itoh Y, Suzuki H, Muraki Y, Hayashi H, Onozaki K, Wood IC, Beech DJ, and Muraki K (2012). Hypoxia-inducible factor-1α (HIF 1α) switches on transient receptor potential ankyrin repeat 1 (TRPA1) gene expression via a hypoxia response element-like motif to modulate cytokine release. J. Biol. Chem 287, 31962–31972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. (2009). Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennessy BT, Lu Y, Gonzalez-Angulo AM, Carey MS, Myhre S, Ju Z, Davies MA, Liu W, Coombes K, Meric-Bernstam F, et al. (2010). A technical assessment of the utility of reverse phase protein arrays for the study of the functional proteome in non-microdissected human breast cancers. Clin. Proteomics 6, 129–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinman A, Chuang HH, Bautista DM, and Julius D (2006). TRP channel activation by reversible covalent modification. Proc. Natl. Acad. Sci. USA 103, 19564–19568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Shestov AA, Swain P, Yang C, Parker SJ, Wang QA, Terada LS, Adams ND, McCabe MT, Pietrak B, et al. (2016). Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 532, 255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SF, Cruz C, Greifenberg AK, Dust S, Stover DG, Chi D, Primack B, Cao S, Bernhardy AJ, Coulson R, et al. (2016). CDK12 inhibition reverses de novo and acquired PARP inhibitor resistance in BRCA wild-type and mutated models of triple-negative breast cancer. Cell Rep. 17, 2367–2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordt SE, Bautista DM, Chuang H, McKemy DD, Zygmunt PM, Högestätt ED, Meng ID, and Julius D (2004). Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature 427, 260–265. [DOI] [PubMed] [Google Scholar]
- Koivisto A, Hukkanen M, Saarnilehto M, Chapman H, Kuokkanen K, Wei H, Viisanen H, Kerman KE, Lindstedt K, and Pertovaara A (2012). Inhibiting TRPA1 ion channel reduces loss of cutaneous nerve fiber function in diabetic animals: sustained activation of the TRPA1 channel contributes to the pathogenesis of peripheral diabetic neuropathy. Pharmacol. Res 65, 149–158. [DOI] [PubMed] [Google Scholar]
- Liu Y, Li Q, Zhou L, Xie N, Nice EC, Zhang H, Huang C, and Lei Y (2016). Cancer drug resistance: redox resetting renders a way. Oncotarget 7, 42740–42761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macpherson LJ, Dubin AE, Evans MJ, Marr F, Schultz PG, Cravatt BF, and Patapoutian A (2007). Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature 445, 541–545. [DOI] [PubMed] [Google Scholar]
- Mailleux AA, Overholtzer M, Schmelzle T, Bouillet P, Strasser A, and Brugge JS (2007). BIM regulates apoptosis during mammary ductal morphogenesis, and its absence reveals alternative cell death mechanisms. Dev. Cell 12, 221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EW, Tulyathan O, Isacoff EY, and Chang CJ (2007). Molecular imaging of hydrogen peroxide produced for cell signaling. Nat. Chem. Biol 3, 263–267. [DOI] [PubMed] [Google Scholar]
- Miller SJ, Jessen WJ, Mehta J, Hardiman A, Sites E, Kaiser S, Jegga AG, Li H, Upadhyaya M, Giovannini M, et al. (2009). Integrative genomic analyses of neurofibromatosis tumours identify SOX9 as a biomarker and survival gene. EM BO Mol. Med 1, 236–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, Yamamoto M, and Motohashi H (2012). Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 22, 66–79. [DOI] [PubMed] [Google Scholar]
- Nassini R, Gees M, Harrison S, De Siena G, Materazzi S, Moretto N, Failli P, Preti D, Marchetti N, Cavazzini A, et al. (2011). Oxaliplatin elicits mechanical and cold allodynia in rodents via TRPA1 receptor stimulation. Pain 152, 1621–1631. [DOI] [PubMed] [Google Scholar]
- Nie Y, Huang C, Zhong S, Wortley MA, Luo Y, Luo W, Xie Y, Lai K, and Zhong N (2016). Cigarette smoke extract (CSE) induces transient receptor potential ankyrin 1 (TRPA1) expression via activation of HIF1α in A549 cells. Free Radic. Biol. Med 99, 498–507. [DOI] [PubMed] [Google Scholar]
- Park YR, Chun JN, So I, Kim HJ, Baek S, Jeon JH, and Shin SY (2016). Data-driven analysis of TRP channels in cancer: linking variation in gene expression to clinical significance. Cancer Genomics Proteomics 13, 83–90. [PubMed] [Google Scholar]
- Patel BP, Rawal UM, Dave TK, Rawal RM, Shukla SN, Shah PM, and Patel PS (2007). Lipid peroxidation, total antioxidant status, and total thiol levels predict overall survival in patients with oral squamous cell carcinoma. Integr. Cancer Ther 6, 365–372. [DOI] [PubMed] [Google Scholar]
- Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, Leitch AM, Johnson TM, DeBerardinis RJ, and Morrison SJ (2015). Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 527, 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reginato MJ, Mills KR, Paulus JK, Lynch DK, Sgroi DC, Debnath J, Muthuswamy SK, and Brugge JS (2003). Integrins and EGFR coordinately regulate the pro-apoptotic protein Bim to prevent anoikis. Nat. Cell Biol 5, 733–740. [DOI] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, Gao S, Puigserver P, and Brugge JS (2009). Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 461, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenkel LB, Olivieri PR, Boezio AA, Deak HL, Emkey R, Grace RF, Gunaydin H, Guzman-perez A, Lee JH, Te Y, et al. (2016). Optimization of a novel quinazolinone-based series of transient receptor potential A1 (TRPA1) antagonists demonstrating potent in vivo activity. J. Med. Chem 59, 2794–2809. [DOI] [PubMed] [Google Scholar]
- Schieber M, and Chandel NS (2014). ROS function in redox signaling and oxidative stress. Curr. Biol 24, R453–R462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu S, Takahashi N, and Mori Y (2014). TRPs as chemosensors (ROS, RNS, RCS, gasotransmitters). Handb. Exp. Pharmacol 223, 767–794. [DOI] [PubMed] [Google Scholar]
- Singh A, Venkannagari S, Oh KH, Zhang YQ, Rohde JM, Liu L, Nimmagadda S, Sudini K, Brimacombe KR, Gajghate S, et al. (2016). Small molecule inhibitor of NRF2 selectively intervenes therapeutic resistance in KEAP1-deficient NSCLC tumors. ACS Chem. Biol 11, 3214–3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soini Y, Haapasaari K-M, Vaarala MH, Turpeenniemi-Hujanen T, Kärjä V, and Karihtala P (2011). 8-hydroxydeguanosine and nitrotyrosine are prognostic factors in urinary bladder carcinoma. Int. J. Clin. Exp. Pathol 4, 267–275. [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Motohashi H, and Yamamoto M (2013). Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharmacol. Sci 34, 340–346. [DOI] [PubMed] [Google Scholar]
- Takahashi N, Kuwaki T, Kiyonaka S, Numata T, Kozai D, Mizuno Y, Yamamoto S, Naito S, Knevels E, Carmeliet P, et al. (2011). TRPA1 underlies a sensing mechanism for 02. Nat. Chem. Biol 7, 701–711. [DOI] [PubMed] [Google Scholar]
- Tennant DA, Durán RV, and Gottlieb E (2010). Targeting metabolic transformation for cancer therapy. Nat. Rev. Cancer 10, 267–277. [DOI] [PubMed] [Google Scholar]
- Wang Y, Li X, and Hu H (2014). Genomics H3K4me2 reliably defines transcription factor binding regions in different cells. Genomics 103, 222–228. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Hsu JL, and Hung MC (2012). Regulation of ubiquitination-mediated protein degradation by survival kinases in cancer. Front. Oncol 2, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong S, Nie YC, Gan ZY, Liu XD, Fang ZF, Zhong BN, Tian J, Huang CQ, Lai KF, and Zhong NS (2015). Effects of Schisandra chinensis extracts on cough and pulmonary inflammation in a cough hypersensitivity guinea pig model induced by cigarette smoke exposure. J. Ethnopharmacol 165, 73–82. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.