

Abstract
The choreography between RNA synthesis and degradation is a key determinant in biology. Engineered systems such as CRISPR have been developed to rid a cell of RNAs. Here, we show that a small molecule can recruit a nuclease to a specific transcript, triggering its destruction. A small molecule that selectively binds the oncogenic microRNA(miR)-96 hairpin precursor was appended with a short 2′-5′ poly(A) oligonucleotide. The conjugate locally activated endogenous, latent ribonuclease (RNase L), which selectively cleaved the miR-96 precursor in cancer cells in a catalytic and sub- stoichiometric fashion. Silencing miR-96 derepressed pro- apoptotic FOXO1 transcription factor, triggering apoptosis in breast cancer, but not healthy breast, cells. These results demonstrate that small molecules can be programmed to selectively cleave RNA via nuclease recruitment and has broad implications.
The ENCODE project showed that only 1−2% of the genome encodes for protein, yet 70−80% is transcribed into RNA.1 Not surprisingly, noncoding RNAs play a myriad of roles in cellular biology including regulating protein production and gene expression.2,3 As key regulators of cellular function, RNA production and destruction is tightly controlled.4
Proteolysis targeting chimeras (PROTACs) are a proven approach for targeted protein degradation by using small molecules.5,6 A potential approach to mediate RNA decay is to exploit ribonucleases (RNases) that naturally regulate RNA lifetime and recruit them to specific transcripts via a small molecule, or ribonuclease targeting chimeras (RIBOTACs). RNase L, an integral part of the antiviral immune response, is present in minute quantities in all cells as an inactive monomer. Upon activation of the immune system, RNase L is upregulated and 2′-5′ oligoadenylate [2′-5′poly(A)] is synthesized; binding of 2′-5′poly(A) dimerizes and activates RNase L (Figure 1A).7 Because of the ubiquitous nature of this system, we sought to assemble active RNase L onto a specific RNA target to cleave it, akin to antisense.8
Figure 1.
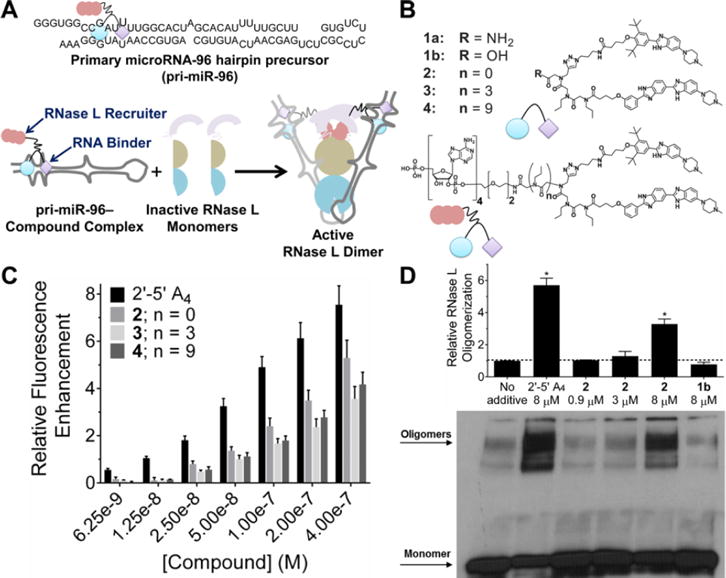
Design and characterization of a transcript-selective RNase L recruiting compound. (A) Top, secondary structure of the primary transcript of microRNA-96 (pri-miR-96). Bottom, schematic depiction of active RNase L recruitment to pri-miR-96 by 2. (B) Structures of compounds used in this study. (C) In vitro cleavage of pri-miR-96 by 2′-5′A4 and chimeric small molecules with different linker lengths; n = 0 linker (2) displayed the highest cleavage capability. (D) Representative Western blot and quantification of cross-linked monomer and oligomer (active) forms of RNase L upon treatment with 2′-5′A4, 2 (selective for pri-miR-96), or lb which lacks 2′-5′A4. Data are expressed as mean ± s.e.m. (n > 3). *p < 0.05, as measured by a two-tailed Student t test.
Previously, our lead identification strategy, Inforna, enabled the design of a small molecule that targets the Drosha endonuclease processing site of microRNA (miR)-96 named Targaprimir-96 (1a).9 This molecule selectively inhibited biogenesis of miR-96, derepressed pro-apoptotic transcription factor FOXO1 (a target of miR-96), and triggered apoptosis selectively in triple negative breast cancer cells (MDA-MB- 231). To study if RNase L could be effiectively recruited to cleave the primary transcript of miR-96 (pri-miR-96), we varied the distance between 1a and a short 2′-5′A4 oligonucleotide, which functions as an RNase L recruiter (2−4, Figure 1B). In vitro fluorescence-based and gel cleavage experiments showed that compounds triggered cleavage of pri-miR-96 and that the shortest spacer (2) was most effiective (Figure S1A, B and Figure 1C). Importantly, addition of the 2′-5′A4 component of 2 did not significantly affiect its avidity for pri-miR-96’s Drosha site, as compared to the parent compound 1a9 (Kd = 20 nM), and no saturable binding was observed to a fully base paired RNA (Figure S1C, D).
We compared recruitment of RNase L by 2 with 2′-5′A4 in vitro. Compared to the natural substrate, 2′-5′ A4, compound 2 assembled RNase L into its active oligomeric species at 50% lower levels at the same concentration (Figure 1D). Next, the capacity of these compounds to cleave pri-miR-96 was studied in vitro. Consistent with the preferred cleavage sites of RNase L, one major cleavage site was observed when 2′-5′A4 alone was added (U12, the Drosha site) (Figure S1E).10 No significant cleavage was observed in the absence of 2′-5′A4 (RNase L alone; Figure S1F). Interestingly, upon addition of the parent compound to a reaction containing 2′-5′A4 and RNase L, the amount of cleavage did not change; rather, the major cleavage site became U35 (Figure S1G, H); that is, the parent compound blocked cleavage by binding the Drosha site as designed and had no inhibitory activity on RNase L. The analogous experiments with 2 revealed that it effiectively activated RNase L and recruited the enzyme to pri-miR-96 to U12, the major cleavage site observed for 2′-5′A4. Further, cleavage at U12 was inhibited by addition of the parent compound (1a, Figure S1G, H), as expected. In contrast to 2′-5′A4, the small molecule, which drives affinity for the Drosha site, did not allow 2 to recruit RNase L to other sites, i.e., U35 for 2′-5′A4, indicating that 2 is selective (Figure S1G, H). This observation was further bolstered by studying cleavage of pri-miR-96 in the presence of increasing concentrations of tRNA. Indeed, addition of tRNA more significantly inhibited cleavage of pri- miR-96 by 2′-5′A4 than by 2 (Figure S1I).
We next sought to determine whether selective recruitment of RNase L by 2 to pri-miR-96 was operational in cells. As oligonucleotides are generally not cell permeable, the cellular permeability of 2 was measured by flow cytometry. Although conjugation of 2′-5′A4 reduced permeability of 2 by ∼50% as compared to 1a, it still entered cells unaided and in significant amounts (Figure S2A). Though distribution of RNase L is mostly cytosolic, nuclear fractions of RNase L have also been observed.11 Therefore, if 2 recruits RNase L to pri-miR-96 in the nucleus, a reduction of both pri-miR-96 and mature miR-96 levels is expected. Indeed, both levels were reduced in MDA- MB-231 cells (Figure 2A, B), which overexpress miR-96.12 These results were recapitulated in other cancer cell lines that express miR-96, suggesting broad applicability (Figure S2B). In contrast, addition of 1a increased the amount of pri-miR-96 and reduced levels of mature miR-96 (Figure 2A, B), as expected since the compound inhibits cleavage by Drosha endonuclease.9 Importantly, these results indicate that cleavage of miRNAs by enzymes other than their canonical processing enzymes directs them to an RNA decay pathway. That is, cleavage by RNase L degrades the pri-miRNA, resulting in decreased mature miRNA, rather than acting as an alternative Drosha endonuclease, which would have led to an increase in mature miRNA levels.
Figure 2.
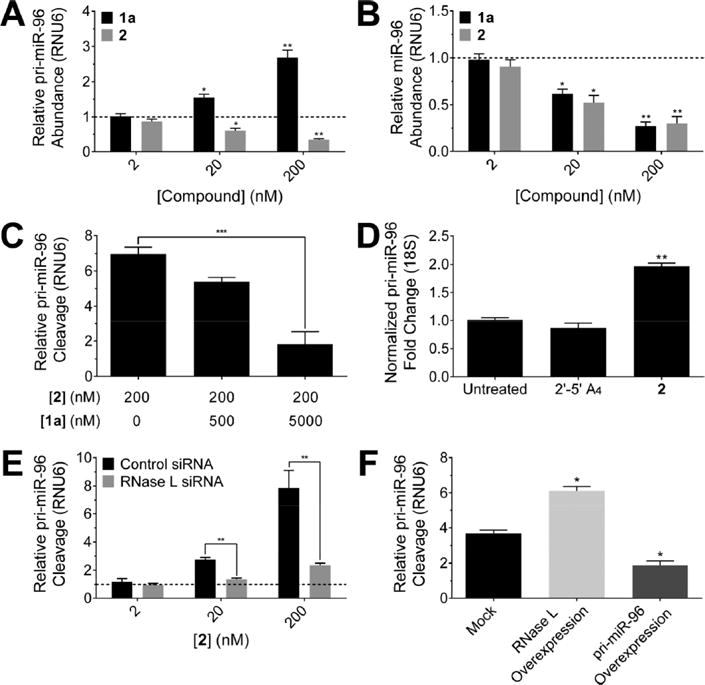
Small molecule RNase L recruitment shows on-target effects in cells. (A) Treatment of MDA-MB-231 triple negative breast cancer cells with 2 decreased abundance of pri-miR-96 via cleavage, as measured by RT-qPCR In contrast, la boosted levels of pri-miR-96 by inhibiting Drosha processing (lacks 2,-5′A4). (B) Effect of la and 2 on mature miR-96 levels. (C) 2-mediated cleavage of pri-miR-96 is reduced by addition of la. The effect of la itself was accounted for in the calculation. (D) RT-qPCR of RNAs isolated from immunopreci- pitated RNase L protein after cells were treated with 2′-5′A4 or 2 (200 nM). RNAs bound to RNase L treated with 2 show enrichment of the pri-miR-96 transcript normalized to RNA immunoprecipitated from ¡5- actin. (E) Relative cleavage of pri-miR-96 by 2 upon knock down of RNase L by siRNA (F) RNase L overexpression resulted in increased cleavage activity of 2 (20 nM), whereas overexpression of pri-miR-96 resulted in decreased cleavage activity. Data are expressed as mean ± s.e.m. (n > 3). *p < 0.05, **p < 0.01, as measured by a two-tailed Student t test.
To ensure that the effiect of 2 on pri-miR-96 levels was due to recruitment of RNase L, a series of experiments were completed. The addition of 2′-5′A4 alone, either directly to cell culture or by forced cellular uptake (transfection), had no effiect on pri-miR-96 levels (Figure S2C). These results suggest that cleavage of pri-miR-96 by 2 is due to specific recruitment of RNase L in cells, as opposed to general stimulation of the RNase L pathway. Further, addition of 2 had no effiect on RNase L mRNA levels (Figure S2D). We cotreated cells with a constant concentration of 2 and increasing concentrations of compound 1a, which targets the same site on pri-miR-96 but does not recruit RNase L, and measured the effiect on pri-miR- 96 cleavage levels. As expected, addition of 1a decreased pri- miR-96 cleavage levels in a dose dependent fashion (Figure 2C). To validate that an RNase L-2-pri-miR-96 ternary complex forms, RNase L was immunoprecipitated from cells treated with 2 or 2′-5′A4. A ∼2-fold enhancement of the pri- miR-96 transcript was observed from cells treated with 2 as compared to cells treated 2′-5′A4 (both normalized relative to background pull-down of β-actin; Figure 2D). Indeed, 2 is selective for formation of the ternary complex with pri-miR-96 as pri-miR-210 is not pulled down (Figure S2E; see note in figure caption for rationale for selecting pri-miR-210).
Next, RNase L was knocked down by using siRNA (Figure S2F), which should reduce 2’s activity. In cells treated with siRNA directed at RNase L, but not a scrambled siRNA, 2’s activity was reduced to levels observed without treatment, as determined by measuring pri-miR-96 cleavage activity (Figure 2E and Figure S2F). Conversely, forced expression of RNase L enhanced 2’s cleavage activity whereas forced expression of pri- miR-96 decreased cleavage activity (Figure 2F and Figure S2G). These gain- and loss-of-function experiments further support the assertion that 2 cleaves a specific RNA transcript through recruitment of RNase L in cells. Additionally, to confirm that 2 does not act as mediator of payload delivery (i.e., 2′-5′A4), cells were treated with 2, and the cellular fraction was analyzed by mass spectrometry. Indeed, 2 is stable in cells (Figure S3A) and thus cleavage of pri-miR-96 occurs through the chimeric properties of 2, rather than through the separate effiects of 2′- 5′A4 and 1a.
Elevated levels of miR-96 contributes to an invasive phenotype in various cancers due to repression of forkhead box protein O1 (FOXO1), a pro-apoptotic transcription factor required for transcription of pro-apoptotic Bcl-xl proteins.13 Addition of 2 (200 nM) to MDA-MB-231 cells increased expression of FOXO1 by ∼2-fold while having no effiect on a protein not regulated by miR-96 (Figure 3A). Although FOXO1 mRNA is regulated by miR-182, miR-27a, and miR- 96, previous studies have shown that inhibition of miR-96 alone is sufficient to enhance FOXO1 expression.9 Importantly 2 did not affiect levels of miR-27a, miR-182, or other miRNAs predicted to regulate FOXO1 mRNA by TargetScan (Figure 3B), supporting that the increase in FOXO1 protein expression is mediated through inhibition of miR-96. To further study selectivity, we assessed the effiect of 2 on all measurable mature miRNA levels in MDA-MB-231 cells, of which the most significantly inhibited was miR-96 (p < 0.01) (Figure 3C). Because FOXO1 is pro-apoptotic, its derepression via inhibition of miR-96 should trigger apoptosis. At both 20 and 200 nM, 2 induced significant apoptosis in MDA-MB-231 cells, and 2’s ability to trigger apoptosis is ablated upon forced expression of pri-miR-96 (Figure 3D). Notably, direct treatment or transfection of 2′-5′A4 alone, at effiective concen- trations, does not significantly induce apoptosis (Figure 3D and Figure S3B), which is observed upon global activation of the RNase L surveillance system. Additionally, 2 did not induce apoptosis in healthy breast epithelial cells (MCF10a) (Figure S3C). Thus, 2 is a precision chemical probe affiecting the biology of cells that express high levels of miR-96. Notably, 2 stimulates apoptosis to the same extent as 1a but at a 2.5-fold lower dose. Given that 2 is taken up at half the amount of 1a (Figure S2A), recruitment of RNase L enhances the activity by at least 5-fold.
Figure 3.
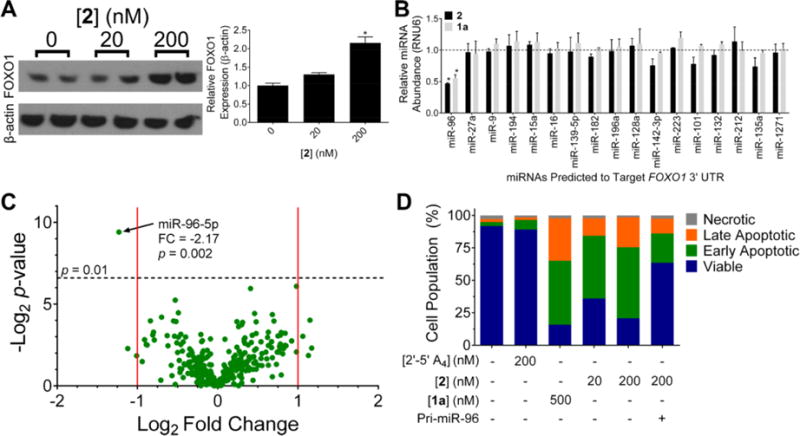
Apoptotic stimulation through selective recruitment of RNase L to pri-miR-96 by 2. (A) FOXOl is a tumor suppressor protein down- regulated by miR-96. Treatment with 2 caused derepression of FOXOl as measured by Western blotting. (B) Selective cleavage of miR-96 with 2 treatment (20 nM) among predicted FOXO 1-targeting miRNAs, as measured by RT-qPCR. (C) Nonhypothesis driven RT-qPCR analysis of validated miRNAs indicated inhibition of miR-96 by 2 (200 nM) with the most magnitude and significance. (D) Treatment with 2 (200 nM) triggered apoptosis in MDA-MB-231 cells, as measured by Annexin V/PI staining. “Pri-miR-96” indicates plasmid overexpression of a pri-miR-96 hairpin, which diminished the percentage of cells undergoing apoptosis upon 2-treatment. Data are expressed as mean ± s.e.m. (n > 3). *p < 0.05, as measured by a two-tailed Student’s t test.
There is the potential of 2 to catalytically cleave pri-miR-96 in cells. The absolute levels of cellular pri-miR-96 were measured by RT-qPCR and compared to the amount of 2 in cells. These studies showed that a mole of 2 cleaves, on average, 3.1 mol of pri-miR-96 (Table S2). The observed turnover agrees well with previous studies of in vitro catalysis of PROTACs.14 Thus, cleavage occurs catalytically and sub- stoichiometrically with targeted recruitment in cells.
In summary, we described the development of a system to endow small molecules with the ability to affiect RNA lifetime by recruiting endogenous ribonucleases (RIBOTACs), induc- ing their cleavage akin to antisense and CRISPR. The ability to custom recruit nucleases is likely to broaden the view of RNA as viable small molecule targets, and parallels can be made to the activities in leveraging PROTACs as chemical probes and lead medicines. Further endeavors will include broadening the nucleases that can be recruited and also to medicinally optimize the recruitment moiety.
Supplementary Material
Acknowledgments
We thank H. Park of the X-ray Crystallography Core at Scripps Florida for Pymol modeling and J. Childs-Disney for helpful conversations and editing the paper. We thank R. H. Silverman (Cleveland Clinic) for kindly providing the pGEX-4T-RNaseL and pcDNA3-RNaseL plasmids. This work was funded by the Scheller Graduate Student Fellowship to M.G.C. and the National Institutes of Health (5R01GM097455) to M.D.D.
Footnotes
Supporting Information:
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b01233.
Supplementary tables and figures, methods for chemical synthesis, supplementary methods, RNA cleavage and binding assays, in vitro selectivity studies and controls, cell uptake, cell line efficacy, immunoprecipitation and siRNA qPCR controls, compound stability, and Caspase 3/7 assays (PDF)
ORCID
Matthew D. Disney: 0000-0001-8486-1796
Notes
The authors declare no competing financial interest.
References
- 1.The Encode Project Consortium. Nature. 2012;489:57. [Google Scholar]
- 2.Mandal M, Breaker RR. Nat Rev Mol. Cell Biol. 2004;5:451. doi: 10.1038/nrm1403. [DOI] [PubMed] [Google Scholar]
- 3.Cech TR, Steitz JA. Cell. 2014;157:77. doi: 10.1016/j.cell.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 4.Moore MJ. Science. 2005;309:1514. doi: 10.1126/science.1111443. [DOI] [PubMed] [Google Scholar]
- 5.Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Proc Natl Acad Sci U S A. 2001;98:8554. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, Bradner JE. Science. 2015;348:1376. doi: 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Silverman RH. J Virol. 2007;81:12720. doi: 10.1128/JVI.01471-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Torrence PF, Xiao W, Li G, Cramer H, Player MR, Silverman RH. Antisense Nucleic Acid Drug Dev. 1997;7:203. doi: 10.1089/oli.1.1997.7.203. [DOI] [PubMed] [Google Scholar]
- 9.Velagapudi SP, Cameron MD, Haga CL, Rosenberg LH, Lafitte M, Duckett DR, Phinney DG, Disney MD. Proc Natl Acad Sci U S A. 2016;113:5898. doi: 10.1073/pnas.1523975113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Floyd-Smith G, Slattery E, Lengyel P. Science. 1981;212:1030. doi: 10.1126/science.6165080. [DOI] [PubMed] [Google Scholar]
- 11.Al-Ahmadi W, al-Haj L, Al-Mohanna FA, Silverman RH, Khabar KSA. Oncogene. 2009;28:1782. doi: 10.1038/onc.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin H, Dai T, Xiong H, Zhao X, Chen X, Yu C, Li J, Wang X, Song L. PLoS One. 2010;5:e15797. doi: 10.1371/journal.pone.0015797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fu Z, Tindall DJ. Oncogene. 2008;27:2312. doi: 10.1038/onc.2008.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bondeson DP, Mares A, Smith IED, Ko E, Campos S, Miah AH, Mulholland KE, Routly N, Buckley DL, Gustafson JL, Zinn N, Grandi P, Shimamura S, Bergamini G, Faelth-Savitski M, Bantscheff M, Cox C, Gordon DA, Willard RR, Flanagan JJ, Casillas LN, Votta BJ, den Besten W, Famm K, Kruidenier L, Carter PS, Harling JD, Churcher I, Crews CM. Nat Chem Biol. 2015;11:611. doi: 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.