

Abstract
Tumorigenicity is the capability of cancer cells to form a tumor mass. A widely used approach to determine if the cells are tumorigenic is by injecting immunodeficient mice subcutaneously with cancer cells and measuring the tumor mass after it becomes visible and palpable. Orthotopic injections of cancer cells aim to introduce the xenograft in the microenvironment that most closely resembles the tissue of origin of the tumor being studied. Brain cancer research requires intracranial injection of cancer cells to allow the tumor formation and analysis in the unique microenvironment of the brain. The in vivo imaging of intracranial xenografts monitors instantaneously the tumor mass of orthotopically engrafted mice. Here we report the use of fluorescence molecular tomography (FMT) of brain tumor xenografts. The cancer cells are first transduced with near infrared fluorescent proteins and then injected in the brain of immunocompromised mice. The animals are then scanned to obtain quantitative information about the tumor mass over an extended period of time. Cell pre-labeling allows for cost effective, reproducible, and reliable quantification of the tumor burden within each mouse. We eliminated the need for injecting imaging substrates, and thus reduced the stress on the animals. A limitation of this approach is represented by the inability to detect very small masses; however, it has better resolution for larger masses than other techniques. It can be applied to evaluate the efficacy of a drug treatment or genetic alterations of glioma cell lines and patient-derived samples.
Keywords: Cancer Research, Issue 134, Fluorescence molecular tomography (FMT), in vivo imaging, tumor heterogeneity, glioblastoma, intracranial orthotopic xenograft, optical imaging
Introduction
Cancer is one of the leading causes of illness-related deaths in humans in the industrialized world. With an extremely high death toll, new treatments are urgently required. Glioblastoma multiforme (GBM) is an extremely lethal type of brain cancer, composed of heterogeneous populations of brain tumor, stromal, and immune cells. According to the Central Brain tumor registry of the USA, the incidence of primary malignant and non-malignant brain tumors is approximately 22 cases per 100,000. Approximately 11,000 new cases are expected to be diagnosed in the USA in 20171.
Preclinical studies investigate the likelihood of a drug, procedure, or treatment to be effective prior to testing in humans. One of the earliest laboratory steps in preclinical studies is identifying potential molecular targets for drug treatment by using cancer cells implanted in a host organism, defined as human xenograft models. Within this context, intracranial brain tumor xenograft models using patient-derived xenografts (PDXs) have been widely used to study brain tumor biology, progression, evolution, and therapeutic response, and more recently for biomarkers development, drug screening, and personalized medicine2,3,4.
One of the most affordable and non-invasive in vivo imaging methods to monitor intracranial xenografts is bioluminescence imaging (BLI)5,6,7,8. However, some BLI limitations include substrate administration and availability, enzyme stability, and light quenching and scattering during imaging acquisition9. Here we report the infrared FMT as an alternative imaging method to monitor preclinical glioblastoma models. In this method, signal acquisition and quantification of intracranially implanted PDXs, expressing a near-infrared fluorescent protein iRFP72010,11 (henceforth termed as FP720) or turboFP635 (henceforth termed as FP635), is performed with a FMT imaging system. Using the FMT technology, the orthotopic tumors can be monitored in vivo before, during, or after treatment, in a non-invasive, substrate-free, and quantitative manner for preclinical observations.
Protocol
The use of experimental research animals and infectious agents, such as lentivirus to transduce the cancer cells, require prior approval by the institutional animal care program and by the institutional biosafety committee. This protocol follows the animal care guidelines of the University of California San Diego (UCSD).
1. Labeling of Glioblastoma Cells with FP635 or FP720 Construct
Produce and purify lentivirus according to the protocol described by Tiscornia et al.12
Culture approximately 2.0 x 106 cells from a human glioma cell line with DMEM plus 10% fetal bovine serum (FBS) in a 15-cm dish; or culture 2.0 x 106 human glioblastoma patient-derived (GBM-PDX) spheres with DMEM/F12 1:1 medium with B27 supplement plus human recombinant epidermal growth factor (EGF; 20 ng/mL) and FGF (10 ng/mL) in a T75 flask.
Maintain all cells at 37 °C, 5% CO2, and 100% relative humidity.
- Dissociate the cells.
- For the glioma cells: remove the supernatant from the plates and adding 3-5 mL of trypsin 1X to the glioma cells.
- For the GBM spheres: collect the cells by pipetting the cells into a 15-mL tube.
- Spin down the GBM spheres at 200 x g for 5 min using a tabletop centrifuge at room temperature.
- Remove the supernatant and add 3-5 mL of cell detachment solution.
- Incubate all cells at 37 °C for 10-15 min.
Carefully dissociate the cells by pipetting up and down several times (ensure that spheres and glioma cells are completed dissociated) and spin down at 200 x g for 5 min.
Remove the supernatant and resuspend the cells with 5 mL of medium by pipetting up and down several times. Determine the cell viability by trypan blue exclusion.
Plate 1.0 x 106 of target cells in a 10-cm dish with 5 mL of medium by pipetting.
Transduce cells with lentivirus expressing fluorescent proteins at multiplicity of infection (MOI) 5 as is described by Tiscornia et al.12 and incubate at 37 °C.
After 24 h, remove the medium from the transduced cells, add 5 mL of fresh medium, and incubate for additional 48 h at 37 °C.
Dissociate the infected cells into a single-cell suspension as described in steps 1.4-1.6. Spin down the cells at 200 x g for 5 min and resuspend in 500 µL of sorting solution (phosphate-buffered saline (PBS) with 1% FBS).
Pipette the cell suspension into FACS tubes. Co-stain the cells with 1 µL/mL of 4',6-diamidino-2-phenylindole (DAPI) dihydrochloride to exclude dead cells. Negative controls are required to set up the flow cytometer gates (non-transduced cells and non-transduced cells/ DAPI stained).
Sort the fluorescent-positive/DAPI-negative cells into sorting solution, as described by Basu et al.13, and collect the sorted cells into a 15-mL conical tube.
Spin down the sorted cells at 200 x g for 5 min, remove the supernatant, and resuspend in 5 mL of culture medium. Seed the sorted cells in a 10-cm dish by pipetting. Incubate at 37 °C for at least 48-72 h.
Expand the sorted cells by dissociating the cells following steps 1.4-1.6 and plating into multiple dishes for in vivo experiments. NOTE: For more details about cell sorting preparation and purification, see Basu et al.13
2. Intracranial Injection of iRFP-tagged Glioblastoma Cells into Immunodeficient Mice
NOTE: Before starting the surgery, make sure that the surgical room and tools are clean for the procedure. Use immunodeficient athymic nude (Foxn1nu) male or female mice, between 4-5 weeks old and 17-19 g for intracranial injections. Animals should be housed for at least 3 days after arrival and before surgery.
- Cell preparation
- Harvest and dissociate fluorescent positive-glioma cells into a single-cell suspension (steps 1.4-1.6) and resuspend 0.5 or 1.0 x 106 cells in 2-5 µL of PBS per injection per animal. Pipette the cell suspension into a 1.5-mL microcentrifuge tube and place on ice.
- Anesthesia preparation and administration
- Prepare 200 µL of saline solution containing ketamine (100 mg/kg) and xylazine (10 mg/kg) per 20 grams of mouse body weight. Weigh the animals and calculate the appropriate dose of anesthesia.
- Provide an intraperitoneal injection of anesthesia and apply ophthalmic ointment to the eyes to prevent dehydration. For more details about this step see Kathleen et al.14 Place the animal on a pre-warmed water thermal pad at 37 °C.
- Check that the animals are anesthetized by monitoring the toe pinch response (pedal reflex), usually within 5-10 min.
- Intracranial injection
- Load a 5 µL Hamilton syringe (blunt tip needle) with cell suspension and mount in the probe holder.
- Place the mouse in a small animal stereotactic frame and fix the head using the ear bars and the incisor adapter following the described details by Cetin et al.15
- Disinfect the head with 70% ethanol and betadine solution. Make sure that the surgical site is complete sterile. Perform a middle incision with a small scalpel and separate the skin and connective tissues.
- Place the Hamilton syringe on the bregma point using the micromanipulator.
- Move the probe holder 1.0 mm anteroposterior and 2.0 mm lateral from the bregma point. Mark this position with a pencil.
- At this location, carefully make a hole in the skull with a micromotor hand-held drill by applying slight pressure downward until the blood vessels become visible. Do not disrupt the arachnoid mater and brain parenchyma.
- Introduce the needle into the burr hole and advance 3.0 mm below the pial surface.
- Set up the volume (2-5 µL) and flow rate (1 µL/min) of injection using a motorized stereotaxic injector or manually perform the injection.
- After the injection, remove the needle gradually by ascending 1.0 mm every 1 min and clean the injection site with 70% ethanol.
- Close the skin wound with surgical staples or by making surgical sutures (often referred to as stitches) and see Kathleen et al.14 for more details.
- Animal recovery
3. FMT Imaging
NOTE: According to the experimental aim, the iRFP-tagged glioblastoma cells can be monitored in vivo before, during, or after treatment. For imaging purpose, anesthetize animals using an isoflurane induction chamber, maintain in an imaging cassette during the scanning, and image in the docking station of the FMT imager.
Remove the animal from the cage and place in the isoflurane induction chamber. Close the chamber and turn on oxygen flow and vaporizer to 3-5%.
Monitor the animal until it is completely anesthetized, usually within 1-2 min. Unconscious animals should not respond to external stimuli.
Remove the anesthetized animal from the chamber and put the animal in the imaging cassette by placing the head adaptor first, followed by placing the animal facing down. Make sure that the head is located in the center of the cassette.
Place the top plate of the cassette on top and tighten the adjustment knobs until 17 mm.
Once that the animal is secure in the imaging cassette, proceed to imaging by inserting the cassette into the internal docking station of the FMT imager, where the isoflurane is pumped to keep the animal anesthetized.
Run the imager and analyzer software by double-clicking the software icon.
In the "Experimental tab" window, select the "Database" and "Study group".
Go to the "Scan tab" window and select the subject to image by clicking "select subject". Also, select the laser channel in the "Laser channel" panel. For the FP635-labeled cells, select "laser channel 635 nm", and for FP720-labeled cells, select "laser channel 680 nm".
Acquire an image by clicking the "Capture" option in the "Scan tab" window. Then, adjust the scan field by clicking and dragging the scan field in the captured image to a determined number of source locations, usually within 20-25 sources for the ipsilateral side.
Check the "Add to reconstruction queue" option and hit "Scan" in the "Scan tab" window.
Wait until the scanning is completed and then remove the imaging cassette from the docking station.
Remove the top plate of the cassette and place the animal back into the cage.
Repeat the steps 3.1-3.12 for the rest of the animals.
4. Image Analysis
From the imager and analyzer software, select the "Analysis tab" window.
Load the dataset and the subject to analyze by clicking the "+" button in the "Dataset selection" panel of the "Analysis tab" window.
Once the image has been loaded in the "Analysis tab" window, perform the region of interest (ROI) analysis by selecting the ellipsoid icon, located in the upper left corner of the "Analysis tab" window.
Adjust the threshold to zero in the "statistic data" panel by right clicking the "threshold" column in the "Analysis tab" window.
The total value in the "Statistic data" panel represents the signal from the fluorescent-tagged glioblastoma cells implanted in the mouse brain.
Repeat steps 4.2-4.4 for the rest of the animals. Load or remove a new subject by clicking the "+" or "−" button, respectively, in the "Dataset selection" panel of the "Analysis tab" window. NOTE: For more information about the FMT imaging and software operation, see20.
Representative Results
Glioblastoma cells U87EGFRvIII (U87 cells over-expressing the EGF receptor variant III) were cultured according to the step 1.2. Lentivirus was produced and purified according to step 1.1. The viral concentration was determined by p24 ELISA analysis. Cells were transduced with lentivirus carrying infrared fluorescent proteins according to step 1.8. The plasmid encoding FP72010,11 was kindly provided by Dr. V.V. Verkhusha and the FP635 vector was purchased from a commercial vendor. Target cells were FACS sorted to enrich for the top 10% most fluorescent cell population (Figure 1A). Increasing the fluorescent signal improves the sensitivity of the fluorescence tomography system. To validate the intensity of the signals of fluorescently-labeled cells prior to orthotopic implantation, we used the phantom cassette of the FMT imager. Specifically, U87EGFRvIII-TurboFP635 and U87EGFRvIII-iRFP720 cells were dissociated with trypsin, and 1 x 105, 1 x 106, and 1 x 107 cells were resuspended in 100 µL of ice cold PBS, respectively and analyzed. The FMT imager allows for detection in 4 channels: 635 nm, 680 nm, 750 nm, and 780 nm. Far infrared channels 750 nm and 780 nm are not suitable for this analysis because there is no emission at such wavelengths from the near-infrared fluorescent proteins used (data not shown). Therefore, we analyzed the emission signals at 635 nm and 680 nm. Reported in Figure 1, the fluorescent signal image and intensity quantification for both FP635 (Figure 1B, C) and FP720 (Figure 1D, E), which increases proportionally as the cell number increases. Of note, the emission for FP635 is visible in the 635 nm channel but not in the 680 nm channel. In contrast, the emission for FP720 is visible in the 680 nm channel but not in the 635 nm channel. This makes these two candidate fluorescent proteins suitable for studies where two populations of cells can be labeled and monitored simultaneously using two near infrared fluorescent labels.

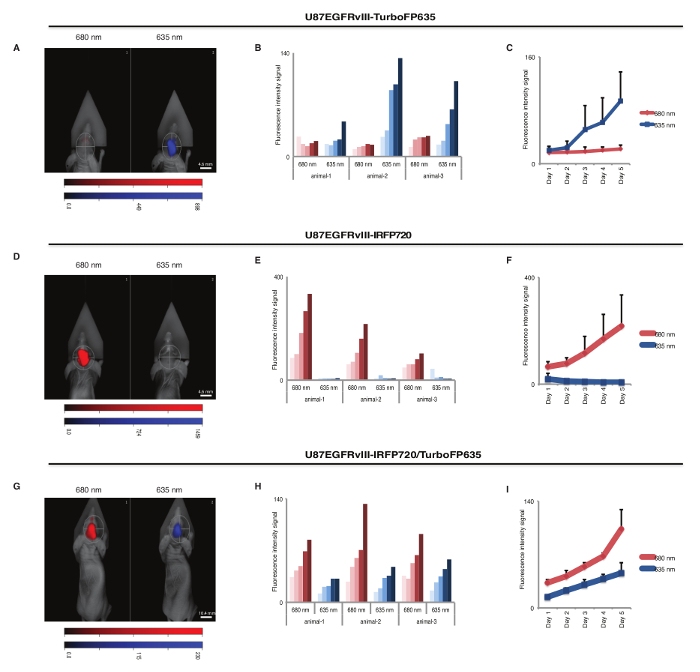
U87EGFRvIII cells labeled with near infrared fluorescent proteins were then used for orthotopic injection and in vivo tumor growth monitoring, as described in steps 2.1-2.4. We used the same cell line, i.e., U87EGFRvIII, labeled with different fluorescent tags, to exclude tumor growth difference between different cancer cell types. The mice were injected with U87EGFRvIII-TurboFP635 cells, U87EGFRvIII-iRFP720 cells, or an equal combination (1:1) of the two cell lines. A total of 5 x 105 cells in a 5 µL volume of PBS were injected intracranially into 4-5 weeks old immunocompromised athymic nude mice using a stereotactic system and a Hamilton syringe, following steps 2.3-2.4. Anesthesia was accomplished by intraperitoneal injection of ketamine/xylazine. The animals were kept warm throughout the injection using a water thermal pad and checked for any signs of distress. Tumor engraftment and growth was then monitored by the FMT imaging system. During the scanning sessions, the mice were temporarily anesthetized using a 3% isoflurane chamber (steps 3.1-3.12). The mice were then placed in an imaging cassette and imaged inside the laser scanning camera. Each mouse was scanned using the 635 nm and 680 nm channels. The signal quantification was recorded over time and mice were euthanized at the onset of neurological symptoms, according to the Institutional Animal Care and Use Committee (IACUC) guidelines. As indicated in Figure 2A, U87EGFRvIII-TurboFP635 emission signal was evident in the 635 nm channel and minimal background signal was recorded in the 680 nm channel (Figures 2B, C). In contrast, U87EGFRvIII-iRFP720 cells emitted in the 680 nm channel showing little background in the 635 nm channel (Figures 2D-F). Finally, mice injected with a 1:1 mix of U87EGFRvIII-TurboFP635 cells and U87EGFRvIII-iRFP720 cells showed increased signal in both channels for the duration of the experiment (Figures 2G-I). These data show that it is possible to utilize infrared fluorescent proteins for dual in vivo imaging of cancer cell populations. The data also indicate that different fluorescent proteins have a different intensity in their signal emission. Here, FP720 signal intensity was superior to FP635, with increased sensitivity in detecting smaller populations of cancer cells in a non-invasive monitoring setting.
The effects of gene silencing of cancer-related genes on tumorigenicity in preclinical models can also be study with this protocol. For this aim, GBM-spheres (GSC23 and GSC11) were cultured (step 1.2) and transduced with FP720-lentivirus (step 1.8). Cells expressing FP720 were FACS sorted as described previously in steps 1.11-1.13. FP720-labeled cells were co-transduced with lentivirus-encoding shRNA targeting DAXX (shDaxx,) or shRNA control (shLuc) at MOI 516. Cells were cultured for 5 days, dissociated into a single cell suspension, and 1 x 106 cells were resuspended in 3 µL of PBS for intracranial injection. The efficacy of the shRNA targeting DAXX was confirmed by reverse transcription quantitative polymerase chain reaction (RT-qPCR) in GSC23 (Figure 3A) and GSC11 (Figure 3C). GBM-iRFP720/shRNA cells were stereotactically injected into the striatum of immunodeficient mice, according to steps 2.3-2.4. Animals were observed for neurological signs and the relative fluorescence signal of the xenografts were analyzed by the FMT imaging system, quantified using analysis software, according to steps 3.1-4.6. Representative images of FMT show a decrease fluorescence signal in mice engrafted with shDAXX/GBM-spheres in comparison with animals implanted with shControl (Figure 3B andFigure 2D) for both GBM-spheres models, confirming our previous finding16 that DAXX inhibition represses tumor growth in preclinical glioblastoma models.
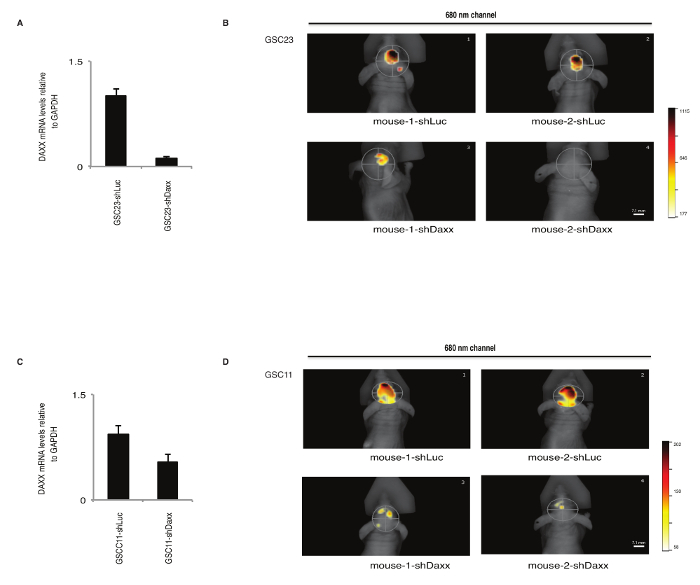
Figure 1: Standard curves using cells for phantom and FACS analysis. (A) Representative FACS histograms of FP720- and FP635-U87EGFRvIII expressing cells (left and right panel, respectively). Glioma cells were infected with lentivirus encoding infrared fluorescent proteins and FASC sorted to enrich for the highest expression of the FP720 or FP635 proteins. Representative images obtained from the imager and analyzer software of U87EGFRvIII-TurboFP635 (B) and U87EGFRvIII-iRFP720 (D) cell suspensions using the phantom cassette. Upper panels represent the signal in the 635 nm channel and bottom panels in the 680 nm channel. Increase in the cell number corresponded to an increase in signal visualization. Signal quantification of U87EGFRvIII-TurboFP635 (C) and U87EGFRvIII-iRFP720 (E) cells in 635 nm and 680 nm channels. Relative fluorescence units are indicated in the color bar. Error bars represent S.E.M. from 3 different scans per cell suspension. Please click here to view a larger version of this figure.
Figure 2: Brain tumor xenograft model using U87EGFRvIII cells scanned in different channels. Representative images of fluorescent signal in 635 nm and 680 nm channels from animals intracranially implanted with U87EGFRvIII-turboFP635 (A), U87EGFRvIII-iRFP720 (D), and a mix combination (1:1) of U87EGFRvIII-iRFP720:U87EGFRvIII-turboFP635 cells (G). Images were acquired at day 5 after first scanning. (B, E, H) Signal intensity monitoring for each mouse over a period of 5 days. Each mouse was scanned in the 635 nm channel (blue bars) and in the 635 nm channel (red bars). The lightest shade of the bar color represents day 1 of scanning and the darkest shade represents day 5. (C, F, I) Relative fluorescence quantification showing a direct comparison of signal intensity from the 635 nm channel (blue) and 680 nm channel (red) in the entire cohort of mice. Relative fluorescence units are indicated in the color bar. Data show the mean values with standard deviation from 3 different animals. Please click here to view a larger version of this figure.
Figure 3: Gene silencing in preclinical glioblastoma models using FMT. Gene expression analysis of DAXX by RT-qPCR in GSC23 (A) and GSC11 (C) glioma patient-derived cells transduced with lentivirus-encoded shRNAs targeting DAXX (shDaxx) or shRNA Control (shLuc) and FP720. Representative images of fluorescent signal from the 680 nm channel of orthotopically engrafted mice implanted with GSC23-iRFP720 (B) and GSC11-iRFP720 (D) patient-derived cells. Strong fluorescence signal is observed in animals implanted with shRNA Control (shLuc) (top part of the panel) in comparison with mice implanted with shRNA-DAXX (shDaxx) (bottom part of the panel), indicating that DAXX inhibition suppresses tumor growth in preclinical glioma models determined by fluorescence molecular tomography. Relative fluorescence units are indicated in the color bar. Please click here to view a larger version of this figure.
Discussion
Tumor xenografts have been extensively used in cancer research and a number of well-established imaging techniques has been developed: BLI; magnetic resonance imaging (MRI); positron emission tomography (PET), computed tomography (CT); FMT. Each of these approaches comes with pros and cons, but ultimately complement each other with the type of information provided. One of the most commonly used in vivo imaging technology is BLI5,6,7,8. BLI and FMT both require cell engineering (with luciferase enzymes or infrared fluorescent proteins) that can affect gene expression. BLI is more sensitive for small tumor masses but requires intraperitoneal injection of a substrate (luciferin), which reaches maximum distribution in the body in 10-12 min; but light quenching and scattering during imaging acquisition, and substrate clearance frequently occur9. FMT, instead, does not require any additional substrate administration to the animals and its signal is stable and independent of time or enzyme stability. Additionally, FMT, when used in the same experimental conditions, offers reproducible semi-quantitative data. BLI and FMT both do not offer spatial resolution; however, coupling these methods with CT scan addresses this issue.
A CT scan measures the attenuation of photons when its signal crosses the body of an animal and it is ideal in identifying body structures. However, soft tissue contrast, which might interfere with the detection of a small intracranial tumor, is a limitation for this approach17,18. One additional disadvantage of CT is the use of radiation and contrast media, which can induce molecular changes in the target cells. A CT scan can be combined with PET, which uses radiolabeled tracers like the glucose analogue, fluorodeoxyglucose 18F-FDG; but this can also interfere with some physiological processes in small animals. In contrast, FMT does not need any injected substrate to measure the tumor mass and it is possible to measure the tumor metabolism, inflammation, angiogenesis, apoptosis, and other markers using fluorescently labeled biomarkers19.
Overall one of the most useful and versatile techniques for in vivo imaging is the MRI. Minor limitations of the MRI can be found in the low acquisition time, lower sensitivity than PET, and some instrument-related variations18,20.
Here, we report the FMT method as an alternative method for preclinical glioblastoma models. Pre-labeling of target cells with infrared fluorescent proteins (Figure 1A-E) and scanning post-implantation in immunocompromised animals can be achieved with a fluorescence tomography imaging system. We reported the FMT method as an alternative method for preclinical glioblastoma models. With this method imaging is applied in a substrate-free and non-invasive manner, animals are kept in a low stress environment, and deep-tissue quantification can be performed (Figure 2A-I, Figure 3A, B). This protocol can be applied to study the efficacy of combinatorial treatments in preclinical glioma models21, to identify new therapeutic targets for brain tumors16, and to investigate new druggable metabolic molecules22, epigenetic pathways23, or new chemotherapeutic agents in combination with radiotherapy (unpublished data), and applied to other preclinical studies. We also propose here the use of the FMT as a dual in vivo imaging approach, when two co-existing, but different, tumor cell populations are meant to be analyzed.
Some limitations with FMT must be taken into consideration, such as auto-fluorescence generated from some organs that can interfere with the signal. For example, murine spleen and liver emit auto-fluorescence in the 680 nm channel but not at 635 nm. Therefore, FMT-related experimental procedures involving these organs must be performed using other infrared fluorescent proteins, like FP635. We used nude foxn1-mutated nude mice to avoid interference in the fluorescence signal recording; if a different mouse model must be used, it is recommended to shave the area of interest of the animals. Additionally, this approach is also limited in detecting very small tumor masses. In our experience, the signal-to-noise ratio improves as the tumor mass becomes larger, yet before the onset of any neurological symptom in the mice. In fact, in previous experiments conducted on orthotopically injected PDXs, we were able to detect tumors during a drug regimen for at least 2 weeks in mice without any sign of distress due to tumor burden or drug administration21. Ultimately, the aggressiveness of each cell line and the number of injected cells will determine the length of each experiment. The data show a linear proportional relationship between number of cells and signal (Figure 1B-E), and tumor mass and signal (Figure 2); but a general signal correlation between tumor mass and number of cells cannot be applied across different cell lines and infrared proteins. The signal for each cell line/infrared fluorescent protein combination must be determined empirically. Moreover, the signal obtained from the analysis of the cells using the phantom (Figure 1B, D) does not correspond to the signal from intracranial xenografts as an attenuation in signal occurs due to the skull of the mouse.
The imager and analyzer software allow for thresholding. Although we did not use any threshold here, scanning animals without any implanted cells gives background noise which tends to be eliminated by reconstruction in presence of a true signal. A few factors influence the signal intensity of the xenograft and must be kept in consideration: each infrared fluorescent protein has different emission spectrum and intensity, with FP720 being one of the most fluorescent proteins used experimentally10,11; each cell line allows for an abundant, yet limited, amount of protein production, thus influencing the maximum fluorescence obtainable. Finally, the FMT imager is setup with internal standard curves for specific compounds, therefore, it is important to consider which compound has the closest emission spectrum to the infrared fluorescent protein used to label the cells. Although well tolerated, it is recommended to determine potential cytotoxic effects mediated by the overexpression of the infrared proteins8,9. Moreover, we recommend using cohorts of mice of at least 8 per experimental group, as loss of data and mice, due to surgical procedure or variability in the cancer cell engraftment, might occur; nonetheless, this adds statistical significance to the study. We also recommend to serially scan the animal cohort for more than one day and analyze the signal emission at the same time, since variations in the imaging cassette depth and fluorescent signal reconstruction might interfere with the final signal quantification. To solve this issue, the analyzer software allows for minor changes in laser intensity (from normal to high and very high), although in our experience the final outcome is not dramatically different. One final note regarding cost-effectiveness is that the FMT imaging system can be easily shared as a core instrument among different laboratories, which helps defray its purchase and maintenance cost. In conclusion, the FMT is a valuable resource that makes experimental and preclinical evaluation of in vivo tumor growth easy and reliable.
Disclosures
The authors declare no conflicts of interests.
Acknowledgments
We thank Dr. Frederick Lang, MD Anderson Cancer Center for GBM-PDX neurospheres. This work was supported by the Defeat GBM Research Collaborative, a subsidiary of National Brain Tumor Society (Frank Furnari), R01-NS080939 (Frank Furnari), the James S. McDonnell Foundation (Frank Furnari); Jorge Benitez was supported by an award from the American Brain Tumor Association (ABTA); Ciro Zanca was partially supported by an American-Italian Cancer Foundation postdoctoral research fellowship. Frank Furnari receives salary and additional support from the Ludwig Institute for Cancer Research.
References
- Ostrom QT, et al. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2009-2013. Neuro-Oncology. 2016;18(suppl_5):v1–v75. doi: 10.1093/neuonc/now207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauli C, et al. Personalized In Vitro and In Vivo Cancer Models to Guide Precision Medicine. Cancer Discovery. 2017;7(5):462–477. doi: 10.1158/2159-8290.CD-16-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart EL, et al. Clinical Utility of Patient-Derived Xenografts to Determine Biomarkers of Prognosis and Map Resistance Pathways in EGFR-Mutant Lung Adenocarcinoma. Journal of Clinical Oncology. 2015;33(22):2472–2480. doi: 10.1200/JCO.2014.60.1492. [DOI] [PubMed] [Google Scholar]
- Gao H, et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med. 2015;21(11):1318–1325. doi: 10.1038/nm.3954. [DOI] [PubMed] [Google Scholar]
- Ozawa T, James CD. Establishing Intracranial Brain Tumor Xenografts With Subsequent Analysis of Tumor Growth and Response to Therapy using Bioluminescence Imaging. J. Vis. Exp. 2010. p. e1986. [DOI] [PMC free article] [PubMed]
- Kondo A, et al. An experimental brainstem tumor model using in vivo bioluminescence imaging in rat. Child's Nervous System. 2009;25(5):527–533. doi: 10.1007/s00381-008-0783-5. [DOI] [PubMed] [Google Scholar]
- Nyati S, Young G, Ross BD, Rehemtulla A. In: ATM Kinase: Methods and Protocols. Kozlov SV, editor. New York: Springer; 2017. pp. 97–111. [Google Scholar]
- Kondo A, et al. Longitudinal assessment of regional directed delivery in a rodent malignant glioma model. J Neurosurg Pediatr. 2009;4(6):592–598. doi: 10.3171/2009.7.PEDS09186. [DOI] [PubMed] [Google Scholar]
- Badr CE. In: Bioluminescent Imaging: Methods and Protocols. Badr CE, editor. Humana Press; 2014. pp. 1–18. [Google Scholar]
- Shcherbakova DM, Verkhusha VV. Near-infrared fluorescent proteins for multicolor in vivo imaging. Nat Meth. 2013;10(8):751–754. doi: 10.1038/nmeth.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filonov GS, et al. Bright and stable near-infrared fluorescent protein for in vivo imaging. Nat Biotech. 2011;29(8):757–761. doi: 10.1038/nbt.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiscornia G, Singer O, Verma IM. Production and purification of lentiviral vectors. Nat. Protocols. 2006;1(1):241–245. doi: 10.1038/nprot.2006.37. [DOI] [PubMed] [Google Scholar]
- Basu S, Campbell HM, Dittel BN, Ray A. Purification of Specific Cell Population by Fluorescence Activated Cell Sorting (FACS) J. Vis. Exp. 2010. p. e1546. [DOI] [PMC free article] [PubMed]
- Pritchett-Corning KR, Luo Y, Mulder GB, White WJ. Principles of rodent surgery for the new surgeon. J Vis Exp. 2011. [DOI] [PMC free article] [PubMed]
- Cetin A, Komai S, Eliava M, Seeburg PH, Osten P. Stereotaxic gene delivery in the rodent brain. Nat. Protocols. 2007;1(6):3166–3173. doi: 10.1038/nprot.2006.450. [DOI] [PubMed] [Google Scholar]
- Benitez JA, et al. PTEN regulates glioblastoma oncogenesis through chromatin-associated complexes of DAXX and histone H3.3. Nature Communications. 2017;8:15223. doi: 10.1038/ncomms15223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner S, et al. Imaging of Orthotopic Glioblastoma Xenografts in Mice Using a Clinical CT Scanner: Comparison with Micro-CT and Histology. PLOS ONE. 2016;11(11):e0165994. doi: 10.1371/journal.pone.0165994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannheim JG, et al. Standardization of Small Animal Imaging-Current Status and Future Prospects. Molecular Imaging and Biology. 2017. [DOI] [PubMed]
- Engblom C, et al. Osteoblasts remotely supply lung tumors with cancer-promoting SiglecFhigh neutrophils. Science. 2017;358(6367) doi: 10.1126/science.aal5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauber DT, et al. State of the art in vivo imaging techniques for laboratory animals. Laboratory Animals. 2017;51(5):465–478. doi: 10.1177/0023677217695852. [DOI] [PubMed] [Google Scholar]
- Zanca C, et al. Glioblastoma cellular cross-talk converges on NF-κB to attenuate EGFR inhibitor sensitivity. Genes & Development. 2017;31(12):1212–1227. doi: 10.1101/gad.300079.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa GR, et al. An LXR-Cholesterol Axis Creates a Metabolic Co-Dependency for Brain Cancers. Cancer Cell. 2016;30(5):683–693. doi: 10.1016/j.ccell.2016.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, et al. EGFR Mutation Promotes Glioblastoma through Epigenome and Transcription Factor Network Remodeling. Molecular Cell. 2015;60(2):307–318. doi: 10.1016/j.molcel.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]