

Abstract
There is a great need for treatment that arrests progression of chronic kidney disease. Increased albumin in primary urine leads to apoptosis and fibrosis of podocytes and tubular cells and is a major cause of functional deterioration. There have been many attempts to target fibrosis but few to target apoptosis, because of lack of appropriate agents. Our group has described an ouabain activated Na,K-ATPase/IP3R signalosome, which protects from apoptosis. Here we show that albumin uptake in primary rat renal epithelial cells is accompanied by a time and dose dependent mitochondrial accumulation of the apoptotic factor Bax, down-regulation of the anti-apoptotic factor Bcl-xL and mitochondrial membrane depolarization. Ouabain opposes these effects and protects from apoptoss in albumin-exposed proximal tubule cells and podocytes. The efficacy of ouabain as an anti-apoptotic and kidney-protective therapeutic tool is tested in rats with passive Heymann nephritis, a model of proteinuric chronic kidney disease. Chronic ouabain treatment preserves renal function, protects from renal cortical apoptosis, up-regulation of Bax, down-regulation of Bcl-xL and rescues from glomerular tubular disconnection and podocyte loss. Thus we have identified a novel clinically feasible therapeutic tool, which has the potential to protect from apoptosis and rescue from loss of functional tissue in chronic proteinuric kidney disease.
Keywords: chronic kidney disease, apoptosis, albuminuria, cell signaling, proximal tubule, podocyte, ouabain, sodium potassium ATPase, a-tubular glomeruli
Introduction
Chronic kidney disease (CKD) is a rapidly increasing world-wide public health problem1. CKD results from many causes, including diabetes, glomerulonephritis, hypoxia, hypertension, infections and polycystic kidney disease. Most forms of CKD are progressive1–3 and characterized by disrupted glomerular perm-selectivity, albuminuria, loss of podocytes, interstitial fibrosis and glomerular-tubular disconnection4–9. Albuminuria, a well-documented predictor of progressive loss of kidney function, is considered a cause of kidney damage and loss of function10–13. The nature of albumin toxicity has been extensively studied in the past decade, and it is well recognized that prolonged exposure of renal tubular cells to albumin results in apoptosis and fibrosis14–18, but their interrelationship is not yet fully understood. There are several ongoing trials aimed at halting progression of CKD using drugs targeted to inhibit pro-fibrotic and/or stimulate anti-fibrotic molecular pathways19–22 but there are few attempts to target the apoptotic process, mainly because of lack of non-toxic agents.
Apoptosis is triggered either via an extrinsic pathway stimulated by activation of plasma membrane death receptors or via an intrinsic mitochondrial pathway. The intrinsic apoptotic pathway is controlled by the family of Bcl-2 proteins. The mitochondrial apoptotic pathway is initiated by activation of Bax, a prominent pro-apoptotic member of the Bcl-2 family, to the outer mitochondrial membrane, where it oligomerizes and penetrates the inner mitochondrial membrane. This results in release of cytochrome C and caspase activation, the apoptotic executors. Bcl-xL, a prominent anti-apoptotic member of the Bcl-2 family, counteracts Bax accumulation on the mitochondria and Bax-induced permeabilization of the mitochondrial membranes.23,24
Our group has identified an anti-apoptotic signal activated by the cardiotonic steroid ouabain, which involves interaction between Na, K-ATPase and the inositol 1,4,5-triphosphate receptor (IP3R) and triggering of slow intracellular calcium oscillations. We have shown that the ouabain signal may interfere with the apoptotic process by down-regulation of the apoptotic factor Bax and up-regulation of the anti-apoptotic factor Bcl-xl. 25–30 Studies from us and other investigators have provided evidence for a tissue protective effect of ouabain30–36.
The aim of this study has been to test the hypothesis that activation of the ouabain signal can, via down-regulation of Bax and up-regulation of Bcl-xL rescue from the onset of albumin-triggered apoptotic process and thereby halt the progression of CKD. If this would be the case, ouabain may be a good candidate for a clinically feasible anti-apoptotic drug. Proximal tubular cells (PTC) are the main target for the toxic effects of albumin overload17, and loss of early PTC will result in glomerular—tubular disconnection, and irreversible renal damage5,7.
To assess at which stage ouabain interferes with the apoptotic process, Bax recruitment to the mitochondria, changes of the mitochondrial membrane potential and cellular abundance and localization of Bcl-xL were sequentially studied in a homogenous preparation of primary rat PTC (RPTC). Podocytes, which constitute a well-recognized locus minoris resistentiae in CKD37,38 were also examined with regard to their apoptotic response to albumin and the rescuing effect of ouabain. To obtain the first proof of principle that ouabain may protect from apoptosis and progressive renal damage in proteinuric CKD, we used a well-established rat model of human proteinuric kidney disease, passive Heymann nephritis (PHN)39,40.
Results
Albumin uptake into primary renal cells triggers apoptosis followed by increased expression of TGF-beta 1. Protective effect of ouabain
It is well documented that excessive uptake of albumin into RPTC triggers apoptosis30,41–43 and generation of pro-fibrotic factors, such as TGF-beta44. To determine the order by which these processes are initiated, we incubated RPTC with fatty acid and endotoxin-free albumin (10 mg/mL) for 2, 4, 8 and 18 hours. The level of apoptosis triggered by albumin was determined with TUNEL staining45 and expression of the pro-fibrotic TGF-β1 precursor with immunoblotting (Figure 1A-D). Apoptotic index (AI) was significantly increased after 2 hours of albumin incubation, and continued to increase during the following 16 hours. In contrast, expression of the TGF-beta1 precursor was not increased until after 8 hours of albumin incubation.
Figure 1. Albumin uptake into primary renal cells triggers apoptosis followed by increased expression of TGF-beta 1. Protective effect of ouabain.
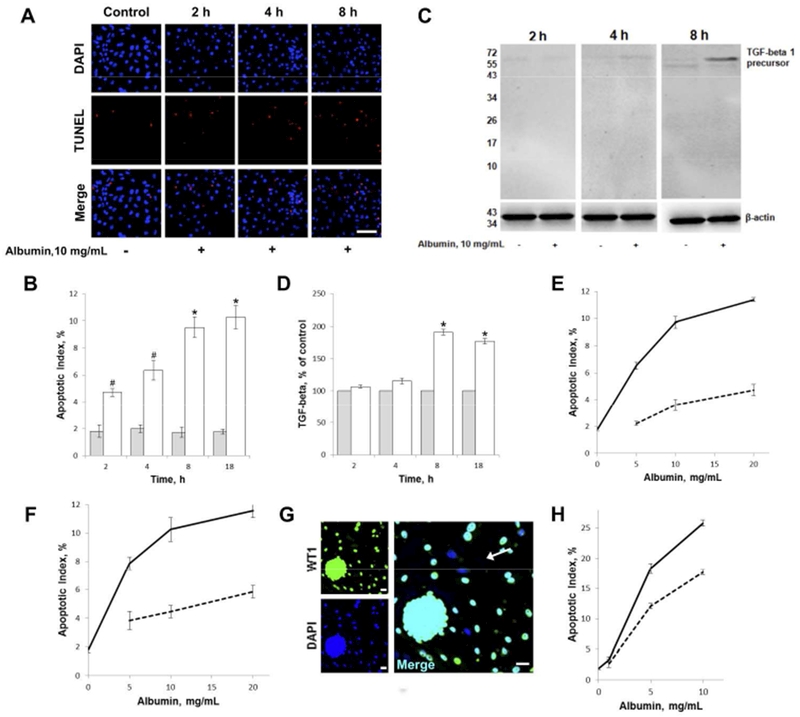
A) Albumin-induced apoptosis in RPTC incubated with 10 mg/mL albumin for 2, 4 or 8 hours and vehicle control. RPTC were TUNEL-stained (red) to detect apoptotic cells and counterstained with DAPI (blue). Scale bar for all images = 40μm.
B) Quantification of albumin-induced apoptosis in RPTC treated with 10 mg/mL albumin (open bars) or vehicle (grey bars) for 2, 4, 8 or 18 hours. Histograms show mean ± SEM. Experiments were repeated four times. The AI in vehicle-treated (control) cells was stable at 2% for the duration of the 18 hours. AI for 10mg/mL albumin was significantly increased vs control *p<0.001, #p<0.01.
C) Albumin-induced TGF-beta 1 expression in RPTC detected by immunoblot. RPTC were incubated with 10 mg/ml albumin in serum-free medium for 2, 4 or 8 hours.
D) Quantification of albumin-induced TGF-beta 1 expression in RPTC incubated with 10 mg/mL albumin (open bars) or vehicle (grey bars) for 2, 4, 8 or 18 hours. Experiments were repeated four times and vehicle control for each time point was set to 100%. Histograms show mean ± SEM, *p<0.001 vs control.
E, F) Dose dependence of albumin-induced apoptosis in RPTC. RPTC were incubated with 0 (vehicle control), 5, 10 or 20 mg/mL albumin alone (solid line), and in combination with 5 nM ouabain (dashed line) for 8 hours in E) or 18 hours in F). Plots represent mean ± SEM. Experiments were repeated four times. p<0.001 for 5, 10 and 20 mg/mL albumin vs vehicle control (0 mg/mL), at both 8 and 18 hours. p<0.01 for 5nM ouabain with 5, 10 and 20 mg/mL albumin vs the corresponding albumin treatment alone, at both 8 and 18 hours.
G) Primary culture of podocytes; images show isolated rat glomeruli plated and cultured for three days. Staining with the podocyte specific transcriptional factor WT1 shown in green and DAPI stained nuclei shown in blue. Podocytes that migrate out from the glomerulus were used for the apoptosis study shown in H). Note that a subset of cells (indicated by arrow) are not WT1 positive and therefore not included in the analysis. All scale bars = 20μm.
H) Dose dependence of albumin-induced apoptosis in podocytes. Podocytes were incubated with 0 (vehicle control), 1, 5 or 10 mg/ml albumin alone (solid line), and in combination with 5 nM ouabain (dashed line) for 18 hours. Only cells positive for the podocyte marker WT1 was included in the analysis. Plots represent mean ± SEM. Experiments were repeated four times. p<0.001 for 5 and 10 mg/mL albumin vs vehicle control (0 mg/mL), p<0.01 for ouabain treatment together with 5 and 10 mg/mL albumin vs the corresponding albumin treatment alone.
The apoptotic effect of albumin was dose-dependent and co-incubation with ouabain (5nM) resulted in a robust reduction of AI with all tested albumin concentrations (5, 10 or 20 mg/mL for 8 or 18 hours; Figure 1E and F respectively). The expression of TGF-beta precursor in RPTC exposed to albumin for 8 or 18 hours was also attenuated by ouabain (5nM) (Supplemental Figure (FigS1B). Ouabain did not interfere with albumin uptake in RPTC (FigS1C).
To test whether albumin might trigger apoptosis of primary rat podocytes, isolated glomeruli were plated, cultured for three days, and stained with the podocyte specific transcriptional factor WT146. AI was determined in cells that had migrated out from the glomerulus and were WT1 positive (Figure 1G). Incubation with albumin for 18 hours triggered apoptosis in adose-dependent manner. Co-incubation with ouabain resulted in significant reduction of AI (Figure 1H).
Ouabain interferes with the albumin triggered intrinsic apoptotic pathway
In RPTC exposed to albumin (10 mg/ml) for 8 or 18 hours, Bcl-xL abundance was decreased and the Bax abundance was increased as measured by immunoblotting (Figure 2B-C). The abundance of cleaved caspase-3 was increased, indicating that the apoptotic process was reaching the point of no return. Ouabain (5nM) partially rescued from Bcl-xL down-regulation, Bax up-regulation and increase of cleaved caspase-3.
Figure 2. Ouabain intervenes with the intrinsic apoptotic pathway triggered by albumin, but not with LPS stimulated cytokine expression.
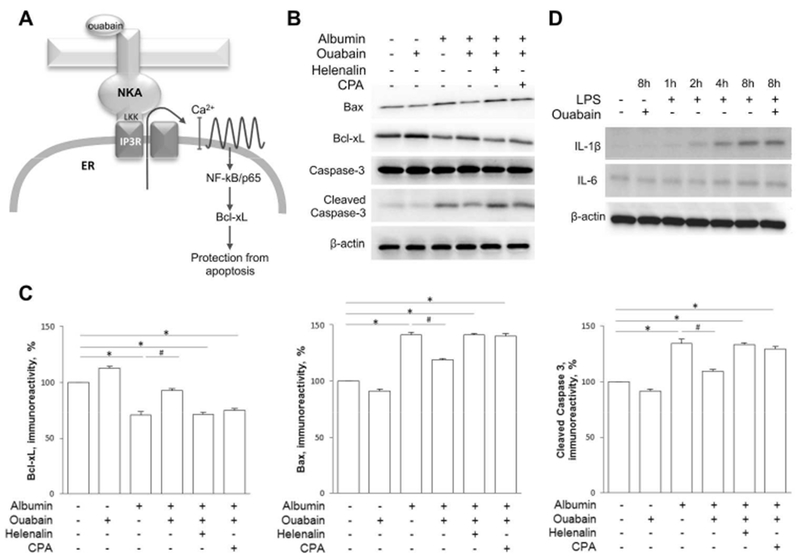
A) Cartoon illustrating the ouabain/Na,K-ATPase/IP3R signaling pathway. Ouabain binds to Na/K-ATPase which triggers interaction between the Na/K-ATPase and the IP3R through the amino acid residues LKK in the N-terminus of the catalytic α subunit of the Na/K-ATPase. The interaction activates the IP3R causing calcium to release from the ER. The slow calcium oscillations promote activation the NF-κB p65 subunit which leads to protection from apoptosis through stimulation of the anti-apoptotic protein Bcl-xL.
B) Western blot showing expression of Bax, Bcl-xL, caspase-3 and cleaved caspase-3 in RPTC after 8 hours incubation with 10 mg/mL albumin, 5 nM ouabain, 1 μM Helenalin and 1μM CPA as indicated.
C) Densitometric quantification of Bcl-xL (left), Bax (center) and cleaved caspase-3 (right) after 8 hours incubation with 10 mg/mL albumin, 5 nM ouabain, 1 μM Helenalin and 1μM CPA as indicated. The density of the band from control cells was set to 100%. Histograms show the mean ± SEM. Experiments were repeated five times. *p<0.001, #p<0.01.
D) Expression of the inflammatory cytokines, IL-1β and IL-6, after incubation with LPS 1 μg/mL for 0, 1, 2, 4 or 8 hours, and for 8 hours together with 5 nM ouabain as indicated.
The ouabain signaling pathway includes activation of the IP3R, release of calcium from endoplasmatic reticulum (ER) via the IP3R and activation of the prosurvival NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) p65 subunit (Fig 2A)25,27,47. When cells co-incubated with albumin and ouabain were depleted of calcium from ER stores by SERCA pump inhibition with cyclopiazonic acid (CPA) or co-incubated with helenalin, a specific NF-κB p65 subunit inhibitor, the rescuing effects of ouabain were abolished (Figure 2B-C). Ouabain had little effect on Bax and Bcl-xL expression in control cells. The extrinsic apoptotic pathway can be triggered by incubation with lipopolysaccharide (LPS). Ouabain (5nM) did not protect against LPS-triggered cytokine release (Figure 2D, FigS2).
Ouabain protects from albumin triggered mitochondrial dysfunction
To further characterize the involvement of mitochondria in albumin toxicity, time-sequence studies were performed with regard to mitochondrial membrane potential, Bax co-localization with mitochondria and Bcl-xL abundance (Figure 3A-C). Albumin (2.5 mg/mL) caused time-dependent depolarization of the mitochondrial membrane (Figure 3G). Co-localization between Bax and mitochondria in albumin-exposed cells increased in a time-dependent manner and the increase was significant after two hours (Figure 3G). The intensity of the fluorescent signal from Bcl-xL in albumin incubated RPTC decreased in a time-dependent manner. Changes were significant after two hours incubation with albumin (Figure 3G). Albumin had no effect on co-localization between Bcl-xL and mitochondria (FigS3A). In RPTC co-incubated with albumin (2.5mg/mL) and ouabain (5nM) for 8 hours, the effects on mitochondrial accumulation of Bax, mitochondrial membrane potential and Bcl-xL abundance were greatly attenuated (Figure 3D-F). Incubation of RPTC with 10 mg/mL albumin resulted in a more pronounced effect (FigS3B-E) and was seen after 30 minutes (FigS4A-C).
Figure 3. Ouabain protects from albumin triggered mitochondrial dysfunction.
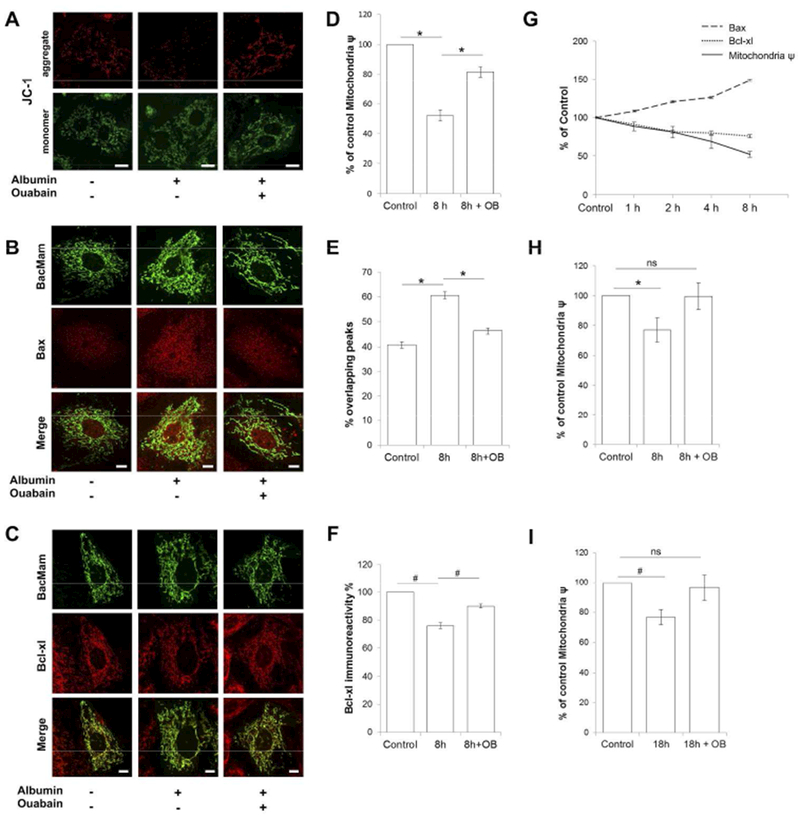
A) Changes in mitochondrial membrane potential in RPTC incubated with vehicle, 2.5 mg/ml albumin or 2.5 mg/ml albumin and 5nM of ouabain for 8h, visualized using JC-1 dye. Aggregation of JC-1 indicate high-Δψ mitochondria in red fluorescence. Monomeric dye indicate low-Δψ mitochondria in green fluorescence. Mitochondrial depolarization is shown as a decrease in the red/green fluorescence intensity ratio. All scale bars = 10μm.
B) Localization of Bax to mitochondria in RPTC incubated with vehicle, 2.5 mg/ml albumin or 2.5 mg/ml albumin and 5nM of ouabain for 8h. Representative confocal images of Bax immunofluorescence staining shown in red and mitochondria in green, by using the mitochondrial marker BacMam 2.0. All scale bars = 5μm.
C) Bcl-xL expression in RPTC incubated with vehicle, 2.5 mg/ml albumin or 2.5 mg/ml albumin and 5nM of ouabain for 8h. Representative confocal images of Bcl-xL immunofluorescence signal in red and mitochondria in green, by using the mitochondrial marker BacMam 2.0. All scale bars = 5μm.
D) Quantification mitochondrial membrane potential change in RPTC incubated with vehicle (control) or 2.5 mg/ml albumin in the presence and absence of 5nM ouabain for 8h. *p<0.001. Data are shown as % of control, mean ± SEM. Experiments were repeated three times.
E) Quantification of Bax localization to mitochondria in RPTC incubated with vehicle (control) or 2.5 mg/ml albumin in the presence and absence of 5nM ouabain for 8h. Co-localization was assessed along two perpendicular line traces across the nucleus. Overlap of mitochondria and Bax fluorescence signal peaks along the lines were analyzed. Peaks were considered to overlap if spaced by no more than 140 nm. *p<0.001. Data are shown as % overlapping Bax/mitochondrial peaks, mean ± SEM. Experiments were repeated four times.
F) Quantification of Bcl-xL expression in RPTC incubated with vehicle (control) or 2.5 mg/ml albumin in the presence and absence of 5nM ouabain for 8h. #p<0.01. All data are shown as % Bcl-xL immune reactivity when control was set to 100%, mean ± SEM. Experiments were repeated four times.
G) Plot shows time dependent change as % of control for mitochondrial membrane potential (solid line), localization of Bax to mitochondria (dashed line) and Bcl-xL expression (dotted line) in response to 2.5mg/ml albumin.
H, I) Quantification of mitochondrial membrane potential change in RPTC incubated with vehicle (control) or 0.2 mg/ml albumin in presence and absence of 5nM of ouabain for 8 hours H) and 18 hours I). Data are shown as % of control, mean ± SEM #p<0.05. Experiments were repeated three times.
For all experiments Mann-Whitney U test was used to determine whether differences were statistically significant.
To study whether albumin concentrations in the range of those reported in primary filtrate in proteinuric disease48 can cause mitochondrial dysfunction, cells were incubated with 0.2 mg/mL albumin. Significant reduction of mitochondrial membrane potential was observed at both 8 and 18 hours. In cells co-incubated with ouabain (5nM) the mitochondrial membrane potential was maintained at control levels (Figure 3H-I).
Chronic ouabain treatment protects from apoptosis in rats with PHN
To assess the nephroprotective effect of ouabain in vivo, rats were induced with PHN, a well-studied model of chronic proteinuric kidney disease with a relatively slow progressive course39,40. PHN rats were treated with ouabain (15 μg/kg/day) or vehicle, and followed for four months. Control rats were treated with vehicle. Treatment with this concentration of ouabain does not affect heart rate (FigS5A) and arterial pressure (FigS5B). Significant albuminuria was present two weeks after PHN induction and was lower in ouabain–treated than in vehicle–treated PHN rats from the fourth week of observation. At the end of the follow-up period serum creatinine was significantly higher in vehicle-treated than in ouabain-treated PHN rats (FigS5C).
At time of sacrifice kidneys were prepared for immunohistochemistry and morphometric analysis. Analysis of apoptosis and expression of Bax and Bcl-xL was performed in the outer cortex where the vast majority of tubular cells are PTC49 as seen in FigS6. To assess the level of apoptosis we quantified the number of cells with a condensed (pyknotic) nucleus, a marker of apoptosis that correlate well with TUNEL staining (FigS7). Vehicle-treated PHN animals had a significantly larger number of condensed nuclei than control animals. The number of condensed nuclei in ouabain-treated PHN animals was not different from that of controls (Figure 4A). Bcl-xL and Bax expression was visualized by immunohistochemistry and semi-quantitative evaluation of the fluorescence signal was performed. Renal cortical expression of Bcl-xL and Bax was detected in all groups studied (Figure 4B-C). The Bax immunofluorescence signal was significantly higher and the Bcl-xL signal was significantly lower in sections from vehicle-treated PHN rats than in sections from control rats. In contrast, neither the Bax nor the Bcl-xL immunofluorescence signal was significantly different between ouabain-treated PHN rats and control rats.
Figure 4. Long-term treatment with ouabain attenuates apoptosis of renal cortical cells in the PHN rat.
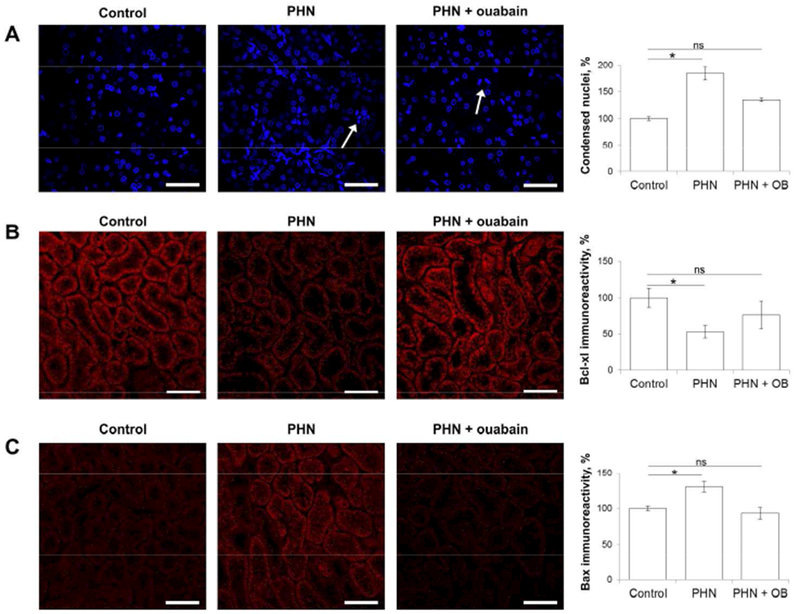
A) Representative DAPI staining of nuclei in renal cortex of control rats, PHN rats and ouabain-treated PHN rats at four months after PHN induction. The arrows indicate typical condensed nuclei. For quantification control was set to 100%. All scale bars = 40μm.
B) Representative immunostaining for Bcl-xL in renal cortex of control rats, PHN rats and ouabain-treated PHN rats at four months after PHN induction. For semi-quantitative evaluation of Bcl-xL immunoreactivity signal, control was set to 100%. All scale bars = 40μm.
C) Representative immunostaining for Bax in renal cortex of control rats, PHN rats and ouabain-treated PHN rats at four months after PHN induction. For semi-quantitative evaluation of Bax immunoreactivity signal, control was set to 100%. All scale bars = 40μm.
For all experiments, analysis was done in two sections from each kidney and in five (condensed nuclei) or six (Bcl-xl and Bax) randomly selected areas of the outer cortex. Histograms show the mean ± SEM. Statistical analysis was performed using ANOVA followed by t-test. *p<0.05
Chronic ouabain treatment protects from glomerular-tubular disconnection in PHN rats
Early PTC are most exposed to increase in filtered albumin. The number of apoptotic cells at the level of the glomerular-tubular junction was 3-fold higher in kidneys from vehicle-treated than in kidneys from ouabain-treated PHN rats (Figure 5A). Apoptotic cells in the early PTC were rarely observed in control rats (data not shown).
Figure 5. Kidneys from ouabain-treated PHN rats have fewer disconnected proximal tubules than kidneys from vehicle-treated PHN rats.
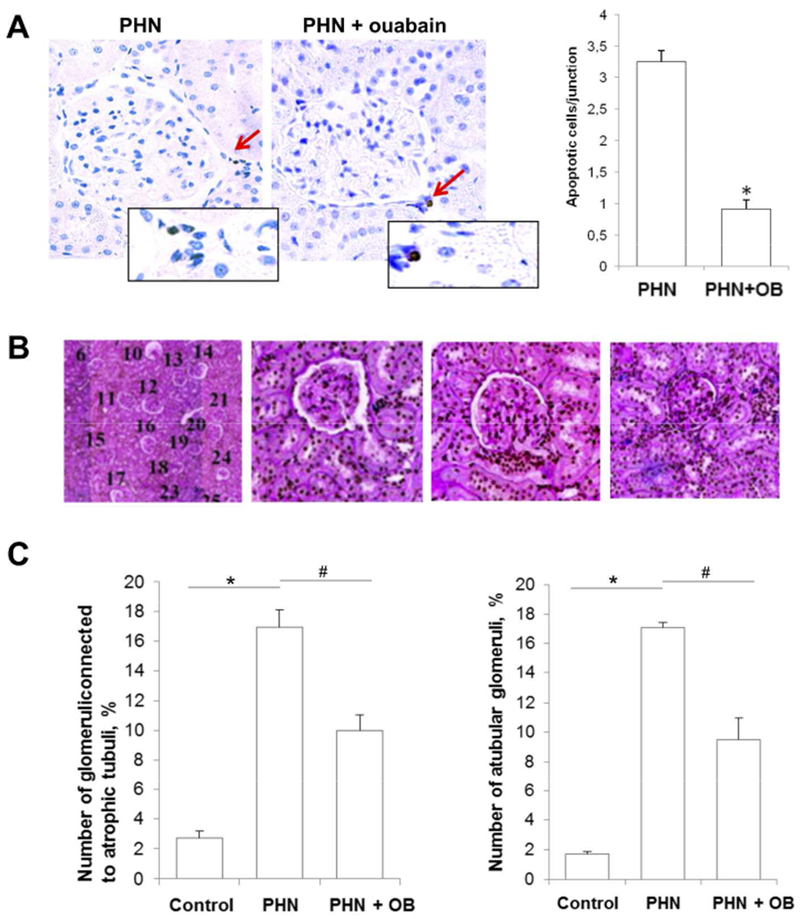
A) Representative TUNEL staining of early proximal tubules from vehicle- and ouabain-treated PHN rats at four months after PHN induction. Control rats rarely display any positive TUNEL stain in this region. Histograms show quantitative determination of apoptotic PTC, shown as mean ± SEM. *p<0.01.
B) Typical PAS staining of slices for morphometric studies, evaluating the glomerular-tubular connections in individual glomeruli. Pictures illustrate the pattern of glomerular-tubular connections, each glomerulus in the slide is counted (left) and evaluated as connected to a normal proximal tubule (middle left), to an atrophic proximal tubule (middle right), or without a tubular connection, i.e a-tubular glomeruli (right).
C) Summary of morphometric studies. Quantitative determination of glomeruli connected to an atrophic proximal tubule (left) and a-tubular glomeruli (right). Histograms represent mean ± SEM. *p<0.001, #p<0.01
For all experiments Mann-Whitney U test was used to determine significant differences.
Apoptosis and atrophy of early PTC may result in glomerular-tubular disconnection7. The frequency of existing and ongoing glomerular-tubular disconnection was studied with morphometric analysis in each animal using on average 75 serial sections (3–4 μm thickness). The number of a-tubular glomeruli and atrophic tubuli were assessed as described by Benigini et al.50. The incidence of a-tubular glomeruli and glomeruli connected to atrophic tubuli was low in control rats. We found a large increase in both a-tubular glomeruli (16%) and glomeruli connected to atrophic tubules (17%) in vehicle-treated PHN rats. The number of a-tubular glomeruli and glomeruli connected to atrophic tubules was significantly less pronounced in ouabain treated PHN rats (8.5% and 11% respectively) (Figure 5B-C).
Chronic ouabain treatment protects from loss of podocytes in PHN rats
Loss of podocytes is generally considered a sign of permanent renal damage38,51–54. Kidney sections were stained for the podocyte marker WT1. The AI of WT1 positive cells was increased in both vehicle and ouabain-treated PHN rats, but the increase was significantly less pronounced in ouabain-treated PHN rats (Figure 6A). There was a large reduction of WT1 positive cells per glomerulus in vehicle-treated PHN rats compared to control rats. Significantly more WT1 positive cells were preserved in ouabain-treated PHN animals (Figure 6B).
Figure 6. Kidneys from ouabain-treated PHN rats have more preserved WT1 positive glomerular cells than kidneys from vehicle-treated PHN rats.
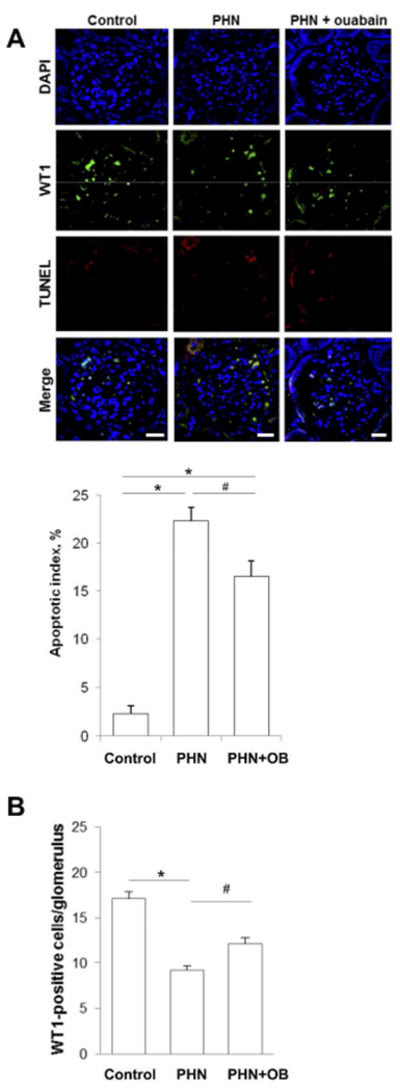
A) Representative micrographs showing TUNEL stain in WT1 positive glomerular cells from control rats, vehicle-treated and ouabain-treated PHN rats. Sections were TUNEL-stained (red) to detect apoptotic cells and stained for WT1 (green) to detect podocytes, DAPI stained nuclei are shown in blue. All scale bars = 20μm. Histogram shows quantification of apoptotic podocytes, for control rats, PHN rats and ouabain-treated PHN rats. Histograms show mean ± SEM.
B) Histogram shows quantification of WT1 positive cells per glomerulus for control rats, PHN rats and ouabain-treated PHN rats. Histograms show mean ± SEM.
For all experiments Mann-Whitney U test was used to determine significant differences *p<0.01, #p<0.05
Kidney fibrosis and glomerular collagen accumulation is less pronounced in ouabain-treated than in vehicle-treated PHN rats
Fibrosis is a typical feature of CKD14,16. We found up-regulation of the signal from the profibrotic factor TGF-beta 1 in vehicle-treated PHN rats compared to control. TGF-beta 1 up-regulation was not observed in ouabain-treated PHN rats (FigS8B). Glomerular collagen IV accumulation was observed in both vehicle and ouabain-treated PHN rats, but was more pronounced in vehicle-treated PHN rats (FigS8C). Kidneys from adult rats have a regenerative capacity, which may be preserved in CKD55,56. Vehicle-treated PHN rats displayed more proliferating cells, detected by markers Ki-67 and PCNA, than control rats. The number of proliferating cells was significantly lower in ouabain-treated than in vehicle-treated PHN rats (FigS8A).
Discussion
The pathogenesis of CKD is multifactorial. Here we confirm the observation from many previous studies that apoptosis contributes to the progressive course of the disease and demonstrate that the progression can be slowed down by ouabain, a compound that acts by interfering with the early phase of the apoptotic process.
There is a great need to develop novel approaches to halt the progression of CKD and specifically target the various factors that contribute to the disease process. Although many studies have pointed to the importance of apoptosis in CKD, there is as yet no anti-apoptotic drug available. Caspase inhibitors have been tested but have failed to reach the market57. Caspase inhibitors interfere with a late stage in the apoptotic pathway and may therefore prevent death of cells where DNA damage has already occurred.
To test if ouabain protects from the early phase of apoptosis, before the process is irreversible, we focused this study on the effect of ouabain on two important members of the Bcl family, the apoptotic protein Bax, and the anti-apoptotic protein Bcl-xL. Bax exists in equilibrium between cytosolic and mitochondria-associated forms, and shifts toward the latter when activated by a stress stimulus to induce cell death. Activated Bax accumulates on mitochondria, oligomerizes and permeabilizes the mitochondrial outer membrane. This results in release of cytochrome c, and marks the point of no return in the apoptotic process. Bcl-xl prevents Bax recruitment to the mitochondria and its oligomerization and permeabilization of the outer mitochondrial membrane. During the course of the apoptotic process Bax expression increases and Bcl-xl expression decreases. Here we demonstrated that ouabain decreases Bax and increases Bcl-xl expression and that ouabain prevents mitochondrial Bax accumulation and depolarization of the outer mitochondrial membrane in primary PTC challenged with an excessive load of albumin. These findings indicate that ouabain protects against the onset of albumin-triggered apoptosis. A cartoon illustrating the ouabain effect on the apoptotic pathway is shown in FigS9.
The molecular mechanism by which ouabain exerts its early antiapoptotic effect remains to be elucidated, but are likely dependent on a signaling pathway involving calcium release from the IP3R. It is possible that ouabain can exert both an acute and a chronic effect. In a previous study27 we have presented evidence that ouabain may exert a long-term anti-apoptotic effect by activating the Nf-κB factor p65, a transcriptional regulator of Bcl-xl58. Our observation that the Bcl-xl abundance is down-regulated in albumin triggered cells, maintained in cells treated with ouabain but not in cells treated with ouabain and helenalin, an inhibitor of the transcriptional effects of NF-κB, suggests that this may be the case.
Kidney sections from rats with untreated PHN showed signs of ongoing proximal tubular apoptosis, as indicated by increased Bax and decreased Bcl-xL abundance as well as elevated number of apoptotic cells. In contrast a comparison between slices from ouabain treated PHN rats and control rats did not reveal significant differences with regard to these parameters. A study of the proximal tubules adjacent to the glomeruli showed significantly increased number of apoptotic cells in kidneys from both untreated and ouabain treated PHN rats as compared to controls. However, the increase was much more pronounced in untreated than in treated kidneys from PHN rats. Massive apoptosis of early PTC leads to glomerular-tubular disconnection. Oliver first described the occurrence of a-tubular glomeruli in CKD in 193759. A-tubular glomeruli have more recently been reported in human transplanted kidneys, kidneys from rats with PHN, and in mouse models for obstructive kidney disease and polycystic kidney disease49,60–62. Yet glomerular-tubular disconnection has remained an under-estimated cause of irreversible loss of renal function in CKD. Notably, the loss of functional nephrons observed in the untreated PHN rats, correlated well to the increase in serum creatinine (Supplemental table 1). Renal tubular cells have a relatively high regenerative capacity55,56. The number of newly formed epithelial cells was increased in kidneys from both vehicle and ouabain-treated PHN rats, but the increase was less pronounced in kidneys from ouabain-treated rats. This regenerative capacity is likely of importance for the preservation of tubular function, but not sufficient to repair a well-advanced or complete glomerular-tubular disconnection.
Excessive exposure to albumin did also trigger apoptosis in primary rat podocytes. Podocytes have low regenerative capacity and the importance of podocyte damage for the progressive course of CKD is well documented. Ouabain protected from podocyte apoptosis both in vitro and in vivo. The podocyte AI was higher and there were fewer WT1 positive cells per glomeruli in kidneys from untreated than in ouabain treated PHN rats. PHN is induced by injection of anti-Fx1A antibody, which mainly targets podocyte foot processes. We cannot exclude that complement dependent cell death may to some extent contribute to podocytes loss. Cell death due to immune reaction is however mainly mediated via the extrinsic apoptotic pathway63,64 which does not involve Bax and Bcl-xl proteins to the same extent as the mitochondrial apoptotic pathway. It is therefore unlikely that ouabain would have interfered with this pathway.
Tubulointerstitial fibrosis is generally considered as the final common pathway of the majority of progressive CKD65,66. Apoptosis is regularly observed in fibrotic tissue, and mounting evidence suggest that apoptosis contributes to the fibrotic process31,67–69. It was recently reported that expression of reticulon 1, a protein that induces apoptosis, attenuates the fibrotic process and the severity and progression of CKD in mice and humans70. In the albumin exposed PTC apoptosis preceded the increase in expression of the multifunctional protein TGFβ which triggers formation of extracellular matrix and fibrosis as well as apoptosis (likely via the extrinsic apoptotic pathway)71. Ouabain down-regulated TGFβ expression, both in the in vitro and in vivo study, suggesting that execution of apoptosis via the intrinsic mitochondrial pathway precedes the increased TGFβ expression. Taken together, available information indicates that a drug targeting the onset of the mitochondrial apoptotic pathway should be highly beneficial in CKD.
A large number of studies performed on rodents have suggested that albumin is a major contributor to the progressive course of CKD. Yet many patients with minimal change disease (MCD) or membranous glomerular nephritis can have stable nephrotic range proteinuria for many years without apparent loss of renal function. These observations are however not necessarily contradictory. Most patients with MCD are very young, and it is well documented that the regenerative capacity of renal tubular cells is age dependent and younger patients have a more favorable prognosis than older patients72,73. It is also important to consider that the progression of CKD to renal failure is generally a slow process3, that often takes more than a decade and that most patients are relatively asymptomatic as long as 50% of the renal functional capacity is preserved. Currently kidney biopsy specimens from humans are rarely examined for apoptosis and expression of pro-apoptotic proteins. More information about the incidence of apoptosis in human kidneys would likely increase the awareness that clinically relevant drugs targeting the apoptotic process before the point of no return would be beneficial.
In conclusion, this study highlights apoptosis as a cause of albumin toxicity and as a contributor to the progressive loss of functional renal tissue in CKD, and provides a basis for the development of the cardiotonic steroid ouabain as a complementary therapy in CKD.
Condensed Material and Methods
The complete material and methods including a list of antibodies used can be found in supplemental material.
Animals and Rat primary cultures
Forty-day-old male Sprague-Dawley rats were used for the PHN, model. For cell preparation twenty-day-old male Sprague-Dawley rats were used. All experiments were performed according to Karolinska Institutet regulations concerning care and use of laboratory animals, and approved by the Stockholm North ethical evaluation board for animal research.
Primary culture of rat proximal tubular cells (RPTC) was prepared as previously described74. Characterization RPTC show, on day 3 in culture 99% of the cells express SGLT-2 a marker for proximal tubular cells (FigS1A). Glomeruli isolation and podocytes culture were performed as previously described75. Albumin treatment of primary cultures was started on day two or three in vitro for all experiments, cells were exposed either vehicle (PBS) or fatty acid and endotoxin-free bovine albumin in the growth media for up to 18 hours, with or without presence of ouabain 5 nM.
Detection of apoptosis in primary culture
TUNEL assay by use of ApopTag Red In Situ Apoptosis Detection kit (Merk Millipore) was used according to the manufacturer’s instructions. Nuclei were counterstained with DAPI. For determination of AI in podocytes, podocytes were detected by anti-WT1 antibody.
Mitochondrial membrane potential determination
The integrity of the mitochondrial membrane potential was measured in RPTC using the JC-1 dye (Life Technologies). Mitochondrial depolarization is detected by a shift in fluorescence emission from red (~590 nm) to green (~527 nm). Cells were incubated with 2.5 μg/ml JC-1 dye in cultured medium for 15 min at 37°C and then analyzed. The mitochondrial membrane potential change was quantified by calculating the ratio of red to green fluorescence intensity.
Immunocytochemistry
RPTC and podocytes in primary culture were fixed in 4% Paraformaldehyde for 10 minutes, washed and treated with 0.3% Triton X-100 for 10 minutes. Then incubated with, 5% BSA, 0.1% Triton X-100, for 1 hour. Primary antibodies were applied overnight at 4°C. Following three washes, cells were incubated with secondary antibodies for 1 hour at room temperature. Cells were washed, and mounted for analysis.
Bax, Bcl-xL translocation assessment
RPTC on day two in vitro were labeled with CellLight® Mitochondria-GFP BacMam (Life Technologies) overnight. The following day, RPTC were incubated with albumin with or without ouabain or vehicle for up to 8 h. Following cells were immunostained for Bax or Bcl-xL and analysis of Bax/Bcl-xL translocation to the mitochondria was done. To assess co-localization between immune-labeled Bax/Bcl-xL and GFP expressing mitochondria, the method of Edlich el al. 201123 was used. Results are shown as fraction of Bax/Bcl-xL – mitochondria overlap to total number of mitochondria.
Animal model, passive Heymann nephritis
Animals were divided into control group and PHN group. PHN was induced by a single injection of rabbit anti-Fx1A antibody (a kind gift from Professor David J Salant, Boston University Medical Center), controls were given vehicle. On day 0 after PHN injection half the group was started on ouabain treatment (15 μg/mL/day) and the other half was given vehicle (sterile PBS) delivered by mini-pumps implanted subcutaneously. All animals were followed for 4 months. Spot urine samples were collected every second week and albuminuria was measured using Nephrat II (Exocell). At sacrifice blood samples were collected for serum creatinine determination and kidneys were removed from for histological and morphological studies.
Renal histology and morphometric analysis
Dissected kidneys were fixed in Dubosq-Brazil, dehydrated in alcohol and embedded in paraffin. Kidney sections 3–4 μm were stained with periodic acid-Schiff reagent (PAS), sections including superficial and juxtamedullary glomeruli were evaluated.
To assess glomerular-tubular disconnection, on average 75 serial sections (3–4 μm) were examined for each animal. For each group of animals, 300–320 glomeruli were examined. Only glomeruli located entirely within the serially sectioned tissue were included in the analysis. The glomeruli were classified as connected to a normal proximal tubule, connected to an atrophic proximal tubule, or without a tubular connection.
Detection of apoptosis in renal tissue
To detect apoptotic PTC kidney sections were deparaffinized, rehydrated and subjected to antigen retrieval, nuclei were stained with DAPI and sections mounted with Immu-Mount. All samples were stained under identical conditions and analyzed using identical microscopy settings. Cells were classified as being apoptotic if they had a condensed nucleus, where the nuclei was shrunk and had abnormal morphology with no visible interior.
To detect apoptotic podocytes, sections were consecutive immunostained using WT1 antibodies, a podocyte marker, followed by ApopTag Red In Situ Apoptosis Detection kit according to the manufacturer’s instructions.
To evaluate apoptotic PTC at the level of the glomerular-tubular junction, sections were stained with Peroxidase In Situ Apoptosis Detection kit (Merk Millipore) according to manufacturer’s instructions, and counterstained with Harris hematoxylin (Richard Allan Scientific). Sections were assessed and the number of apoptotic cells per glomerular-tubular junction was determined. In each slice, 25 to 30 randomly selected glomeruli were examined.
Statistical Analysis
Results are presented as mean ± SEM. To determine differences among groups, two-way ANOVA followed by Fisher-LSD post-hoc test was used. If the distribution of the variables was not parametric, the non-parametric Mann-Whitney test was used. Comparisons between groups were made using Kruskal-Wallis one-way ANOVA on ranks with pair-wise multiple comparisons made by Dunn’s method.
Supplementary Material
Acknowledgement
This study was supported by the Swedish Research Council and the Erling-Persson Family Foundation to AA and the Karolinska Insitute KIRT fellowship to EB. The authors wish to thank Doctor Tommy Linné, Karolinska Institutet for valuable advice and Kristoffer Bernhem, Karolinka Institutet for assistance with data analysis. Parts of this work have been presented at the 2014 ASN meeting. This work was also presented by AA as part of the 2015 Hugh Davson Distinguished Lectureship of the Cell and Molecular Physiology Section at the Experimental Biology meeting in Boston 2015.
Acknowledgment of support:
The Swedish Research Council and the Erling-Persson Family Foundation to AA
The Karolinska Insitute KIRT fellowship to EB.
Footnotes
Disclosures
The authors have nothing to disclose.
References
- 1.United States Renal Data System. USRDS 2013 Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States. National Institutes of Health. National Institute of Diabetes and Digestive and Kidney Diseases; Bethesda, MD, 2013. [Google Scholar]
- 2.Anderson S, Halter JB, Hazzard WR, et al. High KP and for the workshop participants. Prediction, progression, and outcomes of chronic kidney disease in older adults. J Am Soc Nephrol 2009; 20:1199–1209. [DOI] [PubMed] [Google Scholar]
- 3.Inker LA, Lambers Heerspink HJ, Mondal H, et al. GFR decline as an alternative end point to kidney failure in clinical trials: a meta-analysis of treatment effects from 37 randomized trials. Am J Kidney Dis 2014; 64(6):848–859. [DOI] [PubMed] [Google Scholar]
- 4.Haraldsson B, Nyström J, Deen WM. Properties of the glomerular barrier and mechanisms of proteinuria. Physiol Rev 2008; 88:451–487. [DOI] [PubMed] [Google Scholar]
- 5.Remuzzi G, Perico N, Macia M, et al. The role of renin-angiotensin-aldosterone system in the progression of chronic kidney disease. Kidney Int Suppl 2005; 99:57–65. [DOI] [PubMed] [Google Scholar]
- 6.Chevalier RL, Forbes MS. Generation and evolution of atubular glomeruli in the progression of renal disorders. J Am Soc Nephrol 2008; 19:197–206. [DOI] [PubMed] [Google Scholar]
- 7.Lindop GB, Gibson IW, Downie TT, et al. The glomerulo-tubular junction: a target in renal diseases. J Pathol 2002; 197:1–3. [DOI] [PubMed] [Google Scholar]
- 8.Fukuda A, Wickman LT, Venkatareddy M, et al. Progression is caused by angiotensin II-dependent persistent podocytes loss from destabilized glomeruli. Kidney Int 2012; 81:40–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weening JJ, Ronco P, Remuzzi G. Advances in the pathology of glomerular diseases. Contrib Nephrol 2013; 181:12–21. [DOI] [PubMed] [Google Scholar]
- 10.Tojo A, Kinugasa S. Mechanisms of glomerular albumin filtration and tubular reabsorption. Int J Nephrol 2012; 2012:481520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perkins BA, Ficociello LH, Ostrander BE, et al. Microalbuminuria and the risk for early progressive renal function decline in type 1 diabetes. J Am Soc Nephrol 2007; 18:1353–1361. [DOI] [PubMed] [Google Scholar]
- 12.Zoja C, Abbate M, Remuzzi G. Progression of renal injury toward interstitial inflammation and glomerular sclerosis is dependent on abnormal protein filtration. Nephrol Dial Transplant 2015; 30(5):706–712. [DOI] [PubMed] [Google Scholar]
- 13.Birn H, Christensen EI. Renal albumin absorption in physiology. Kidney Int 2006; 69(3):440–449. [DOI] [PubMed] [Google Scholar]
- 14.Brezniceanu M-L, Liu F, Wei C-C, et al. Attenuation of interstitial fibrosis and tubular apoptosis in db/db transgenic mice overexpressing catalase in renal proximal tubular cells. Diabetes 2008; 57:451–459. [DOI] [PubMed] [Google Scholar]
- 15.Sanz AB, Santamaría B, Ruiz-Ortega M, et al. Mechanisms of renal apoptosis in health and disease. J Am Soc Nephrol 2008; 19(9):1634–1642. [DOI] [PubMed] [Google Scholar]
- 16.Eddy AA. Molecular basis of renal fibrosis. Pediatr Nephrol 2000; 15:290–301. [DOI] [PubMed] [Google Scholar]
- 17.Erkan E, Devarajan P, Schwartz GJ. Mitochondria are the major targets in albumin-induced apoptosis in proximal tubule cells. J Am Soc Nephrol 2007; 18:1199–1208. [DOI] [PubMed] [Google Scholar]
- 18.Erkan E, Garcia CD, Patterson LT, et al. Induction of renal tubular cell apoptosis in focal segmental glomerulosclerosis: roles of proteinuria and Fas-dependent pathways. J Am Soc Nephrol 2005; 16:398–407. [DOI] [PubMed] [Google Scholar]
- 19.Jafar TH, Stark PC, Schmid CH, et al. The effect of angiotensin-converting-enzyme inhibitors on progression of advanced polycystic kidney disease. Kidney Int 2005; 67:265–271. [DOI] [PubMed] [Google Scholar]
- 20.Essien E, Goel N, Melamed ML. Role of vitamin D receptor activation in racial disparities in kidney disease outcomes. Semin Nephrol 2013; 33:416–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akhurst RJ, Hata A. Targeting the TGFβ signaling pathway in disease. Nat Rev Drug Discov 2012; 11:790–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Zeeuw D, Agarwal R, Amdahl M, et al. Selective vitamin D receptor activation with paricalcitol for reduction of albuminuria in patients with type 2 diabetes (VITAL study): a randomised controlled trial. Lancet 2010; 376:1543–1551. [DOI] [PubMed] [Google Scholar]
- 23.Edlich F, Banerjee S, Suzuki M, et al. Bcl-x(L) retrotranslocates Bax from the mitochondria into the cytosol. Cell 2011; 145(1):104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen H-C, Kanai M, Inoue-Yamauchi A, et al. An interconnected hierarchical model of cell death regulation by the BCL-2 family. Nat Cell Biol 2015; 17(10):1270–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miyakawa-Naito A, Uhlen P, Lal M, et al. Cell signaling microdomain with Na,K-ATPase and inositol 1,4,5-trisphosphate receptor generates calcium oscillations. J Biol Chem 2003; 27:50355–50361. [DOI] [PubMed] [Google Scholar]
- 26.Aizman O, Uhlen P, Lal M, et al. Ouabain, a steroid hormone that signals with slow calcium oscillations. Proc Natl Acad Sci USA 2001; 98:13420–13424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li J, Zelenin S, Aperia A, et al. Low doses of ouabain protect from serum deprivation-triggered apoptosis and stimulate kidney cell proliferation via activation of NF-κB. J Am Soc Nephrol 2006; 17:1848–1857. [DOI] [PubMed] [Google Scholar]
- 28.Fontana JM, Burlaka I, Khodus G, et al. Calcium oscillations triggered by cardiotonic steroids. FEBS J 2013; 280:5450–5455. [DOI] [PubMed] [Google Scholar]
- 29.Aperia A 2011 Homer Smith Award: To serve and protect: classic and novel roles for Na+, K+ -adenosine triphosphatase. J Am Soc Nephrol 2012; 23:1283–1290. [DOI] [PubMed] [Google Scholar]
- 30.Burlaka I, Liu XL, Rebetz J, et al. Ouabain protects from Shiga toxin-triggered apoptosis by reversing the imbalance between Bax and Bcl-xL. J Am Soc Nephrol 2013; 24:1413–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aperia AC, Akkuratov EE, Fontana JM, et al. Na+, K+-ATPase, a new class of plasma membrane receptors. Am J Physiol Cell Physiol 2016; doi: 10.1152/ajpcell.00359.2015. [DOI] [PubMed] [Google Scholar]
- 32.Dvela-Levitt M, Cohen-Ben Ami H, Rosen H, et al. Reduction in maternal circulating ouabain impair offspring growth and kidney development. J Am Soc Nephrol 2015; 26:1103–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jacobs BE, Liu Y, Pulina MV, et al. Normal pregnancy: mechanisms underlying the paradox of a ouabain-resistant state with elevated endogenous ouabain, suppressed arterial sodium calcium exchange, and low blood pressure. Am J Physiol Heart Circ Physiol 2012; 302:H1317–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li J, Khodus GR, Kruusmägi M, et al. Ouabain protects against adverse developmental programming of the kidney. Nat Commun 2010; 1(4):1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pasdois P, Quinlan CL, Rissa A, et al. Ouabain protects rat hearts against ischemia-reperfusion injury via pathway involving src kinase, mitoKATP, and ROS. Am J Physiol Heart Circ Physiol 2007; 292:H1470–1478. [DOI] [PubMed] [Google Scholar]
- 36.Dvela-Levitt M, Ami HC-B, Rosen H, et al. Ouabain improves functional recovery following traumatic brain injury. J Neurotrauma 2014; 31:1942–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peired A, Angelotti ML, Ronconi E, et al. Proteinuria impairs podocytes regeneration by sequestering retinoic acid. J Am Soc Nephrol 2013; 24(11):1756–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mundel P, Reiser J. Proteinuria: an enzymatic disease of the podocyte? Kidney Int 2010; 77(7):571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jefferson JA, Pippin JW, Shankland SJ. Experimental models of membranous nephropathy. Drug Discov Today Dis Models 2010; 7:27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cybulsky AV, Quigg RJ, Salant DJ. Experimental membranous nephropathy redux. Am J Physiol Renal Physiol 2005; 289(4):F660–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Amsellem S, Gburek J, Hamard G, et al. Cubulin is essential for albumin reabsorption in the renal proximal tubule. J Am Soc Nephrol 2010; 21:1859–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khan S, Abu Jawdeh BG, Goel M, et al. Lipotoxic disruption of NHE1 interaction with PI(4,5)P2 expedites proximal tubule apoptosis. J Clin Invest 2014; 124:1057–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lau GJ, Godin N, Maachi H, et al. Bcl-2-modifying factor induces renal proximal tubular cell apoptosis in diabetic mice. Diabetes 2012; 61(2):474–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dizin E, Hasler U, Nlandu-Khodo S, et al. Albuminuria induces a proinflammatory and profibrotic response in cortical collecting ducts via the 24p3 receptor. Am J Physiol Renal Physiol 2013; 305:1053–1063. [DOI] [PubMed] [Google Scholar]
- 45.Galluzzi L, Aaronson SA, Abrams J, et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ 2009; 16(8):1093–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Su J, Li SH, Chen ZH, et al. Evaluation of podocytes lesion in patients with diabetic nephropathy. Wilms’ tumor-1 protein used as a podocyte marker. Diabetes Res Clin Pract 2010; 87(2):167–175. [DOI] [PubMed] [Google Scholar]
- 47.Zhang S, Malmersjo S, Li J, et al. Distinct role of the N-terminal tail of the Na,K-ATPase catalytic subunit as a signal transducer. J Biol Chem 2006; 281:21954–21962. [DOI] [PubMed] [Google Scholar]
- 48.Oken DE, Flamenbaum W. Micropuncture studies of proximal tubule albumin concentrations in normal and nephrotic cells. J Clin Invest 1971; 50:1498–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Larsson SH, Larsson L, Lechene C, et al. Studies of terminal differentiation of electrolyte transport in the renal proximal tubule using short-term primary cultures. Pediatr Nephrol 1989; 3:363–368. [DOI] [PubMed] [Google Scholar]
- 50.Benigni A, Gagliardini E, Remuzzi A, et al. Angiotensin-converting enzyme inhibition prevents glomerular-tubule disconnection and atrophy in passive Heymann nephritis, an effect not observed with a calcium antagonist. Am J Pathol 2001; 159:1743–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chang AM, Ohse T, Krofft RD, et al. Albumin-induced apoptosis of glomerular parietal epithelial cells is modulated by extracellular signal-regulated kinase 1/2. Nephrol Dial Transplant 2012; 27:1330–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Okamura K, Dummer P, Kopp J, et al. Endocytosis of albumin by podocytes elicits an inflammatory response and induces apoptotic cell death. PLoS One 2013; 8(1):e5481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Matsusaka T, Kobayashi K, Kon V, et al. Glomerular sclerosis is prevented during urinary tract obstruction due to podocyte protection. Am J Physiol Renal Physiol 2011; 300:792–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wiggins RC. The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int 2007; 71:1205–1214. [DOI] [PubMed] [Google Scholar]
- 55.Kale S, Karihaloo A, Clark P, et al. Bone marrow stem cells contribute to repair of the ischemically injured renal tubule. J Clin Invest 2003; 112:42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Duffield JS, Park KM, Hsiao L-L, et al. Restoration of tubular epithelial cells during repair of the postischemic kidney occurs independently of bone marrow-derived stem cells. J Clin Invest 2005; 115:1743–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.MacKenzie SH, Schipper JL, Clark AC. The potential for caspases in drug discovery. Curr Opin Drug Discov Devel 2010; 13:568–576. [PMC free article] [PubMed] [Google Scholar]
- 58.del Nogal M, Luengo A, Olmos G, et al. Balance between apoptosis or survival induced by changes in extracellular matrix composition in human mesangial cells: a key role for ILK-NFκB pathway. Apoptosis 2012; 17(12):1261–1274. [DOI] [PubMed] [Google Scholar]
- 59.Oliver J The aglomerular nephrons of terminal Bright’s disease, in Architecture of the kidney in chronic Bright’s disease Paul B. Hoeber. New York NY, 1937, pp 43–57. [Google Scholar]
- 60.Gibson IW, Downie TT, More IAR, et al. Atubuli glomeruli and glomerular cysts – a possible pathway for nephron loss in the human kidney? J Pathol 1996; 179:421–426. [DOI] [PubMed] [Google Scholar]
- 61.Forbes MS, Thornhill BA, Galarreta CI, et al. Chronic unilateral obstruction in the neonatal mouse delays maturation of both kidneys and leads to late formation of atubular glomeruli. Am J Physiol Renal Physiol 2013; 305:F1736–F1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Galarreta CI, Grantham JJ, Forbes MS, et al. Tubular obstruction leads to progressive proximal tubular injury and atubular injury and atubular glomeruli in polycystic kidney disease. Am J Pathol 2014; 184:1957–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov 2006; 5(9):769–784. [DOI] [PubMed] [Google Scholar]
- 64.Czaja AJ. Targeting apoptosis in autoimmune hepatitis. Dig Dis Sci 2014; 59(12):2890–2904. [DOI] [PubMed] [Google Scholar]
- 65.Eddy AA. The origin of scar-forming kidney myofibroblasts. Nat Med 2013; 19(8):964–966. [DOI] [PubMed] [Google Scholar]
- 66.Papasotiriou M, Genovese F, Klinkhammer BM, et al. Serum and urine markers of collagen degradation reflect renal fibrosis in experimental kidney diseases. Nephrol Dial Transplant 2015; 30:1112–1121. [DOI] [PubMed] [Google Scholar]
- 67.Johnson A, Di Pietro LA. Apoptosis and angiogenesis: an evolving mechanism for fibrosis. FASEB J 2013; 27:3893–3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang K, Lin B, Brems JJ, et al. Hepatic apoptosis can modulate liver fibrosis through TIMP1 pathway. Apoptosis 2013; 18:566–577. [DOI] [PubMed] [Google Scholar]
- 69.Spender LC, Carter MJ, O’Brien DI, et al. Transforming growth factor-β directly induces p53-upregulated modulator of apoptosis (PUMA) during the rapid induction of apoptosis in Myc-driven B-cell lymphomas. J Biol Chem 2013; 288:5198–5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fan Y, Xiao W, Li Z, et al. RTN1 mediates progression of kidney disease by inducing ER stress. Nat Commun 2015; 6:7841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gentle ME, Shi S, Daehn I, et al. Epithelial cell TGFβ signaling induces acute tubular injury and interstitial inflammation. J Am Soc Nephrol 2013; 24(5):787–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Celsi G, Jakobsson B, Aperia A. Influence of age on compensatory renal growth in rats. Pediatr Res 1986; 20(4):347–350. [DOI] [PubMed] [Google Scholar]
- 73.Mendonça AC, Oliveira EA, Fróes BP, et al. A predictive model of progressive chronic kidney disease in idiopathic nephrotic syndrome. Pediatr Nephrol 2015; 30:2011–2020. [DOI] [PubMed] [Google Scholar]
- 74.Khan F, Spicarová Z, Zelenin S, et al. Negative reciprocity between angiotensin II type 1 and dopamine D1 receptors in rat renal proximal tubule cells. Am J Physiol Renal Physiol 2008; 295:F1110–1116. [DOI] [PubMed] [Google Scholar]
- 75.Lal MA, Andersson AC, Katayama K, et al. Rhophilin-1 is a key regulator of the podocytes cytoskeleton and is essential for glomerular filtration. J Am Soc Nephrol 2015; 26:647–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.