

Abstract
DNA replication timing is an important cellular characteristic, exhibiting significant relationships with chromatin structure, transcription, and DNA mutation rates. Changes in replication timing occur during development and in cancer, but the role replication timing plays in development and disease is not known. Zebrafish were recently established as an in vivo model system to study replication timing. Here is detailed the protocols for using the zebrafish to determine DNA replication timing. After sorting cells from embryos and adult zebrafish, high-resolution genome-wide DNA replication timing patterns can be constructed by determining changes in DNA copy number through analysis of next generation sequencing data. The zebrafish model system allows for evaluation of the replication timing changes that occur in vivo throughout development, and can also be used to assess changes in individual cell types, disease models, or mutant lines. These methods will enable studies investigating the mechanisms and determinants of replication timing establishment and maintenance during development, the role replication timing plays in mutations and tumorigenesis, and the effects of perturbing replication timing on development and disease.
Keywords: Genetics, Issue 134, DNA replication, replication timing, zebrafish, in vivo, development, transcription, chromatin, next generation sequencing, copy number variations, mutations, cancer, FACS
Introduction
For cells to successfully divide, they must first accurately and faithfully replicate their entire genome. Genome duplication occurs in a reproducible pattern, known as the DNA replication timing program1. DNA replication timing is correlated with chromatin organization, epigenetic marks, and gene expression2,3. Changes in replication timing occur throughout development, and are significantly related to transcriptional programs and alterations to chromatin marks and organization4,5. Furthermore, replication timing is correlated with mutational frequencies, and changes in timing are observed in various types of cancer6,7,8. Despite these observations, the mechanisms and determinants of replication timing establishment and regulation are still largely unknown, and the role it plays in development and disease is undetermined. In addition, until recently the genome-wide replication timing changes that occur throughout vertebrate development had only been examined in cell culture models.
Zebrafish, Danio rerio, are well suited to study replication timing in vivo during development, as a single mating pair can yield of hundreds of embryos that develop rapidly with many similarities to mammalian development9,10. Furthermore, throughout zebrafish development, there are changes to the cell cycle, chromatin organization, and transcriptional programs that share relationships with DNA replication timing11. Zebrafish are also an excellent genetic model, as they are particularly amenable to manipulation by transgenesis, mutagenesis, and targeted mutations, and genetic screens have identified many genes required for vertebrate development12. Therefore, zebrafish can be used to identify genes involved in replication timing establishment and maintenance and to observe the effects of deregulating replication timing on vertebrate development. Transgenic lines can also be used to assess replication timing from individual cell types isolated at different developmental timepoints or in disease conditions. Importantly, there are various zebrafish models of human disease that can be used to investigate the role of replication timing in disease formation and progression9,13,14.
Recently, the first replication timing profiles were generated from zebrafish, establishing it as a model system to study replication timing in vivo15. To accomplish this, cells were collected from zebrafish embryos at multiple stages of development and in a cell type isolated from adult zebrafish. Cells were then sorted by FACS (fluorescence-activated cell sorting) based on DNA content to isolate G1 and S phase populations. Using the G1 sample as a copy number control, copy number variations in S phase populations were determined and used to infer relative replication timing16. Changes in replication timing can then be directly compared between different developmental samples and cell types and this was used to determine changes in replication timing that occur in vivo throughout vertebrate development. This method offers several advantages over other genomic methods, chiefly that it does not require labeling with thymidine analogs or immunoprecipitation of DNA4,6.
Here is detailed the protocols to profile genome-wide DNA replication timing at high-resolution in zebrafish. These protocols have been used to determine relationships with genomic and epigenetic features in the zebrafish genome, as well as profiling changes in these relationships that occur throughout development. These protocols are also easily adapted to study changes in replication timing in mutant strains of zebrafish and in disease models. Additionally, these methods provide a foundation that can be expanded upon to study replication timing in specific cell types, by first sorting out the individual cell types from the zebrafish. The zebrafish can serve as an excellent in vivo model system to study replication timing and to ultimately reveal the biological functions of this important epigenetic trait.
Protocol
All animals were handled in strict accordance with protocols approved by the Oklahoma Medical Research Foundation Institutional Animal Care and Use Committee.
1. Setting up adult zebrafish for breeding
Use a large cohort of adult male and female zebrafish of a single strain for breeding. There are small differences in the genetic makeup of zebrafish of even a single strain, use a large cohort to ensure results are representative of the genetic variability of the population and not restricted to a small subset of the population.
- The night before embryos will be collected, place dozens of breeding adults into breeding tanks, use approximately equal numbers of males and females. Depending on the experiment, use one of the following breeding strategies.
- Breeding strategy 1: Place a few male and female zebrafish in individual breeding tanks, separate the males from the females with a divider. If this approach is used, setup many different breeding tanks and pool the embryos from each to ensure the biological variability is representative of the population.
- Breeding strategy 2: Combine dozens of male and female zebrafish in a large breeding tank. Use this strategy as long as there are a sufficiently large number of embryos, it is reasonable to assume they came from a variety of founders and are therefore genetically representative of the population.
2. Timed matings - collecting, sorting, and housing zebrafish embryos for experiments
Perform timed matings, beginning as soon as the light cycles being in the morning. Discard any embryos generated overnight.
Allow fish to breed in shallow water (3-5 cm) for a period of 10 min, with a false bottom for embryos to escape through.
After 10 min, collect embryos by pouring through a strainer and rinsing into a 10-cm plate with a wash bottle. Pool all embryos collected at the same time point, mark them with time of collection, and place immediately in an incubator at 28.5 °C.
Hundreds of embryos can be expected from a single mating pair in a day. Allow adults to breed in 10 min cycles until the number of embryos obtained is less than twenty.
- Approximately 1-1.5 h after collection, sort embryos to remove dead and unfertilized embryos. To sort embryos, remove from incubator and observe under a dissecting microscope.
- Count embryos and place at a density of 100 embryos per 10 cm plate.
- Add fresh E3 medium (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, 0.33 mM MgSO4) and return embryos to the 28.5 °C incubator.
3. Dechorionate, deyolk, and fix zebrafish embryos
NOTE: This section of the protocol is designed for embryos prior to 48 h post fertilization (hpf). There is no need to remove the chorions of embryos at later stages of development (after 48 hpf), as they often naturally fall off. There is no need to deyolk or remove the chorions of fish older the 5 days post fertilization (dpf).
When embryos reach desired developmental age, verify appropriate stage of development morphologically under a light microscope according to "Stages of Embryonic Development of the Zebrafish"17.
- Transfer up to 1500 staged embryos to a 15-mL conical tube and wash several times with E3.
- Add 1 mL of E3 and 20 µL of Pronase solution (30 mg/mL) for every 100 embryos and gently swirl. Up to 1500 embryos can be dechorionated in a single tube with 15 mL of E3.
- Lay tube on its side and arrange embryos in an even layer to ensure maximum surface to volume ratio. Gently agitate tube every 2-3 min until all chorions have fallen off of the embryos. The total dechorionation time will vary depending on the Pronase activity.
Remove supernatant and gently rinse embryos 3 times with 10 mL of E3. Place embryos on ice for the remainder of this section of the protocol.
- Remove E3 and wash embryos with freshly prepared ice-cold deyolking buffer (0.5x Ginzburg Fish Ringer's solution, without calcium: 55 mM NaCl, 1.8 mM KCl, 1.25 mM NaHCO3).
- Add 1 mL of ice-cold deyolking buffer for up to 500 embryos.
- Using a P1000, gently pipette embryos up and down 5-10 times to disrupt yolk sack.
- Transfer 1 mL to a 1.5 mL microfuge tube and centrifuge at 4 °C and 500 x g for 5 min.
Remove supernatant and wash pellet with 1 mL of ice-cold 1x PBS (phosphate buffered saline, pH 7.0) to remove Ringer's solution. Centrifuge at 4 °C and 500 x g for 5 min.
Carefully remove supernatant and resuspend cells in 1 mL of 1x PBS. Place tube on ice.
- Add 3 mL of ice-cold ethanol (EtOH) to a 15-mL conical tube. Vortex tube of EtOH gently on low setting.
- Using a P1000, slowly (1 drop per second) drip zebrafish embryo cell suspension into EtOH while vortexing gently.
- Add a total of 1 mL of the embryo cell suspension, for a final fixation solution of 75% EtOH.
After 1 mL of cell suspension has been added, gently swirl tube to mix and place at -20 °C for at least 1 h. It is recommended to place the tubes on their side to ensure maximum surface to volume ratio and reduce the number of embryos and cells that stick together. Store embryo cells at -20 °C for 2-3 weeks.
4. Staining DNA and FACS sorting embryos
NOTE: This section of the protocol is designed for embryos at 1 dpf.
- Remove EtOH-fixed embryo cells from -20 °C and centrifuge at 4 °C 1500 x g for 5 min.
- Place tube on ice and carefully remove supernatant. Using a P1000, gently resuspend cells in 1 mL of freshly prepared cold PBS-BSA (1x PBS, 1% bovine serum albumin).
- Centrifuge cells at 4 °C and 1500 x g for 5 min and place on ice.
- Remove supernatant and resuspend cells in propidium iodide (PI) staining solution (50 µg/mL PI, 100 µg/mL RNase A, PBS, pH 7.0), using 200 µL for every 1000 embryos.
- Allow cells to incubate in PI solution for 30 min on ice, gently mixing every 5 min.
- Place a 40 µm nylon cell strainer on a 50-mL conical tube on ice. Gently pipette cells to mix and filter through the mesh. NOTE: If multiple tubes of embryos from the same timepoint were collected in section 3 of the protocol, combine these embryos at this point so that they are all sorted together.
- Using the flat inside end of a syringe plunger, gently disrupt any remaining clumps of cells on the mesh.
- Rinse cells through the filter with 500 µL of cold PBS-BSA for every 1000 embryos.
- Place a 40-µm mesh over a 15-mL conical tube on ice and filter the cell suspension from the 50-mL conical tube a second time into the 15-mL tube.
- Using an appropriate cell sorting instrument, set the laser excitation to 561-nm and emission detection to 610/20 to detect propidium iodide.
- Mix 15 mL tube of stained cells briefly by vortexing and place in instrument at 4 °C.
- Run cells at a slow flow rate, ideally 1.0 mL/min, in order to obtain a clean cell cycle profile and avoid mixing G1 or G2/M cells with the S phase population.
- First, gate for FSC-A x SSC-A (forward scatter area by side scatter area) to remove debris (Figure 1A). NOTE: Cells from the embryos can be varying sizes depending on the stage and cell type. The neutral density (ND) filter may need to be switched between 1.0 and 1.5 depending on the size of cells present. Gate a large area to collect most cells and remove debris.
- Second, gate for SSC-area (SSC-A) by PI-area (PI-A), to remove debris and cell doublets (Figure 1B).
- Third, gate for SSC-A by SSC-W (width) to remove any remaining cell doublets (Figure 1C).
- Display the gated populations on a histogram with cell number (count) on the y-axis and propidium iodide area (PI-A) on the x-axis. For 24 hpf embryos, this should give a stereotypical cell cycle profile as in Figure 1D.
- To sort the S phase population, set the gate from the right side of the G1 peak to the left side of the G2 peak, including only a minimal amount of the G1 and G2 populations. For 24 hpf embryos, this will typically comprise about 10-15% of the gated population (see Figure 1D).
- To sort the G1 population, set the gate on the left side of the G1 peak, not to pass the top of the peak. Adjust the width of the G1 gate as narrow as possible (see Figure 1D).
- Back gate the G1 and S phase populations to ensure proper initial gating (Figure 1E).
Sort the G1 and S phase populations into 15 mL conical tubes with 3 mL of PBS-BSA. The goal should be to obtain between 500,000 and 1 million or more sorted cells per fraction (G1 and S phase).
After sorting is complete, centrifuge tubes at 1500 x g and 4 °C for 10 min. NOTE: The small cell pellet will be difficult to see and it is important to avoid disrupting it, so use preferably a swinging bucket rotor over a fixed-angle rotor.
Carefully remove supernatant and resuspend pellet with 1 mL of 1x PBS. Transfer cells to a 1.5 mL microfuge tube and centrifuge at 6,000 x g and 4 °C for 5 min.
Carefully remove all liquid from tube and freeze dry pellet at -20 °C. Store the pellet at -20 °C for 2-3 weeks.

5. DNA isolation, RNase treatment, and DNA purification
- Remove cell pellet from -20 °C and place on ice.
- Add 400 µL of SDS buffer (50 mM Tris-HCl (pH 8.0), 10 mM EDTA, 1 M NaCl, 0.5% SDS) to pellet and resuspend.
- Add proteinase K to a final concentration of 0.2 mg/mL and mix by vortexing briefly.
- Incubate samples at 56 °C for 2 h, vortex every 15 min.
- After 2 h, perform phenol-chloroform extraction by adding 400 µL of phenol-chloroform (Phenol:Chloroform:Isoamyl Alcohol 25:24:1, saturated with 10 mM Tris, pH 8.0, 1mM EDTA).
- Vortex for 20 s and centrifuge sample at 16,000 x g for 5 min to separate phases.
- Carefully remove the upper aqueous phase (400 µL) and transfer to a 1.5 mL tube.
- Perform EtOH precipitation by adding 40 µL of 3M sodium acetate (pH 5.2), 2 µL of glycogen, and 1 mL of EtOH.
- Vortex to mix thoroughly and place at -20 °C overnight to precipitate DNA. Sample can also be precipitated at -80 °C or on dry ice for 1 h.
- Centrifuge sample at 4 °C and 16,000 x g for 30 min to pellet DNA.
- Discard supernatant and wash pellet with 1 mL 70% EtOH. Centrifuge at 4 °C and 16,000 x g for 5 min.
- Discard supernatant and air-dry pellet at ambient temperature for 2-3 min.
- Dissolve pellet in 96 µL of autoclaved water, and add 4 µL of RNase A (2 mg/mL).
- Mix well by vortexing and incubate at 37 °C for 1 h.
Perform phenol-chloroform extraction by adding 100 µL of phenol-chloroform and following procedure in steps 5.2.1 - 5.2.2.
Perform EtOH precipitation by adding 10 µL of 3M sodium acetate, 1 µL glycogen, and 250 µL of EtOH and following the procedure in steps 5.3.1 - 5.3.5.
Resuspend pellet in 60 µL of TE buffer (10 mM Tris-HCl (pH 8.0), 10 mM EDTA (pH8.0)) and determine DNA concentration using a quantitative fluorescence-based method. Store DNA at -20 °C. NOTE: It is important to quantify DNA using fluorescence-based DNA binding dyes, or other means more reliable than UV spectrometer-based methods.
6. Preparation of DNA libraries and next generation sequencing
NOTE: A G1 copy number reference sample for each biological source is required for each sequencing run (i.e. WT, mutant, transgenic, cell line, etc). Compare all S phase samples from the same biological source in the same sequencing run to the same G1 reference. Run at least two biological replicates of each sample to ensure consistency between samples.
Dilute 1 µg of DNA to a final volume of 50 µL and transfer to a specialized tube for sonication.
Shear genomic DNA for a target of 250-300 bp in a focused ultrasonicator, according to manufacturer's specifications and protocol.
Verify quality and size of sheared genomic DNA using an automated electrophoresis machine, according to manufacturer's specifications and protocol. Ensure there is a large peak of ~250-300 bp for the sheared genomic DNA.
Prepare sequencing libraries using a DNA library preparation kit with multiplex oligos for sequencing on Illumina platform, according to manufacturer's protocol.
Verify quality of library prep using automated electrophoresis machine, according to manufacturer's protocol. Ensure there is a large peak of ~400-450 bp, and there are minimal peaks for primer dimers (80-85 bp) and adapter dimers (~120 bp).
Quantify libraries using a DNA library quantification kit for next-generation DNA sequencing, according to manufacturer's protocol.
Dilute 15 µL of library to a concentration of 5 nM and combine multiplexed libraries as required to achieve desired uniquely mapping read counts per sample. NOTE: The number of uniquely mapping reads per sample will determine the resolution. Use as few as 5 million mapped reads to assess replication timing for the zebrafish genome. It is recommended to start with 10-20 million reads.
Perform paired-end 100 bp whole genome DNA sequencing of samples on a next generation sequencing platform, according to manufacturer's instructions.
7. Analysis of sequencing data
NOTE: The instructions in this section are intended as a guideline for analysis. Use additional methods for sequencing alignment, filtering, processing, etc. This section of the protocol will deal with the preferred method of analysis in this work. If additional methods are used, adjust the parameters and functions to suit those purposes. The commands below are entered in Ubutnu or Mac terminal, with the appropriate packages installed.
- Obtain the most recent versions of the zebrafish genome (danRer10, as of when this protocol was written) from UCSC genome browser using the following commands: wget ftp://hgdownload.cse.ucsc.edu/goldenPath/danRer10/bigZips/danRer10.fa.gz-O danRer10.fa.gz
- Uncompress the fa.gz file with the following command: gunzip danRer10.fa.gz
- Align sequencing data to genome using BWA-mem using the following commands: bwa mem -t 8 danRer10.fa read_1.fastq read_2.fastq > output.sam
- Convert .sam file to .bam file using samtools with the following command: samtools view -bS input.sam > output.bam
- Sort the aligned sequencing files using samtools with the following command: samtools sort output.bam > output_sorted.bam
- Index the .bam file using samtools with the following command: samtools index output.bam
Mark PCR duplicates with Picard using the following commands: java -Xmx2g -jar picard.jar MarkDuplicates \ INPUT=input.bam OUTPUT=output.bam REMOVE_DUPLICATES=false METRICS_FILE=output_metrics.txt
Generate text (.txt) files of reads for each chromosome using the following command: for i in {1.25}; do CHROM="chr"$i; echo $CHROM; samtools view input.bam $CHROM | cut -f 2,4,5,7,8,9 > readsChr${i}.txt; done NOTE: The columns in the resulting .txt file are Flag, Location, mapQ, pair chr, pair location, and fragment size, respectively.
- Read .txt files into Matlab (or any other similar software) using the textscan function.
- Filter out low mapping quality reads by setting a mapQ threshold of 30.
- Remove reads with flags for not primary alignment, PCR duplicates, or pair mapping to a different chromosome.
- Filter out any reads with their pair beyond a distance threshold of 2000 bp.
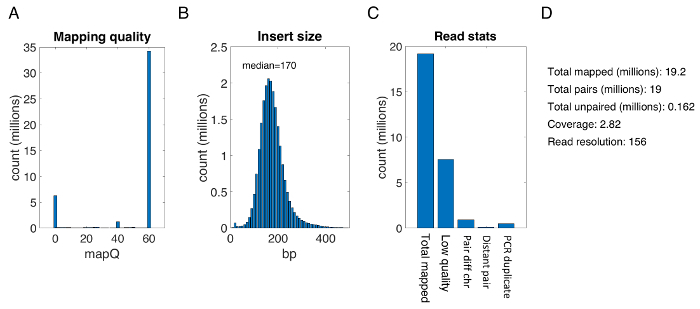
- Evaluate statistics of resulting reads to determine number of read statistics, coverage, insert size, and quality (Figure 2).
- Determine read coverage in 100 bp fixed windows for each sample by counting the number of reads in 100 bp intervals across the length of each chromosome.
- Calculate the median read count for all windows.
- Filter out regions of high coverage by removing windows greater than 5 standard deviations from the median.
- Define regions for analysis in the G1 reference samples by finding segments along each chromosome with 200 reads using sliding windows.
- Determine read depth in each S phase sample by counting the number of S phase reads in the 200-read count windows defined for the accompanying G1 reference.
Smooth the raw data using the csaps function in the software, with an interpolation factor (p) of 10-16.
Normalize smoothed data to z-score with a mean of 0 and standard deviation of 1.
Representative Results
Using published replication timing data, representative replication timing profiles and quality control measures are provided15. The initial steps of processing involve aligning the sequencing data to the genome, calculating read length and genome coverage statistics, and filtering low quality, unpaired, and PCR duplicate reads. Read statistics for a typical zebrafish sequencing sample are shown in Figure 2. After filtering, read counts are determined in variable-sized windows and the data are smoothed and normalized. Typical smoothed/normalized replication timing profiles of a representative zebrafish chromosome for biological replicates are shown in Figure 3A. The profiles for biological and experimental replicates should be visually very similar (see Figure 3A) and also display high correlation along the length of the chromosome (Figure 3B). They should also display high correlation between timing values genome-wide (Figure 3C). Autocorrelation, a method to assess pattern continuity, should be used to evaluate the degree of structure in the data set (Figure 3D). The autocorrelation for structured mature timing programs should be high at short distances and gradually decrease as the distance increases. Samples that show high reproducibility between biological and experimental replicates and a strong autocorrelation signal should be considered as high quality samples and can be combined to obtain a higher coverage sample.
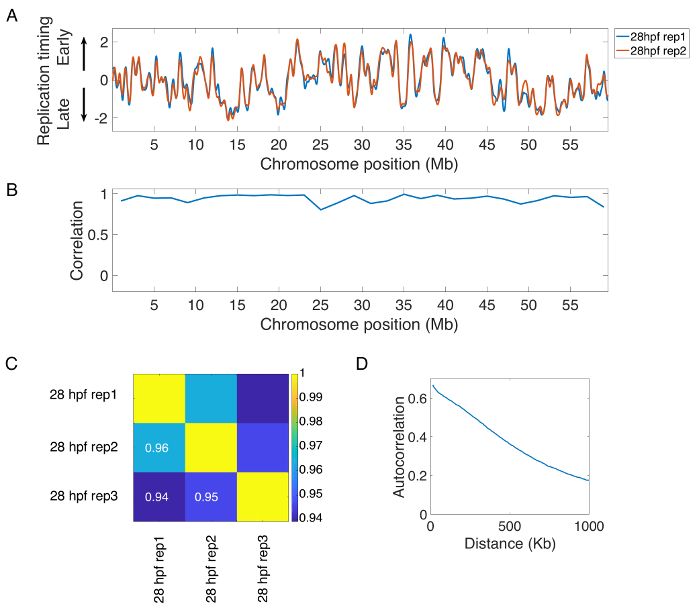
Figure 1: Sorting zebrafish embryos for replication timing analysis. (A) Gated population of all cells sorted for forward scatter area (FSC-A) by side scatter area (SSC-A). The colors represent the density of bins in the dot plot with high-to-low density represented by colors ranging from red-to-green-to-blue. (B) Gated population of the FSC x SSC gated cells sorted for SSC-A by propidium iodide area (PI-A). (C) Gated population of the SSC x PI gated cells sorted for SSC-A by side scatter width (SSC-W). (D) Histogram of all gated populations displaying a stereotypical cell cycle profile. Sort gates for cells in G1 and S phase are displayed by the grey shaded boxed with black lines. (E) Backgated G1 and S phase populations show that the initial gating captured the majority of cells. Please click here to view a larger version of this figure.
Figure 2: Sequencing read statistics for a typical zebrafish sample. (A) Reap mapping quality from statistics of mapQ values. (B) Histogram of the distribution of insert sizes for all sequencing reads. (C) Read statistics for total mapped reads, reads containing low quality flag, reads with pairs mapping to a different chromosome (pair diff chr), reads with pair beyond distance threshold (distant pair), and PCR duplicates. (D) Representative numbers of total mapped, unmapped, and unpaired reads, coverage, and read resolution for a typical sequencing run of a zebrafish sample. Please click here to view a larger version of this figure.
Figure 3: Representative zebrafish replication timing results and quality control measures. (A) Replication timing along the length of a representative chromosome for two biological replicates of 28 hpf embryos (Siefert, 2017). (B) Correlation between replication timing values in 2 Mb windows along the length of a representative chromosome for the biological replicates of 28 hpf embryos shown in Figure 3A. (C) Genome-wide correlation between replication timing values for biological (rep1 and rep2) and experimental (rep3) replicates of 28 hpf embryos. The color map represents Pearson's correlation coefficient. (D) Autocorrelation for a structured mature timing program displays high autocorrelation at close distances that decreases gradually over increasingly longer distances. Please click here to view a larger version of this figure.
Discussion
Zebrafish provide a new and unique in vivo model system to study DNA replication timing. When timed matings are performed as detailed in this experimental protocol, thousands of embryos can be collected in a single day for experiments. These embryos develop synchronously through precisely timed and distinctly characterized stages of development. Zebrafish can be easily and accurately staged by morphology using a stereomicroscope, as zebrafish embryos develop externally and are optically clear. This protocol details the use of zebrafish embryos to study replication timing throughout vertebrate development.
Areas of the protocol that may present problems include ensuring synchronous development of zebrafish embryos, loss of cells during preparation, FACS sorting, and library preparation. The timing of zebrafish development is temperature dependent, therefore use zebrafish adapted to light/dark cycles and housed at 28.5 °C for experiments. To ensure synchronous development, perform precisely timed matings, and collect embryos in very small time-windows (10 min or less). It is critical to maintain embryos at 28.5 °C and ensure they spend minimal time at ambient temperature. The timing of zebrafish development is also highly dependent on the density of embryos in a given area. It is critical to not house embryos at a density higher than 100 embryos per 10 cm plate, or they will not develop properly. The optimal time to remove dead and unfertilized embryos is when they have progressed to the 4-16 cell stage and can easily be identified as fertilized (use "Stages of Embryonic Development of the Zebrafish"17 as a guide). Dead embryos typically appear as black balls and unfertilized embryos appear as 1-cell. Work as quickly as possible to reduce the time at ambient temperature.
When dechorionating embryos, it is important to ensure all chorions are removed from the embryos. Keep embryos in pronase solution until most chorions have come off. Discard any remaining embryos with chorions intact or remove their chorions with forceps. Pipette embryos using a slow and smooth motion. Do not pipette rapidly as it will subject the cells to undue stress and result in loss of cells. It is also important not to use a P200 as this can cause increased shear stress that results in breakage of cells. Conversely, a plastic transfer pipette may not have a small enough bore or provide enough shear stress to adequately disrupt the yolk sack and disaggregate the cells. Alternative pipettes could be used provided the bore and shear would be equivalent to the optimal P1000.
When FACS sorting 28 hpf, if a stereotypical cell cycle profile is not obtained, adjust the voltage on the PI laser so that the G1 peak is centered around 50K. The gains for the FSC and SSC may also need to be adjusted such that the gating and cell populations appear similar to Figure 1. If there is still difficulty, the ND filter should be switched, and the FSC and SSC voltages adjusted to ensure the majority of cells are within the gate. It should be obvious whether there is PI staining, as there will be a range that approximates doubles for the PI-A. The histogram should show two clear peaks, a large peak at 2N DNA for the G1 cells, and a smaller peak at double the intensity for the 4N DNA content of G2/M cells. If this is still not obtained there may either be low numbers of intact cells, problems with the PI staining, or problems with fixation and the protocol will need to be optimized accordingly.
Cells from early embryos (prior to 10 hpf) will not give typical cell cycle profiles, as a high percentage of these cells are in S phase. These samples can be treated as S phase samples, and compared to a G1 reference from 24 hpf embryos. Embryos between 10 hpf and 24 hpf may give intermediate cell cycle profiles and can be sorted if desired. The S-phase population can also be identified and sorted by incorporating thymidine analogs such as EDU (5-Ethynyl-2'-deoxyuridine) or BrdU (Bromodeoxyuridine) in brief pulses prior to fixation, however, this is not necessary for accurate determination of replication timing.
Ideally the library preparation should be started with 1 µg of DNA. Theoretically ~300,000 sorted cells would yield about 1 µg of DNA (estimating ~3.3 pg/nucleus), however, there is always loss throughout the protocol. 500,000 to 1 million sorted cells should give more than 1 µg of DNA. A typical 24 hpf embryo will have approximately 18,000 cells, 10-15% of which will be in S-phase. Embryos at earlier stages of development will have considerably less cells, but will have higher percentages of cells in S-phase.
Performing accurate purification and size selection using magnetic beads is critical to eliminating unwanted elements from the library preparation. Follow the manufacturer's instructions carefully and additional optimization may be required. A small amount of PCR dimers is typically not a major problem, but adapter dimers will sequence very efficiently so they should be minimized. This can be achieved by adjusting the initial adapter to DNA ratio, and performing accurate size selection during library preparation.
Single-end sequencing may also be appropriate for different experimental needs. Single-end is cheaper and provides most of the required information since this approach relies on counting reads; paired-end provides a higher ability to remove PCR and optical duplicates and to resolve areas of repetitive sequences and other structural variations (which are common in the zebrafish genome). The analysis portion below will deal only with paired-end sequencing reads. Shorter sequencing reads may also be appropriate. This will provide a higher number of reads at a lower cost, but the reads may be more difficult to map. The analysis portion below is optimized for 100 bp reads, and adjustments may be necessary if shorter read length is used.
This protocol can also easily be adapted for additional uses of the zebrafish model. It has already been used for analysis of zebrafish tailfin fibroblasts (ZTF cells), a primary cell culture line isolated from adult zebrafish tailfin. To study isolated cell types from zebrafish, transgenic markers can be used to identify and isolate individual cell types before staining and sorting for DNA content. One potential strategy would be to use a transgenic line expressing a fluorescent marker driven by a tissue-specific promoter, and to first isolated cells by FACS sorting, followed fixation and sorting for DNA content. Additionally, the use of cell permeable DNA dyes may allow for simultaneous isolation of individual cell types as well as G1 and S phase populations. These adaptations to this protocol could also enable replication timing studies in other in vivo models.
An exciting possibility for the future use of this protocol is to study the role of replication timing in disease. There are several cancer models in zebrafish, some spontaneous and other inducible, that can be used to determine when changes in replication timing occur during oncogenesis. This will allow assessment of the role replication timing plays in tumorigenesis and disease progression. Furthermore, zebrafish are an excellent model for drug screening and can be used to identify drugs that affect replication timing regulation, which have the potential for use in cancer treatment.
The zebrafish is a promising new model to study replication timing. This protocol will afford many others the opportunity to utilize this model in studying the role of replication timing in development and disease. This protocol can be adapted for use with other in vivo developmental model systems, such as Drosophila melanogaster and Xenopus laevis, and look forward to future findings from these and other organisms.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by National Institute of General Medical Sciences of the National Institutes of Health through grants 5P20GM103636-02 (including Flow Cytometry core support) and 1R01GM121703, as well as awards from the Oklahoma Center for Adult Stem Cell Research.
References
- Rhind N, Gilbert DM. DNA replication timing. Cold Spring Harb Perspect Biol. 2013;5(8):a010132. doi: 10.1101/cshperspect.a010132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope BD, et al. Topologically associating domains are stable units of replication-timing regulation. Nature. 2014;515(7527):402–405. doi: 10.1038/nature13986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Mulia JC, et al. Dynamic changes in replication timing and gene expression during lineage specification of human pluripotent stem cells. Genome Res. 2015;25(8):1091–1103. doi: 10.1101/gr.187989.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiratani I, et al. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 2008;6(10):e245. doi: 10.1371/journal.pbio.0060245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiratani I, et al. Genome-wide dynamics of replication timing revealed by in vitro models of mouse embryogenesis. Genome Res. 2010;20(2):155–169. doi: 10.1101/gr.099796.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren A, et al. Differential relationship of DNA replication timing to different forms of human mutation and variation. Am J Hum Genet. 2012;91(6):1033–1040. doi: 10.1016/j.ajhg.2012.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryba T, et al. Abnormal developmental control of replication-timing domains in pediatric acute lymphoblastic leukemia. Genome Res. 2012;22(10):1833–1844. doi: 10.1101/gr.138511.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sima J, Gilbert DM. Complex correlations: replication timing and mutational landscapes during cancer and genome evolution. Curr Opin Genet Dev. 2014;25:93–100. doi: 10.1016/j.gde.2013.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldman MB, Lin S. Zebrafish as a developmental model organism for pediatric research. Pediatr Res. 2008;64(5):470–476. doi: 10.1203/PDR.0b013e318186e609. [DOI] [PubMed] [Google Scholar]
- Link BA, Megason SG. Zebrafish as a Model for Development. Sourcebook of Models for Biomedical Research. 2008. pp. 103–112.
- Siefert JC, Clowdus EA, Sansam CL. Cell cycle control in the early embryonic development of aquatic animal species. Comp Biochem Physiol C Toxicol Pharmacol. 2015;178:8–15. doi: 10.1016/j.cbpc.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill AJ, Teraoka H, Heideman W, Peterson RE. Zebrafish as a model vertebrate for investigating chemical toxicity. Toxicol Sci. 2005;86(1):6–19. doi: 10.1093/toxsci/kfi110. [DOI] [PubMed] [Google Scholar]
- Dooley K, Zon LI. Zebrafish: a model system for the study of human disease. Curr Opin Genet Dev. 2000;10(3):252–256. doi: 10.1016/s0959-437x(00)00074-5. [DOI] [PubMed] [Google Scholar]
- Santoriello C, Zon LI. Hooked! Modeling human disease in zebrafish. J Clin Invest. 2012;122(7):2337–2343. doi: 10.1172/JCI60434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siefert JC, Georgescu C, Wren JD, Koren A, Sansam CL. DNA replication timing during development anticipates transcriptional programs and parallels enhancer activation. Genome Res. 2017;27(8):1406–1416. doi: 10.1101/gr.218602.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren A, et al. Genetic variation in human DNA replication timing. Cell. 2014;159(5):1015–1026. doi: 10.1016/j.cell.2014.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Dev Dyn. 1995;203(3):253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]