

Abstract
When steady state RNA levels are compared between two conditions, it is not possible to distinguish whether changes are caused by alterations in production or degradation of RNA. This protocol describes a method for measurement of RNA production, using 5-Bromouridine labelling of RNA followed by immunoprecipitation, which enables investigation of RNA synthesized within a short timeframe (e.g., 1 h). The advantage of 5-Bromouridine-labelling and immunoprecipitation over the use of toxic transcriptional inhibitors, such as α-amanitin and actinomycin D, is that there are no or very low effects on cell viability during short-term use. However, because 5-Bromouridine-immunoprecipitation only captures RNA produced within the short labelling time, slowly produced as well as rapidly degraded RNA can be difficult to measure by this method. The 5-Bromouridine-labelled RNA captured by 5-Bromouridine-immunoprecipitation can be analyzed by reverse transcription, quantitative polymerase chain reaction, and next generation sequencing. All types of RNA can be investigated, and the method is not limited to measuring mRNA as is presented in this example.
Keywords: Genetics, Issue 135, RNA synthesis, 5-Bromouridine labelling, immunoprecipitation, phenol-chloroform extraction, reverse transcription, quantitative polymerase chain reaction
Introduction
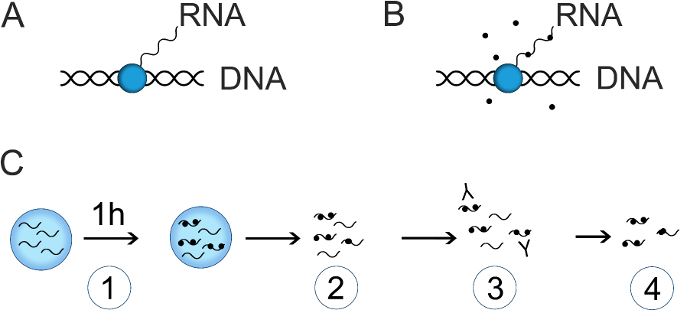
5-Bromouridine (BrU) immunoprecipitation (IP) allows the study of RNA production in cells with no or very limited effects on cell physiology during the brief labelling period1,2. The method is based upon incorporation of the synthetic uridine derivative BrU into newly synthesized RNA followed by IP of labelled RNA using anti-BrU antibodies (Figure 1).
It has been known for decades that protein synthesis can be transcriptionally regulated, and the existence of transcription factors was hypothesized more than 50 years ago3. Today, it is known that several diseases are caused by dysregulation of transcription and RNA stability (Supplementary Figure S1 in reference4), and the ability to measure changes in RNA production is of great importance in understanding disease development.
On the other hand, tools to regulate RNA production provide new possibilities for treating diseases caused by too little or too much protein expression, or protein accumulation such as in Parkinson's disease (PD). The predominant protein accumulating as insoluble aggregates in PD is α-synuclein, and the level of α-synuclein is directly linked to the disease, as gene multiplication of the α-synuclein gene causes familial PD5. Furthermore, α-synuclein aggregates in a concentration dependent manner. Downregulation of α-synuclein mRNA levels is therefore an interesting therapeutic strategy, which has been successfully achieved using RNA interference to decrease neuronal cell loss in PD rodent models6,7.
It is important to be able to measure the changes in RNA production caused by either disease or therapeutic interventions. State of the art methods for measuring steady state levels of RNA, such as reverse transcription followed by quantitative polymerase chain reaction (RT-qPCR), are not capable of distinguishing between changes caused by transcriptional regulation or by altered RNA stability. A widely used method for investigation of RNA decay rates is blocking of the transcriptional machinery using compounds such as α-amanitin or actinomycin D followed by measurements of the decaying RNAs. However, there are some problems associated with an overall blockage of transcription in cells, such as induction of apoptosis8,9. Beside the cytotoxic effects, transcriptional inhibitors also present several technical issues, such as slow cellular uptake of α-amanitin and lack of specificity of actinomycin D (reviewed in reference10).
To avoid the overall blockage of transcription, nucleotide analogues have been used, such as 5-ethynyl uridine (5-EU), 4-thiouridine (4-TU), and BrU. These are readily taken up by mammalian cells and incorporated into newly synthesized RNA, enabling pulse-labelling of RNA within a specific time-frame. BrU is less toxic than 5-EU and 4-TU, making it the preferred analogue of choice11,12.
BrU-IP can be used to investigate both the rate of RNA synthesis and stability, and thus distinguish between the underlying causes of changes in total RNA. This article will focus on the measurement of RNA synthesis and refers to reference2 for details on investigation of RNA stability. To investigate RNA synthesis, cells are briefly labelled with BrU e.g. for 1 h followed by BrU-IP1 (Figure 1C). This enables measurements of RNA synthesized within the short labelling time and observed changes will give a better estimate of regulations in RNA synthesis than by measuring changes in total RNA by RT-qPCR. It should be mentioned, however, that even though the labelling time is, for example, only 1 h, degradation can still have an influence on the RNA levels observed.
RNA from BrU-IP experiments are suitable for downstream analysis by both RT-qPCR1 or next generation sequencing2,13. Other standard RNA detection methods, such as northern blotting or ribonuclease protection assay, may also be applicable for certain RNAs.
Protocol
NOTE: Perform all steps at room temperature unless otherwise stated and keep buffers on ice or in the fridge (except from the Elution buffer containing SDS). The protocol is divided into 5 sections: Preparation of RNA samples; preparation of beads for IP; binding of BrU-labelled RNA to beads; elution and purification of bound BrU-labelled RNA by 3 steps phenol-phenol/chloroform-chloroform extraction; analysis of RNA (in this example using RT-qPCR)
1. Preparation of RNA Samples
Count HEK293T cells using an automated cell counter and seed 3,500,000 cells in a 10 cm petri dish in 10 mL growth media (Dulbecco's Modified Eagle's Medium supplemented with 10% fetal calf serum and 50 µg/mL penicillin/streptomycin) to obtain a total of >40 µg RNA upon extraction 48 h later. Maintain cells at 37 °C and 5% CO2.
48 h after seeding, aspirate 8 mL growth media and leave 2 mL behind to avoid drying out the cells. Add 2 mM BrU and any treatment to the 8 mL aspirated growth media, and remove the residual 2 mL from the cells before reapplying the 8 mL BrU containing growth media. Avoid the use of fresh media upon application of the BrU to the cells, since this can induce changes in gene expression.
Incubate the cells for 1 h with BrU and treatment. Wash the cells in 5 mL Hank's buffer and trypsinize the cells using 2 mL 0.05% trypsin. Add 8 mL growth media to stop the reaction, transfer the cells to a 15 mL tube, and spin down the cells for 2 min at 1,000 x g.
Remove the supernatant and extract total RNA from the cell pellet using an RNA isolation kit (see Table of Materials) or any other preferred method, and elute in RNase-free H2O. Store the RNA at -80 °C until ready to proceed.
2. Preparation of Beads for IP
Prepare beads in batches for the number of samples to be investigated plus one extra to account for loss during pipetting and whirl mixing. Resuspend anti-Mouse IgG magnetic beads thoroughly by whirl mixing and take out 20 µL beads per sample in an RNase-free 1.5 mL tube. Place the 1.5 mL tube in a magnetic stand and leave approximately 20 s for the beads to collect on the side.
Remove the supernatant, take out the tube from the magnetic stand, and resuspend in 400 µL 1x BrU-IP buffer by pipetting. Spin the tube very briefly for 2 s in a table-top centrifuge, place it back in the magnetic stand, and remove the supernatant. Repeat the washing step once.
Block unspecific binding to the beads using heparin. Resuspend the beads in 1 mL 1x BrU-IP+ 1 mg/mL heparin buffer, and leave rotating for 30 min. Wash the beads once, as in step 2.2. NOTE: Heparin is a negatively charged polymer, which will compete with RNAs for binding. tRNA or other irrelevant RNAs can also be used if the downstream application is not sequencing.
Resuspend the beads in 1 mL 1x BrU-IP buffer and add 1.25 µg/sample anti-α-Bromodeoxyuridine antibody, which also recognizes BrU. Incubate the beads for 1 h while rotating, or overnight at room temperature.
Wash the beads three times as in step 2.2. Resuspend the beads in 50 µL/sample 1x BrU-IP buffer supplemented with 1 mM BrU and leave rotating for 30 min to lower the sensitivity of the antibody bound to the beads and hereby the risk of unspecific binding during IP.
Wash the beads three times as in step 2.2, and resuspend in 50 µL/sample 1xBrU-IP buffer.
3. Binding of BrU-Labelled RNA to Beads
Measure RNA concentrations in the RNA extracts from step 1.3 using ultraviolet-visible spectrophotometry, and dilute 40 µg RNA from each sample in RNase-free H2O to a total volume of 200 µL.
Heat RNA samples for 2 min at 80 °C to denature RNA and spin briefly in a tabletop centrifuge. Add 200 µL 2x BrU-IP+BSA/RNAse inhibitor.
Add 50 µL resuspended and prepared beads from step 2.6 to each sample and leave rotating 1 h. Wash the beads four times as in step 2.2.
4. Elution and Purification of RNA by 3 Steps Phenol-phenol/Chloroform-chloroform Extraction
- Elute RNA and perform phenol extraction
- Resuspend the beads in 200 µL Elution buffer to elute RNA, and quickly thereafter add 200 µL phenol, pH 6.6 (CAUTION: Toxic, see note below step 4.1.2) to remove any non-RNA molecules from the sample.
- Whirl mix the sample, and spin 3 min at 25,000 x g in a tabletop centrifuge. NOTE: Phenol is toxic and should be handled in a fume hood wearing skin protection. The slightly acidic pH ensures that only RNA, and not DNA, is extracted. The RNA will remain in the upper aqueous phase due to the hydrophilic nature of charges. The hydrophobic cores of proteins will bind phenol and move to the lower organic phenol-phase.
- Perform phenol-chloroform extraction
- Aspirate as much upper-phase as possible (approximately 190 µL of the 200 µL, depending on experience in handling) and move it to a tube with 200 µL phenol/chloroform, pH 6.6 (CAUTION: Toxic, see note). Make sure to aspirate the same amount across samples.
- Whirl mix the sample and spin for 3 min at 25,000 x g. NOTE: Chloroform is toxic and should be handled in a fume hood wearing skin protection. Chloroform is added to remove phenol, which can affect the downstream analysis of RNA by inhibiting reverse transcription14 and polymerase chain reactions15.
Perform chloroform extraction by aspirating the aqueous upper phase as before (now approximately 180 µL), and transfer to a tube containing 200 µL chloroform. Whirl mix the sample and spin 3 min at 25,000 x g.
Aspirate the aqueous upper phase as before (now approximately 170 µL) and transfer it to a tube containing 17 µL (1/10 volume) 3 M CH3COONa, pH 6.0. To neutralize and precipitate RNA from the aqueous solution, add 2 µL glycogen (20 mg/mL), which precipitates together with RNA and makes the RNA pellet more visible.
Add 500 µL 96% ethanol to increase binding between the salt ions and RNA and mix by inverting the tube a couple of times. Leave the tube overnight at -20 °C or 15 min on dry ice.
Spin the sample at 25,000 x g, 4 °C for 30 min. Discard supernatant without disturbing the pellet and wash the pellet in 185 µL 75% ethanol to remove any residual salt. Spin at 25,000 x g, 4 °C for 5 min.
Discard supernatant completely without disturbing the pellet and leave the pellet to dry 5-10 min with an open lid. Resuspend in 10 µL RNase-free H2O applied on the side of the tube to avoid disturbing the pellet.
5. Analysis of RNA
Prepare cDNA using a cDNA reverse transcription kit (see Table of Materials) or any other preferred method using 2 µL RNA sample and 1 µg yeast total RNA as carrier RNA for the cDNA synthesis reaction (not used if cDNA is used for next generation sequencing). Use 1 µg RNA without yeast RNA for cDNA preparation of corresponding input.
Dilute cDNA 1:25 and mix 9 µL diluted cDNA with 10 µL qPCR Master mix and 1 µL fluorogenic probe, both specified in the Table of Materials. NOTE: The cDNA dilution depends on the cDNA preparation and qPCR method used.
Run the following qPCR program (e.g., on a 7500 Fast Real-time PCR system or equivalent): 2 min at 50 °C, 20 s at 95 °C, 40 cycles of 3 s at 95 °C and 30 s at 60 °C.
Representative Results
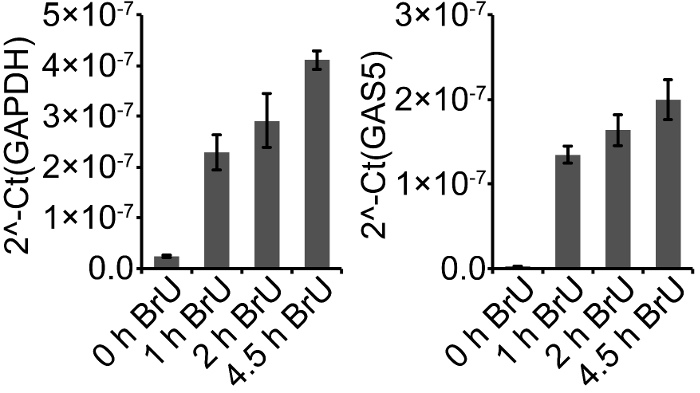
Our initial tests of BrU-labelling time were performed in AsPC-1 cells. The cells were treated with BrU for 0, 1, 2, and 4.5 h, followed by total RNA extraction and BrU-IP. GAS5 and GAPDH were measured in BrU-IP RNA, which demonstrated that 1 h labelling was sufficient to reach an approximately 9- and 44-fold change compared to background (0 h) for GAPDH and GAS5, respectively.
BrU-IP and the analysis described above were used to investigate if the changes discovered in steady state levels of α-synuclein mRNA upon Polo-like kinase 2 (PLK-2) inhibition were caused by changes in mRNA synthesis1. A 1 h labelling time was chosen because this provides sufficient labelling compared to background (Figure 2) and also allows the treatment time to have an effect. 2 mM BrU treatment did not induce any morphologic changes in HEK293T cells for either 1 h or the longer 4 h treatment (Figure 3A).

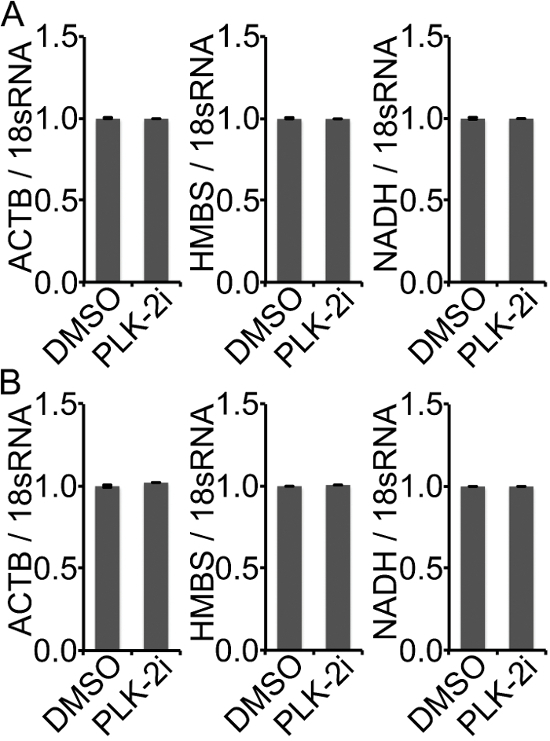
α-Synuclein transfected HEK293T cells were treated for 1 h with DMSO or a specific PLK-2 inhibitor (PLK-2i) in the presence of BrU to label freshly synthesized RNA. Cells were hereafter harvested and total RNA was extracted. BrU-labelled RNA produced within the 1 h treatment was collected using BrU-IP ("BrU-IP" in Figure 3B) from the total pool of RNA ("Input" in Figure 3B). The amount of RNA obtained with BrU-IP from 40 µg starting material varies between different cell lines, and we generally obtain approximately 500 ng from HEK293T cells, but only 10-50 ng from HeLa cells. PLK-2 inhibition caused an increase in α-synuclein mRNA in both the Input and BrU-IP (Figure 3B) suggesting that the increase in the steady state RNA is caused by an increase in synthesis of RNA. To ensure that PLK-2i does not just cause an overall increase in RNA production, 3 additional endogenous genes (ACTB, HMBS, and NADH) were measured (Figure 4). Neither of these genes was increased in the Input (Figure 4A) and BrU-IP (Figure 4B) upon PLK-2i treatment.
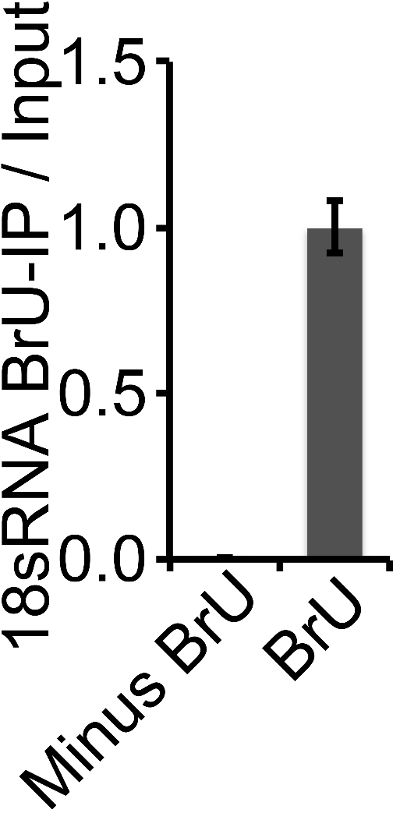
To validate the specificity of the BrU-IP, a control with non-BrU treated cells was included. The levels of 18sRNA captured by BrU-IP from BrU-treated and untreated cells were compared (Figure 5). This clearly demonstrates that the BrU-IP is highly specific and only captures a very small fraction of the non-labelled RNA (Figure 5).
Because the BrU-labelled RNA is only produced within 1 h, it is important to choose a proper reference gene, since some normally suitable genes might be expressed in too low concentrations to be reliable. 18sRNA was chosen as a reference gene for the data presented in Figure 3B and Figure 4 because of its high abundance. Several reference genes were, however, tested, and some showed high threshold cycles (>30) (Figure 6). This data also demonstrates that the difference between BrU-labelled IP'ed RNA and total RNA in the input is approximately 5 Ct's, and some RNAs expressed at low amounts in the cell might therefore not be properly detected following BrU-IP.
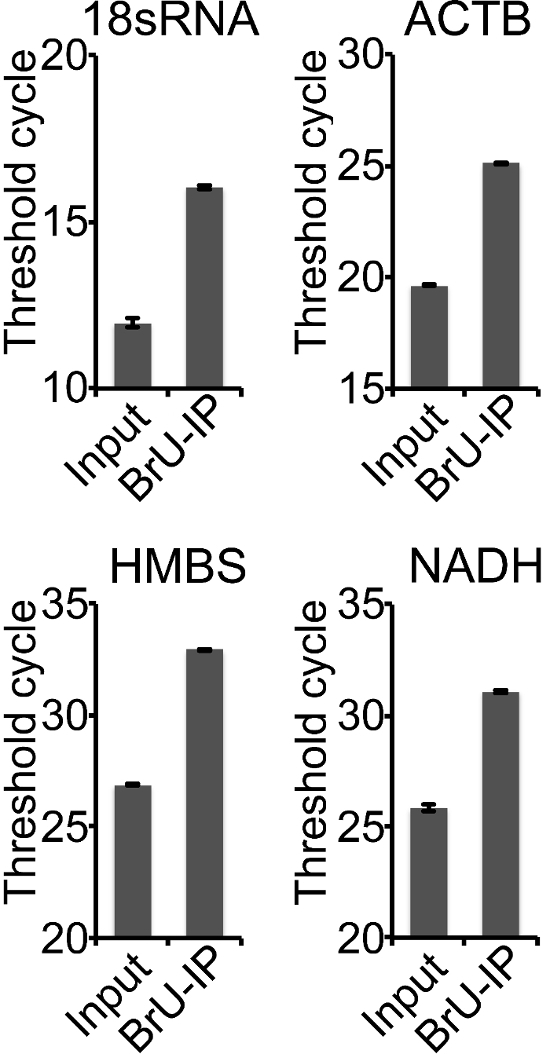
Figure 1: Description of BrU-IP for investigation of RNA synthesis. (A) RNA is transcribed in cells from DNA by RNA polymerases. (B) Upon addition to the media, BrU is taken up by cells and incorporated into the newly transcribed RNA. (C) For measuring RNA synthesis rates: (1) newly synthesized RNA is labelled for 1 h with BrU, (2) RNA is extracted from the cells immediately after labelling, (3) BrU-labelled RNA is immunoprecipitated using anti-BrU antibodies, (4) BrU-labelled (and corresponding input) RNA is analyzed by RT-qPCR or sequencing. Please click here to view a larger version of this figure.
Figure 2: Assessment of labelling time. Example of an initial assessment of BrU-labelling time. AsPC-1 cells grown in Roswell Park Memorial Institute (RPMI) medium supplemented with 10% fetal bovine serum and 50 U/mL / 50 µg/mL penicillin/streptomycin were treated with BrU at a final concentration of 2 mM for 0, 1, 2, and 4.5 h before RNA extraction and analysis by RT-qPCR. Data is quantified as 2-Ct, and is presented as the mean of technical triplicates ± standard deviation, n = 1. Please click here to view a larger version of this figure.
Figure 3: Representative results showing Input and BrU-IP RNA from DMSO and PLK-2i treated cells. (A) 1 h and 4 h 2 mM BrU treatment did not induce any visible morphologic changes in HEK293T cells. Scale bar = 100 µm. (B) Adapted figure from reference1. 3,500,000 HEK293T cells were seeded per 10-cm dish and transfected with a plasmid expressing α-synuclein 24 h after seeding. 24 h post transfection cells were incubated for 1 h with BrU in the presence of DMSO or PLK-2i, followed by RNA extraction and BrU-IP. Analysis of RNA was done by RT-qPCR using probe sets directed against human SNCA (α-synuclein) and 18sRNA. BrU-IP and Input SNCA was quantified relative to BrU-IP and Input 18sRNA, respectively, using the 2-ΔΔCT method. Results were normalized to the DMSO-treated samples. Data is presented as mean ± standard deviations. Statistical comparisons were performed using Student's unpaired t-test, *p <0.001, n = 3. Please click here to view a larger version of this figure.
Figure 4: Measurement of other genes not changed by PLK-2i. PLK-2i changes neither the total nor synthesized RNA of ACTB, HMBS, and NADH. (A) ACTB, HMBS, and NADH RNA from input were measured in triplicate in one experiment and quantified relative to 18sRNA in Input using the 2-ΔΔCT method. Values were normalized to the DMSO treated samples and bars represent mean of triplicates ± standard deviations, n = 1. (B) ACTB, HMBS, and NADH were also measured in the BrU-IP'ed RNA and quantified relative to 18sRNA in the BrU-IP. Values were normalized to DMSO and presented as in A, n = 1. Please click here to view a larger version of this figure.
Figure 5: Specificity of BrU-IP. For comparison of 18sRNA captured by BrU-IP from BrU- and non-BrU-treated cells, RNA from BrU-IP was validated relative to Input RNA to account for differences in starting material and hereafter normalized to BrU-treated samples. Only a very small fraction (0.002) of non-BrU-labelled 18sRNA was immunoprecipitated, demonstrating that the BrU-IP is highly specific. Please click here to view a larger version of this figure.
Figure 6: Levels of different mRNAs in BrU-IP RNA compared to Input RNA. Several potential reference genes were tested because some RNAs will be present in very low concentrations in BrU-IP RNA due to small amounts of produced RNA within the 1 h BrU treatment. Please click here to view a larger version of this figure.
Discussion
This protocol describes the use of BrU-IP for determination of RNA production by labelling of newly produced RNA for 1 h with BrU, followed by immediate RNA extraction and immunoprecipitation of BrU-labelled RNA. This method has previously been described in reference16, and this article includes some additional steps to increase IP specificity and RNA purity, by pre-treating beads with low concentrations of BrU as well as a phenol-chloroform purification step to increase RNA purity following IP. Additionally, we also present data here demonstrating the specificity of the BrU-IP by comparing the RNA captured from BrU and non-BrU treated cells. Phenol-chloroform purification is cheap, but considering the low amounts of RNA labelled with BrU, increased signals in the downstream analysis might be obtained by using RNA clean-up kits instead. Our experience is, however, that the most equal extraction of RNA across samples is obtained using phenol-chloroform purification.
BrU-labelling and -IP are, in contrast to analysis of total RNA levels, capable of investigating the underlying causes of changes in steady state RNA levels. The method has no or limited effects on cell physiology, in contrast to the use of transcriptional inhibitors such as α-amanitin and actinomycin D. It has been suggested that comparison of newly synthesized RNA to steady state levels can be used to calculate RNA decay rates17, and the BrU-IP method described here enables simultaneous determination of both RNA synthesis and decay in one assay.
In Figure 5, we demonstrated that RNA is only IP'ed from BrU treated cells, showing that there is indeed BrU incorporated into the RNA in the cell line used here. However, we suggest conducting an initial test of BrU incorporation into RNA in other cell lines before performing the BrU-IP. This could, for example, be done by using anti-BrU antibodies to quantify BrU in total RNA extracts via dot blot or enzyme-linked immunosorbent assay.
Here we have not presented a test of BrU-IP efficiency across samples, but this can be investigated using BrU-labelled spikes in RNA either produced in vitro or from another organism, such as Drosophila melanogaster. This RNA could then also be used in the downstream quantifications and normalizations.
Correct pipetting techniques are crucial when performing highly sensitive quantitative assays, such as BrU-IP and RT-qPCR, and a particular effort should be made to perform equal pipetting across samples. The phenol-chloroform extractions demand some experience, but upon unsatisfactory pipetting, the aqueous and phenol/chloroform phases can be re-mixed and centrifuged again.
This protocol suggests BrU-labelling for 1 h when measuring RNA synthesis. This labelling time enables a rough estimate of changes in transcription, since the majority of human and mice mRNAs have been estimated to have half-life >6 h18,19. Caution should, however, be exercised when interpreting the data obtained, since many RNAs have half-lives <1 h18,19, such as several transcripts induced by Lipid A, which rapidly increase upon stimulation and decline back to base-line levels within 2 h after induction20. To lower the risk of influences from RNA degradation, the BrU-labelling time can be decreased, however, it is also important to label enough RNA to be able to distinguish RNA obtained by BrU-IP from the background (which was obtained after 1 h labelling in our hands (Figure 2)). Another option is to measure both RNA synthesis and decay of the gene of interest using BrU-labelling and IP, the latter method is described in2. The data from these two approaches supplement each other, and if a change in synthesis between two conditions is measured but no change is seen in decay rates, it can be concluded that the measured changes in RNA were due to regulation of synthesis. However, it is important to remember that both techniques only measure RNA functionality by estimating RNA levels and completely neglect to investigate other mechanisms influencing RNA functions such as chemical modifications14.
Since the cells are incubated with BrU for only 1 h, very slowly produced mRNAs can be difficult to measure using the BrU-IP method. This is one point where the use of transcriptional inhibitors such as α-amanitin and actinomycin D might be preferred over BrU-IP, however, they can only be used to measure changes in degradation and not directly measure RNA production. The low levels of BrU-labelled RNA can also be overcome by increasing the incubation time with BrU, though, long incubation times increase the risk of influences by RNA degradation. In that case, it is no longer possible to know whether changes in BrU-labelled RNA are caused by changes in RNA production or decay.
In this example, the downstream analysis of the captured BrU-labelled RNA is performed using RT-qPCR, however, next generation sequencing is also a widely used downstream method to screen for changes in a large pool of RNAs2,13. Similarly, the method is not restricted to analysis of mRNAs, but may analyze all types of RNAs2.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The Lundbeck Foundation, Aarhus University, Dandrite and Lundbeck A/S supported this work.
References
- Kofoed RH, et al. Polo-like kinase 2 modulates α-synuclein protein levels by regulating its mRNA production. Neurobiol. Dis. 2017;106:49–62. doi: 10.1016/j.nbd.2017.06.014. [DOI] [PubMed] [Google Scholar]
- Imamachi N, et al. BRIC-seq: A genome-wide approach for determining RNA stability in mammalian cells. Methods. 2014;67:55–63. doi: 10.1016/j.ymeth.2013.07.014. [DOI] [PubMed] [Google Scholar]
- Jacob F, Monod J. Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol. 1961;3:318–356. doi: 10.1016/s0022-2836(61)80072-7. [DOI] [PubMed] [Google Scholar]
- Lee TI, Young RA. Transcriptional Regulation and its Misregulation in Disease. Cell. 2014;152:1237–1251. doi: 10.1016/j.cell.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrer M, et al. Comparison of kindreds with parkinsonism and α-synuclein genomic multiplications. Ann. Neurol. 2004;55:174–179. doi: 10.1002/ana.10846. [DOI] [PubMed] [Google Scholar]
- Khodr CE, Becerra A, Han Y, Bohn MC. Targeting alpha-synuclein with a microRNA-embedded silencing vector in the rat substantia nigra: Positive and negative effects. Brain Res. 2014. pp. 47–60. [DOI] [PMC free article] [PubMed]
- Cooper JM, et al. Systemic exosomal siRNA delivery reduced alpha-synuclein aggregates in brains of transgenic mice. Mov. Disord. 2014;29:1476–1485. doi: 10.1002/mds.25978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magdalan J, et al. alpha-Amanitin induced apoptosis in primary cultured dog hepatocytes. Folia Histochem. Cytobiol. 2010;48:58–62. doi: 10.2478/v10042-010-0010-6. [DOI] [PubMed] [Google Scholar]
- Lu DF, et al. Actinomycin D inhibits cell proliferations and promotes apoptosis in osteosarcoma cells. Int. J. Clin. Exp. Med. 2015;8:1904–1911. [PMC free article] [PubMed] [Google Scholar]
- Bensaude O. Inhibiting eukaryotic transcription. Which compound to choose? How to evaluate its activity? Transcription. 2011;2:103–108. doi: 10.4161/trns.2.3.16172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tani H, et al. Nuclear Bodies and Noncoding RNAs. Methods in Molecular Biology. 2015;1262:305–320. doi: 10.1007/978-1-4939-2253-6_19. [DOI] [PubMed] [Google Scholar]
- Meneni S, et al. 5-Alkynyl-2'-deoxyuridines: chromatography-free synthesis and cytotoxicity evaluation against human breast cancer cells. Bioorg. Med. Chem. 2007;15:3082–3088. doi: 10.1016/j.bmc.2007.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meola N, et al. Identification of a Nuclear Exosome Decay Pathway for Processed Transcripts. Mol. Cell. 2016;64:520–533. doi: 10.1016/j.molcel.2016.09.025. [DOI] [PubMed] [Google Scholar]
- Zhao BS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 2016;18:31–42. doi: 10.1038/nrm.2016.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader C, Schielke A, Ellerbroek L, Johne R. PCR inhibitors - occurrence, properties and removal. J. Appl. Microbiol. 2012;113:1014–1026. doi: 10.1111/j.1365-2672.2012.05384.x. [DOI] [PubMed] [Google Scholar]
- Paulsen MT, et al. Use of Bru-Seq and BruChase-Seq for genome-wide assessment of the synthesis and stability of RNA. Methods. 2014;67:45–54. doi: 10.1016/j.ymeth.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dölken L, et al. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. Rna. 2008. pp. 1959–1972. [DOI] [PMC free article] [PubMed]
- Raghavan A, et al. Genome-wide analysis of mRNA decay in resting and activated primary human T lymphocytes. Nucleic Acids Res. 2002;30:5529–5538. doi: 10.1093/nar/gkf682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharova LV, et al. Database for mRNA half-life of 19 977 genes obtained by DNA microarray analysis of pluripotent and differentiating mouse embryonic stem cells. DNA Res. 2009;16:45–58. doi: 10.1093/dnares/dsn030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya-Jones A, et al. Splicing kinetics and transcript release from the chromatin compartment limit the rate of Lipid A-induced gene expression. RNA. 2013;19:811–827. doi: 10.1261/rna.039081.113. [DOI] [PMC free article] [PubMed] [Google Scholar]