

Abstract
Cyclin-dependent kinase 1 (Cdk1) is a master controller for the cell cycle in all eukaryotes and phosphorylates an estimated 8 - 13% of the proteome; however, the number of identified targets for Cdk1, particularly in human cells is still low. The identification of Cdk1-specific phosphorylation sites is important, as they provide mechanistic insights into how Cdk1 controls the cell cycle. Cell cycle regulation is critical for faithful chromosome segregation, and defects in this complicated process lead to chromosomal aberrations and cancer.
Here, we describe an in vitro kinase assay that is used to identify Cdk1-specific phosphorylation sites. In this assay, a purified protein is phosphorylated in vitro by commercially available human Cdk1/cyclin B. Successful phosphorylation is confirmed by SDS-PAGE, and phosphorylation sites are subsequently identified by mass spectrometry. We also describe purification protocols that yield highly pure and homogeneous protein preparations suitable for the kinase assay, and a binding assay for the functional verification of the identified phosphorylation sites, which probes the interaction between a classical nuclear localization signal (cNLS) and its nuclear transport receptor karyopherin α. To aid with experimental design, we review approaches for the prediction of Cdk1-specific phosphorylation sites from protein sequences. Together these protocols present a very powerful approach that yields Cdk1-specific phosphorylation sites and enables mechanistic studies into how Cdk1 controls the cell cycle. Since this method relies on purified proteins, it can be applied to any model organism and yields reliable results, especially when combined with cell functional studies.
Keywords: Biochemistry, Issue 135, Cyclin-dependent kinase 1, nuclear localization signal, kinase assay, phosphorylation, mass spectrometry, analytical gel filtration, karyopherin alpha, CENP-F, G2 phase, cell cycle, kinase, affinity purification
Introduction
Kinases are enzymes that transfer phosphate groups from ATP onto substrates and regulate many cellular processes. This phosphorylation is reversible, fast, adds two negative charges, and stores free energy, and is one of the most common posttranslational modifications used by cells. Cdk1, which is also known as cell division cycle protein 2 homolog (cdc2) is a master controller for the cell cycle in all eukaryotes1,2,3,4,5, and phosphorylates an estimated 8-13% of the proteome6,7.
While recent proteomic studies have identified many phosphorylation sites in proteins, in most cases, the kinase responsible for these modifications is unknown. The number of known Cdk1 targets, particularly in human cells is low7. The identification of Cdk1-specific phosphorylation sites is important, as it enables mechanistic studies that establish how Cdk1 controls the cell cycle. Cell cycle regulation is important for faithful chromosome segregation and cell division, and a myriad of cellular processes need to occur to support this important physiological function. This includes halting transcription and translation prior to the onset of mitosis, as well as a dramatic reorganization in cellular structure and organization, such as disassembly of the nuclear envelope, chromosome condensation, and mitotic spindle assembly. Deregulation and errors in these processes cause cancer, birth defects, or mitotic cell death. Specific inhibitors of Cdk1 such as RO-3306 were developed8, which provide powerful tools for functional studies, and some of these inhibitors are currently in clinical trials for cancer treatment (see9 for review).
Here, we describe an in vitro kinase assay that allows the identification of Cdk1-specific phosphorylation sites. In this assay, commercially available human Cdk1/cyclin B is used to phosphorylate a purified target protein in vitro. Phosphorylation of a substrate increases its mass and adds two negative charges; therefore, successful phosphorylation is confirmed by an upward shift of the protein gel band on SDS-PAGE. Cdk1-specific phosphorylation sites are subsequently identified by mass spectrometry analysis of the in vitro phosphorylated protein. To aid with experimental design, we also review computational tools and references for the prediction of Cdk1-specific phosphorylation sites from the protein sequence. Furthermore, we also describe purification protocols that yield highly pure and homogeneous protein preparations suitable for the kinase assay. Finally, the identified phosphorylation sites must be verified by functional studies, and a simple binding assay is described here for that purpose. Combined, this is a very powerful approach that yields Cdk1-specific phosphorylation sites and enables mechanistic studies into how Cdk1 controls the cell cycle7,10,11. Since this method relies on purified proteins, it can be applied to any model organism and yields reliable results. However, functional verification of the obtained phosphorylation sites in vitro is recommended, as cells have additional regulatory mechanisms in place, such as posttranslational modifications, interaction partners, or cellular localization that may render phosphorylation sites accessible or inaccessible for recognition by Cdk1.
Cdk1 recognizes a consensus phosphorylation site that consists of (Ser/Thr-Pro-X-Lys/Arg), where X is any residue and a serine or threonine is the site of phosphorylation. Especially important for recognition is the presence of the proline in the +1 position. In addition, basic residues are preferred in the +2 or +3 positions, with most Cdk1-specific phosphorylation sites containing a Lys or Arg at the +3 position6,12.
Activation of Cdk1 is tightly regulated and leads to the onset of mitosis1,2,3,4,5. The activity of cyclin-dependent kinases in general depends on their association with distinct cyclins (cyclin A, B, C, D, and E in humans), which are expressed at oscillating levels throughout the cell cycle13. Cdk1 expression is constant across the cell cycle and the regulation of its activity relies on its association with the regulatory subunits cyclin A and cyclin B5,13,14,15, as well as post-translational modifications. Formation of the Cdk1/cyclin B complex is required for the kinase activation5,14,15,16,17,18. In the G2 phase, cyclin B is translated in the cytosol and imported into the nucleus where it binds to Cdk15,14,15,16,17,18; however, Cdk1/cyclin B is held inactivated by phosphorylation at residues Thr14 and Tyr15 by the human Cdk1-inhibitory kinases Myt1 (membrane-associated tyrosine- and threonine-specific cdc2-inhibitory kinase) and Wee1, respectively19,20,21. In the late G2 phase, dephosphorylation of Thr14 and Tyr15 by cell division cycle 25 phosphatase (cdc25) activates the kinase activity of the Cdk1/cyclin B complex and triggers the initiation of mitosis12,14,18,20,22,23. Phosphorylation of Thr161 is also required for Cdk1/cyclin B activation and is mediated by Cdk7, the Cdk-activating kinase (CAK)18. Degradation of cyclin B in anaphase inactivates Cdk1, allowing exit from mitosis24,25. Activation of Cdk1/cyclin B is therefore a complicated process. The protocol presented here is performed with commercially available Cdk1/cyclin B. During recombinant expression of this complex in insect cells, it is activated in vivo by endogenous kinases14,20 and remains active in the purified state. The resulting active, recombinant human Cdk1/cyclin B is suitable for in vitro kinase assays.
Here, we describe a protocol for the identification of Cdk1-specific phosphorylation sites in the human centromere protein F (CENP-F)10. CENP-F is a kinetochore protein that resides in the nucleus during most of interphase (G1 and S-phase) and is exported to the cytosol in the G2 phase26,27,28 in a Cdk1-dependent manner10,11. Nuclear localization is conferred by a bipartite cNLS26. cNLSs are recognized by the nuclear transport factor karyopherin α, which facilitates, together with karyopherin β and RanGDP, the import of cNLS-cargo into the nucleus29. Nuclear export in the G2 phase is facilitated via an unknown export pathway10. Once CENP-F resides in the cytosol, it is recruited to the nuclear envelope and in turn recruits the motor protein complex dynein30,31. This pathway is important to position the nucleus respective to the centrosome during initial stages of mitotic spindle assembly in a dynein-dependent manner, which is important for the correct timing of mitotic entry and for a fundamental process in brain development30,31,32. Starting in the G2 phase, CENP-F is also assembled into the kinetochore where it has important roles for faithful chromosome segregation27,28,33,34,35. A key regulatory step of these pathways is the nuclear export of CENP-F in the G2 phase, which is dependent on Cdk110,11. We describe here a protocol for the identification of Cdk1-specific phosphorylation sites in the cNLS of CENP-F. Phosphomimetic mutations of these sites slow down nuclear import of CENP-F, suggesting that Cdk1/cyclin B directly regulates cellular localization of CENP-F by phosphorylation of its cNLS10.
Overall, this in vitro kinase assay allows the identification of specific substrates for the kinase Cdk1. Purified target proteins are phosphorylated in vitro by the commercially available Cdk1/cyclin B complex and the phosphorylation sites are subsequently identified by mass spectrometry. The identification of Cdk1-specific phosphorylation sites supports mechanistic studies that reveal how Cdk1 controls the cell cycle.
Protocol
1. Prediction of Cdk1-specific Phosphorylation Sites from the Protein Sequence
- Before beginning the kinase assay, analyze the protein sequence for predicted Cdk1-specific phosphorylation sites and search the literature for experimentally established phosphorylation sites with unknown kinase specificity. Use the following tools, databases, and references that are summarized.
- In the software, enter the protein sequence in FASTA format. For kinase specificity, check Serine/Threonine kinase, CMGC, CDK, CDC2, CDK1, and uncheck all other specificities. Select medium as the threshold and click the Submit button.
- Compare the resulting predicted sites with the Cdk1 consensus phosphorylation site (Ser/Thr-Pro-X-Lys/Arg).
- Importantly, check the predicted site for the proline next to the phosphorylated residue (some Cdk1 substrates are phosphorylated at a minimal site (Ser/Thr-Pro)6,12). In addition, check for basic residues, that are preferred in the +2 or +3 positions, with most Cdk1-specific phosphorylation sites containing a Lys or Arg at the +3 position6,12.
- Also check that the site is accessible for recognition, as the presence of a full consensus site is not sufficient for Cdk1 phosphorylation.
- Inspect the surface accessibility and secondary structure prediction annotation in the iGPS output, which states whether the site is predicted to be accessible; N- and C-termini of proteins are in many cases flexible and accessible for phosphorylation.
- If an X-ray structure of the target protein is available, check accessibility of putative phosphorylation sites by inspecting their location in the structure.
- Search the literature for experimentally established phosphorylation sites with unknown kinase specificity using the UniProtKB/Swiss-Prot database38 (http://www.uniprot.org).
- Enter the name of the protein in the search box and click the Search button. Select the correct sequence entry. Phosphorylation sites are annotated and referenced in the amino acid modifications section.
- Identify bipartite cNLS by NLSmapper (optional). NOTE: For proteins that shuttle between the cytosol and the nucleus, a common mechanism for regulation of cellular localization is the phosphorylation of a bipartite cNLS in the -1 position of the major motif by Cdk1. The phosphorylation abolishes nuclear localization in the G2 phase10,41,42,43.
- For such shuttling proteins, identify the cNLS in the protein sequence by NLSmapper43 (http://nls-mapper.iab.keio.ac.jp/cgi-bin/NLS_Mapper_form.cgi). Paste the protein sequence as text in the sequence box, select a cut-off score of 5.0, and elect to search for the NLS in the entire region. Click the Predict NLS button.
- Compare the resulting predicted cNLS against the consensus sequence of a bipartite cNLS, which consists of the minor motif (Lys-Arg), a linker of at least 10 residues, and the major motif (Lys-Lys/Arg-X-Lys/Arg), where X is any residue that is not negatively charged.
- Check if the predicted Cdk1 phosphorylation site (*) is located adjacent to the major motif in the -1 position of the linker: Ser/Thr*-Pro/X-X-Lys-Lys/Arg-X-Lys/Arg. NOTE: The phosphorylation of a bipartite cNLS in the -1 position by Cdk1 was established as a regulatory mechanism for cellular localization for several shuttling proteins10,41,42,43.
2. Expression of Recombinant Proteins in Escherichia Coli
- Use the expression plasmids from steps 2.1.1-2.1.2 for the expression of recombinant proteins in E. coli.
- To create the human CENP-F (residues 2,987 - 3,065) expression construct, generate inserts by amplifying DNA fragments by polymerase chain reaction (PCR) from a full-length CENP-F construct (GenBank accession: U19769.1). Use the following primers: AGTCGGGGATCCCAGCAATCTAAACAAGATTCCCG and AGTCGGCTCGAGTCATTATTCTGCAGGGTGAATACCACTCATG. NOTE: The full-length human CENP-F plasmid was generously provided by Dr. X. Zhu, Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, China.
- Clone the insert into the pGEX6p1 vector with the BamHI and XhoI restriction sites. Use this plasmid to express N-terminal glutathione S-transferase (GST) fusion proteins of CENP-F (residues 2,987-3,065). NOTE: The GST-tag can be cleaved off by the PreScission protease (henceforth referred to as PS protease).
- For the human karyopherin α expression construct, generate inserts by amplifying DNA fragments by PCR from a full-length karyopherin α2 cDNA template (sequence accession NM_002264.3; primers: GCACTACATATGTCCACCAACGAGAATGCTAATA and TCACGCCTCGAGTTATCAAAAGTTAAAGGTCCCAGGAGCC). NOTE: Due to the similarity of the isoforms, this karyopherin α2 construct is referred to in the subsequent text as karyopherin α.
- Clone the insert into the pET28a-pres vector with the NdeI and XhoI restriction sites. NOTE: This is a modified pET28a vector in which the sequence encoding for the thrombin cleavage site was replaced by a sequence that encodes for a PS protease cleavage site. Use this plasmid to express a fusion protein of karyopherin α with an N-terminal His6-tag, where the His6-tag can be cleaved off by PS protease.
To create point mutations of a target protein, perform site-directed mutagenesis with a kit.
- Express the recombinant proteins in E. coli for subsequent purification.
- Make the following stocks: 50 mg/mL kanamycin (1:1,000 stock solution), 35 mg/mL chloramphenicol in 70% Ethanol (1:1,000), 100 mg/mL ampicillin (1:1,000), and 1 M isopropyl β-D-1-thiogalactopyranoside (IPTG; 1:5,000).
- Sterile filter the stocks and store at -20 °C. Avoid repeated freeze-thaw cycles for IPTG.
- Transform 1 µL of expression plasmid into 50 µL of competent cells of the E. coli Rosetta 2(DE3)pLysS strain, according to the manufacturer's instructions.
- Plate the culture on Luria-Bertani (LB) agar with chloramphenicol (35 µg/mL LB) and ampicillin (100 µg/mL LB) for the CENP-F construct, or chloramphenicol and kanamycin (50 µg/mL culture) for the karyopherin α construct. Incubate the plates for 12-20 h at 37 °C.
- Pick a single colony and inoculate 50 mL of LB supplemented with antibiotics (see step 2.3.4). Incubate the preculture while shaking at 160-180 rpm for 12-20 h at 37 °C.
- Prepare 1 L of LB medium in a 2,800 mL baffled Fernbach flask. Make two flasks for each preculture and autoclave them. To allow aeration, do not fill the flasks to more than two thirds the flask volume. Erlenmeyer flasks can be used as well.
- Add antibiotics (step 2.3.4) and 10 mL of a sterile-filtered 40% (w/v) glucose solution per 1 L of cooled LB. Add 20 mL of preculture per 1 L of LB medium. NOTE: The absorbance at 600 nm of a 1:10 dilution of the preculture should be between 0.15-0.2.
- Incubate the flasks while shaking at 160-180 rpm at 37 °C. Measure the absorbance at 600 nm of the bacteria culture regularly. If the absorbance reaches 0.5-0.6, induce protein expression by adding 0.2 mM IPTG (200 µL of 1 M stock per 1 L of LB medium). NOTE: It is important that cells are induced at the correct absorbance. Usually it takes 3-4 h to reach this stage.
- Incubate the flasks while shaking for 3 h at 37 °C. Harvest the cells by centrifugation (15 min, 4,100 x g, 4 °C, swing-out rotor). Discard the supernatant. Resuspend the cell pellets in 20 mL GST-binding buffer (CENP-F) or His6-binding buffer (karyopherin α) per 1 L of culture. Store the cells at -80 °C.
- Prepare the GST-binding buffer (10 mM Tris pH 8.0 at 25 °C, 150 mM NaCl, 1 mM dithiothreitol (DTT), 0.5 mM EDTA) and His6-binding buffer (10 mM Tris pH 8.0 at 25 °C, 250 mM NaCl, 5 mM Imidazole pH 8.0, 5 mM β-mercaptoethanol (BME)) in advance.
3. Purification of Recombinant Protein by Glutathione-affinity Chromatography and Gel Filtration
Make the following stocks: 1 M DTT (1:1,000 stock, sterile filtered with 0.2 µM pore size, and degassed); 250 mM phenylmethylsulfonyl fluoride (PMSF; 1:1,000 stock in dimethylsulfoxide). Store at -20 °C. Avoid repeated freeze-thaw cycles.
- Make the following buffers and cool them to 4 °C.
- Prepare the GST-binding buffer using 10 mM Tris pH 8.0 at 25 °C, 150 mM NaCl, 1 mM DTT, 0.5 mM EDTA.
- Prepare the GST-elution buffer using 10 mM reduced glutathione (0.15 g/50 mL) dissolved in a buffer of 50 mM Tris pH 8.0 at 25 °C, 150 mM NaCl, 1 mM DTT. Confirm that the pH of the final buffer is 8.0 and use on the same day it was made.
- Prepare the Gel-filtration buffer using 20 mM Tris pH 7.5 at 25 °C, 60 mM NaCl, 2 mM DTT. Sterile filter the buffer with a bottle-top filter (0.2 µM pore size). Degas the buffer while stirring under vacuum (-6 psi) for 15 min.
- Add DTT to all above buffers on the day of use.
Thaw the cells from Section 2 that contain the CENP-F construct. Adjust the volume to 40 mL with the GST-binding buffer. Add 1 mM DTT, 0.25 mM PMSF, 0.5 mM EDTA (0.5 M stock solution, pH 8.0), 7 mg benzamidine hydrochloride (1 mM), and 40 mg D/L methionine. NOTE: To reduce proteolytic degradation, perform all steps at 4 °C, preferably in a cold room, unless otherwise noted. Keep cells, lysates, and purified protein fractions on ice at all times, and add protease inhibitors. To prevent oxidation of cysteine and methionine residues, add reductants such as DTT and methionine on the day of the experiment. To reduce proteolytic degradation, work through the steps quickly.
Lyse the cells in a sonicator as follows. Fill a glass beaker with the lysate and immerse it in an ice water bath. Sonicate the culture (1 min 40 s, at 100 W (50%) output/amplitude, with 10 s pulses and 10 s rest cycles).Add 250 µM PMSF after sonication. Inspect the cell lysate, which should look more transparent and no longer be viscous.
Clear the lysate by centrifuging at 12,000-40,000 x g, 25 min, 4 °C. Use round-bottom centrifuge tubes and a fixed angle rotor.
Perform equilibration by adding 2 mL of glutathione agarose 1:1 slurry to a disposable chromatography column. The resulting column will have a volume of 1 mL. Wash the column with 25 mL of ultrapure water and 25 mL of GST-binding buffer.
Perform binding as follows. Working in a cold room or cold box, decant the lysate from the pellets. Filter the lysate (0.2 µm pore size) and pour it onto the plugged column. Cap the column and incubate it while gently nutating for 30 min at 4 °C.
To wash the column, put it on a rack, let the resin settle, and collect the flow through. Wash the column twice with 25 mL of GST-binding buffer.
- Elute by proteolytic cleavage.
- Plug the column. Add 400 µL of GST-binding buffer and 250 µL of PS protease (2 mg/mL stock with an activity of 1,000 U/mg, either purified in the lab or purchased; see Table of Materials).
- Cap the column and gently swirl to resuspend the resin in the buffer. Incubate the columns for 16-20 h at 4 °C. Then add 4 mL of GST-binding buffer to elute the proteolytically cleaved CENP-F fragment from the column.
- Glutathione elution
- Plug the column, add 4 mL of GST-elution buffer (4 column volumes), and incubate for 10 min to elute the bound GST-tag. Collect the eluate. Regenerate the columns before re-use as described by the manufacturer.
Analyze the PS protease elution fraction and the glutathione elution fraction by SDS-PAGE on 16% acrylamide gels. Perform electrophoresis at 25 V/cm for 45 min. Stain the gels by Coomassie Blue. The PS protease fraction will contain the purified CENP-F fragment, whereas the glutathione fraction will contain the cleaved GST-tag. Inspect the gel to assess proteolytic cleavage efficiency.
Concentrate the PS protease eluate to 0.5 mL using centrifugal filter units with a 3 kDa molecular weight cutoff (see Table of Materials). Add the eluate in the upper compartment of the filter, and centrifuge it in 10-15 min increments at 4,100 x g, 4 °C (swing-out rotor) to concentrate the sample. Mix by pipetting after each increment.
Connect a suitable gel filtration column (see Table of Materials for purchasing information) to a fast protein liquid chromatography (FPLC) system, and equilibrate it with 1 column volume of gel filtration buffer.
Centrifuge the concentrated CENP-F fragment in a microcentrifuge (20 min, 21,700 x g, 4 °C). Inject the sample on the column and elute it with 1 column volume of gel filtration buffer. Collect 0.6 mL fractions. NOTE: The gel filtration buffer is optimized for compatibility with the subsequent kinase assay. Test if the target protein is stable in this buffer. If it is not stable, try adding more sodium chloride.
Analyze the peak fractions by SDS-PAGE (step 3.11). Pool the peak fractions that contain pure CENP-F fragments. Concentrate the purified CENP-F fragments to 3.3 mg/mL (step 3.12). Analyze the purified CENP-F fragments by SDS-PAGE (step 3.11).
- Protein concentration determination
- Dilute the sample 1:100 in ultrapure water and place it in a quartz micro-cuvette. Record spectra at a wavelength range of 220-300 nm in a spectrophotometer. NOTE: The protein concentration c (mg/mL) is derived from the following equation:
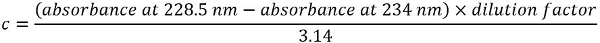
Make 50 µL aliquots of the purified protein in 0.5 mL microtubes and flash-freeze them in liquid nitrogen. Store the aliquots at -80 °C.
4. Purification of Recombinant Protein by Ni-NTA Affinity Chromatography
- Make the following buffers and cool to 4 °C.
- Prepare the His6-binding buffer using 10 mM Tris pH 8.0 at 25 °C, 250 mM NaCl, 5 mM Imidazole pH 8.0, 5 mM BME. Add BME to all buffers on the day of use.
- Prepare the His6-wash buffer in the same manner as the His6-binding buffer but using 20 mM Imidazole.
- Prepare the His6-elution buffer in the same manner as the binding buffer but using 150 mM Imidazole.
- Thaw the cells from Section 2 that contain the karyopherin α construct and adjust the volume to 40 mL with the His6-binding buffer. Add 5 mM BME, 0.250 mM PMSF, 7 mg benzamidine hydrochloride (1 mM), and 40 mg D/L methionine.
- Proceed as described in steps 3.4-3.8 with the following variations: Use His6-binding buffer instead of GST-binding buffer for all steps. Instead of glutathione agarose, use 4 mL of the Nickel affinity gel (see Table of Materials) to make the column, which will have a column volume of 2 mL. NOTE: Nickel affinity columns are incompatible with DTT and EDTA. Instead of the Nickel affinity gel, Ni2+-nitrilotriacetic acid (NTA) agarose can be used, if the imidazole concentration in the elution buffer is increased to 250 mM (see Table of Materials).
Wash the columns with 8 mL of His6-wash buffer.
Elute the karyopherin α with 12 mL of His6-elution buffer and analyze eluate by SDS-PAGE (step 3.11).
Add 250 µL of PS protease (step 3.9) to the eluate and incubate for 16-20 h at 4 °C. In the meantime, dialyze the eluate twice against 1 L of His6-binding buffer for 2-20 h, using a dialysis membrane with a 6-8 kDa molecular weight cutoff.
Regenerate the Nickel affinity gel column as described by the manufacturer. Equilibrate the column with 25 mL of His6-binding buffer. Add the dialyzed protein to the column and perform the binding step as described in step 3.7.
Collect the flow through, which contains the purified karyopherin α (with the His6-tag cleaved off), and elute the remaining protein with an additional 4 mL of His6-binding buffer. Pool these fractions. Then elute the columns with 12 mL of His6-elution buffer. This fraction contains impurities.
Analyze the uncleaved and cleaved karyopherin α from steps 4.4-4.5 and the two elution fractions from this step by SDS-PAGE (step 3.11) to evaluate the efficiency of His6-tag cleavage and purity.
Concentrate the karyopherin α to 8 mg/mL and flash-freeze in liquid nitrogen (steps 3.15-3.17).
5. In Vitro Kinase Assay with Cdk1/Cyclin B
- In vitro kinase assay NOTE: Upon arrival of the kinase, make small aliquots, flash-freeze them in liquid nitrogen and store them at -80 °C. Use within 6 months. While the molecular weight is 34 kDa for Cdk1 and 48 kDa for cyclin B, the apparent molecular weight of cyclin B on SDS-PAGE is about 60 kDa.
- Prepare the 10x PK buffer (supplied with the kinase) using 0.5 M Tris-HCl, 0.1 M MgCl2, 1 mM EDTA, 20 mM DTT, 0.1% Brij 35, pH 7.5 at 25 °C.
- Prepare the 10x ATP stock: Make a solution of 4 mM ATP in 1x PK buffer, and check the final pH of the stock. Store in small aliquots at -20 °C and avoid freeze-thaw cycles.
- Mix 40 µL of CENP-F fragment (at a concentration of 3.3 mg/mL, or 400 µM), 6 µL 10x PK buffer, 3 µL ultrapure water, 6 µL ATP 10x stock, and 5 µL human Cdk1/cyclin B (1 µg, 100 U) in a 0.5 mL microtube. NOTE: The CENP-F fragment has a molar mass of 8.6 kDa and four Cdk1-specific phosphorylation sites. For a new substrate, adjust the kinase concentration in the assay according to the molar concentration of the protein and according to the number of phosphorylation sites. The activity of the human recombinant Cdk1/cyclin B stock is 20,000 U/mL, and the specific activity is 1,000,000 U/mg. 1 U is defined as the amount of Cdk1/cyclin B required to catalyze the transfer of 1 pmol of phosphate to Cdk1 peptide substrate PKTPKKAKKL-NH2 (50 µM) in 1 min at 30 °C (see Table of Materials).
- Prepare a control reaction without Cdk1/cyclin B. Incubate the reactions in a water bath for 1-16 h at 30 °C.
Analyze 2.5 µL of the kinase assay reaction and 2.5 µL of the control by SDS-PAGE (step 3.11), using a gel with 16% acrylamide. To increase the resolution, extend the running time.
Add an equal volume of 6 M guanidine hydrochloride solution to the remaining kinase assay reaction (and to the control). The final guanidine hydrochloride concentration will be 3 M. Analyze these samples by mass spectrometry (Section 6) to identify the phosphorylation sites or send the samples to a mass spectrometry facility for analysis, and then perform function verification (Section 7). NOTE: For the subsequent mass spectrometry analysis, it is very important that the phosphorylation efficiency of the individual sites is as high as possible, preferably 100%, as otherwise the phosphorylation sites cannot be mapped. To greatly enhance phosphorylation efficiencies, add larger amounts of Cdk1/cyclin B and increase the incubation time to 16 h. The concentration of ATP can also be doubled.
For troubleshooting, perform an in vitro kinase assay of CENP-F (residues 2,987-3,065) as a positive control. Use fresh batches of Cdk1/cyclin B with high activities.
6. Identification of Cdk1-specific Phosphorylation Sites by Mass Spectrometry
To denature and desalt the protein samples, perform microbore reverse phase liquid chromatography with a PLRP300 column.
To obtain the number of phosphorylation sites in the CENP-F fragment, analyze the intact in vitro phosphorylated CENP-F 84-mer by electrospray ionization ion trap mass spectrometry (ESI-ITMS)44,45.
To assess the phosphoamino acid site location of CENP-F, prepare the trypsin digests of the in vitro phosphorylated CENP-F protein samples. Desalt the trypsin digests with desalting pipet tips.
Analyze the tryptic digests of CENP-F by electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry (ESI-FTICR MS). Perform analysis on an ESI-FTICR mass spectrometer with an accumulation time of 169 µs. NOTE: The accuracy of masses determined by ESI-FTICR MS is within 0.005 amu. To quantify the ratio of each phosphorylated species respective to the total protein amount, determine the peak heights of the mass spectra.
7. Functional Verification: Testing the Effects of Phosphomimetic Mutations on Protein-protein Interactions by Analytical Size Exclusion Chromatography
NOTE: For functional verification of the identified Cdk1-specific phosphorylation sites, phosphomimetic mutants of CENP-F fragments were created by replacing the identified phosphorylation sites with aspartates. The negative charge of aspartate mimics the effects of phosphorylation. A S3048D mutant of the CENP-F fragment (residues 2,987-3,065) was created.
For functional verification, compare the binding of wild-type and phosphomimetic CENP-F fragments to karyopherin α. Use an analytical gel filtration as the binding assay. For the analytical gel filtration, purify CENP-F fragments by glutathione affinity chromatography (steps 3.1-3.12), and skip the gel filtration step.
Concentrate the protein to 5-6 mg/mL. Flash-freeze protein aliquots in liquid nitrogen (steps 3.15-3.17).
Mix the purified CENP-F fragments (0.1 mg, either wild-type or the S3048D variant) and purified karyopherin α (0.7 mg) in a 1:1 molar ratio. Adjust the sample volume to 200 µL with gel filtration buffer. Incubate the sample for 30 min on ice, filter sample (0.2 µm pore size), and clear the sample by centrifugation (25 min, 21,700 x g, 4 °C)
Separate the sample by size exclusion chromatography (steps 3.13-3.14). Use a gel filtration buffer consisting of 20 mM HEPES, pH 7.5, 150 mM NaCl, and 2 mM DTT. Perform the analytical gel filtration experiments under identical conditions for all samples.
Calibrate the analytical gel filtration column with molecular weight standards of known molar mass as described by the manufacturer.
Analyze the elution fractions by 16% SDS-PAGE (step 3.11).
Representative Results
We have recently used an in vitro kinase assay (Figure 1) to identify Cdk1-specific phosphorylation sites in a CENP-F fragment that contained a cNLS10. This signal confers nuclear localization of CENP-F during most of interphase. In the G2 phase, CENP-F is exported from the nucleus to the cytosol in a Cdk1-dependent manner. To obtain mechanistic insights on how Cdk1 regulates cellular localization of CENP-F, we analyzed the sequence of CENP-F to predict Cdk1-specific phosphorylation sites, using the iGPS server36,37. Within the cNLS of CENP-F, three Cdk1-specific phosphorylation sites were predicted at residues 3,042, 3045, and 3,048 (Table 1). Another Cdk1-specific phosphorylation site was also predicted for residue 3,007 (Table 1)10. Therefore, we hypothesized that Cdk1 regulates CENP-F localization directly by phosphorylation of its cNLS.
To test whether the cNLS of CENP-F is a substrate for Cdk1, we performed an in vitro kinase assay with the purified human CENP-F fragment (residues 2,987-3,065) and human Cdk1/cyclin B. In vitro phosphorylated CENP-F was analyzed on 16% SDS-PAGE, together with a negative control that lacked the kinase Cdk1 (Figure 2A)10. For in vitro phosphorylated CENP-F, a clear upward shift is observed for the band on the gel compared to the negative control. This is typical of phosphorylated proteins as each phosphate group adds two negative charges and additional mass. These results suggest that CENP-F is phosphorylated by Cdk1/cyclin B10.
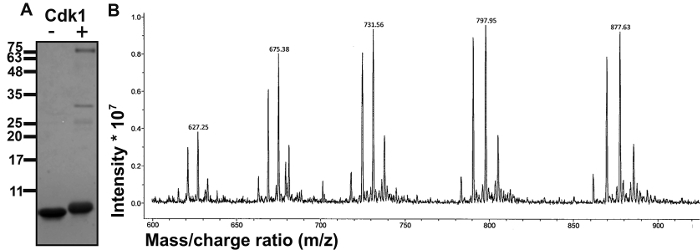
To determine the number of CENP-F phosphorylation sites and to quantify the phosphorylation efficiency of these sites, the intact in vitro phosphorylated CENP-F fragment (84-mer) was analyzed by mass spectrometry (ESI-ITMS)10. The observed ESI-ion trap mass spectrum (Figure 2B) suggests that the intact CENP-F fragment is phosphorylated at four sites (Table 1)10.
The location of the phosphorylated amino acid sites in the sequence of CENP-F was assessed by examining tryptic digests by ESI-FTICR MS. Trypsin cleaves protein chains after lysines and arginines, and a trypsin digest of the CENP-F fragment resulted in several peptides, including one that contained residues 3,031-3,052. Mass spectra of this CENP-F peptide were consistent with phosphorylation at residues T3042, T3045, and S3048, which are located within the cNLS (Table 1)10. Mass spectra of another tryptic peptide (residues 2,995-3,016) were consistent with phosphorylation at S3007 (Table 1)10. All four Cdk1-specific phosphorylation sites were also predicted from the sequence. Cdk1-specific substrates often contain a proline next to the residue that is phosphorylated6, as observed for residues T3042, T3045, and S3007. It should be noted that S3048 is a somewhat unusual Cdk1-specific phosphorylation site, as a phenylalanine is located next to the serine. To conclude, the results suggest that CENP-F is specifically phosphorylated at residues 3,007, 3,042, 3,045, and 3,048 by the kinase Cdk1. Three of these residues are located within the cNLS of CENP-F (Table 1), indicating that Cdk1 may regulate CENP-F cellular localization through phosphorylation of the cNLS in the G2 phase, when Cdk1 becomes active10.
To verify that these Cdk1-specific phosphorylation sites have a physiological function, we created the phosphomimetic variant S3048D10. Phosphomimetic mutations mimic the effects of phosphorylation by negatively charged residues. Next, we purified the phosphomimetic version of the CENP-F fragment that contained the cNLS. The cNLS of CENP-F is recognized by the nuclear transport receptor karyopherin α, which binds with high affinity and is required for nuclear import of CENP-F. To test whether the phosphomimetic mutation weakens the interaction between the cNLS of CENP-F with its nuclear transport receptor karyopherin α, we performed a binding assay (Figure 3)10. In these experiments, a purified CENP-F fragment (either the wild-type or the S3048D variant) was mixed with purified human karyopherin α, and the mixture was separated by analytical size exclusion chromatography. In addition, the proteins were also analyzed individually. SDS-PAGE analysis of the elution fractions revealed that the wild-type CENP-F fragment with the cNLS interacts strongly with karyopherin α, since both proteins co-elute (Figure 3)10. However, only a negligible amount of the CENP-F S3048D fragment binds to karyopherin α, and these proteins elute separately (Figure 3)10. These results suggest that phosphorylation of residue 3,048 of the cNLS of CENP-F would have a strong effect on the interaction with the nuclear transport factor karyopherin α. It should be noted that nuclear import rates are highly dependent on the affinity of a nuclear localization signal towards a nuclear transport factor46, and therefore, it is expected that these mutations slow down nuclear import10.
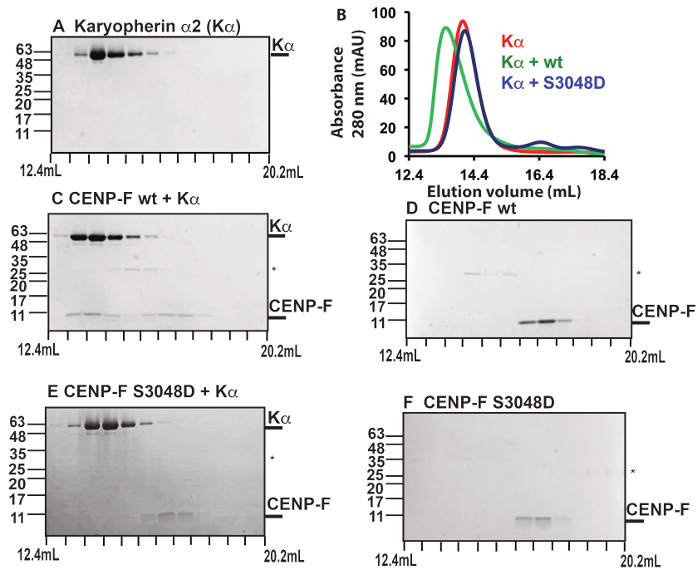
Figure 1: Schematic representation of the Cdk1 in vitro kinase assay. Please click here to view a larger version of this figure.
Figure 2: CENP-F is phosphorylated by Cdk1/cyclin B. (A) To identify Cdk1 specific phosphorylation sites, an in vitro kinase assay was performed with purified CENP-F (residues 2,987-3,065). SDS-PAGE analysis of a negative control without Cdk1/cyclin B is shown in the left lane (-Cdk1), next to in vitro phosphorylated CENP-F in the right lane (+Cdk1). Molecular weight standards are indicated. Note the upward shift of phosphorylated CENP-F on SDS-PAGE, which is consistent with successful phosphorylation. Cdk1 and cyclin B appear as faint bands at 34 kDa and 60 kDa. (B) ESI-ion trap mass spectrometry of intact phosphorylated CENP-F from (A) was used to determine the phosphate load of the intact phosphorylated CENP-F (84-mer). The corresponding mass spectrum is shown. The intensity is plotted versus the mass-to-charge ratio (m/z). The spectrum shows the +10 to +14 charge states of the intact CENP-F fragment after phosphorylation. They indicate a species population with 0, 1, 2, 3, and 4 phosphate esters. To quantify the ratio of each species to total protein amount, the peak heights were determined and compared (Table 1). This figure has been modified from10 and was reproduced with permission by Taylor and Francis publisher. Please click here to view a larger version of this figure.
Figure 3: Functional verification: phosphomimetic mutations of CENP-F strongly diminish the interaction with the nuclear transport receptor karyopherin α. (A-F) Purified CENP-F fragments (residues 2,987-3,065) (0.1 mg) and purified karyopherin α (0.7 mg) and were mixed in 1:1 molar ratio and analyzed by size exclusion chromatography. The CENP-F fragments and karyopherin α were also analyzed individually. SDS-PAGE analysis of the peak fractions is shown. Elution volumes are indicated on the bottom (in 0.6 mL increments). On the left, positions of molecular weight marker bands and their masses in kDa are indicated. An asterisk marks traces of residual GST (glutathione S-transferase). (A) Karyopherin α. (B) Elution profiles. The absorbance at 280 nm is plotted versus the elution volume. The elution profile for karyopherin α (red) is overlaid with elution profiles of mixtures of karyopherin α with CENP-F fragments (wild-type: green; phosphomimetic S3048D mutant: blue). Note that addition of the wild-type CENP-F fragment shifts the elution volume of the karyopherin α peak to higher mass, which is consistent with binding of the CENP-F wild-type fragment. This shift is not observed when the CENP-F fragment with the phosphomimetic mutation S3048D is added, suggesting that the phosphomimetic mutation greatly diminishes the interaction of CENP-F with karyopherin α. (C) CENP-F wild-type fragment (wt) and karyopherin α. (D) CENP-F wt fragment. (E) CENP-F S3048D fragment and karyopherin α. (F) CENP-F S3048D fragment. This figure was created with data from10. Please click here to view a larger version of this figure.
| Table 1A. Sequence of the CENP-F fragment (residues 2987-3065) with predicted Cdk1-specific phosphorylation sites highlighted by larger font size. Tryptic fragments are highlighted in bold and underlined. | ||
| GPLGSQQSKQDSRGSPLLGPVVPGPSPIPSVTEKRLSSGQNKAS | ||
| GKRQRSSGIWENGGGPTPATPESFSKKSKKAVMSGIHPAE | ||
| Table 1B. Mass spectrometry analysis of the intact CENP-F | ||
| 84 mer after in vitro phosphorylation with Cdk1/cyclin B | ||
| # of phosphorylation sites (P) detected | Phosphate load expressed as % of total protein | |
| CENP-F 2987-3065 | 0P | 6% |
| 1P | 37% | |
| 2P | 40% | |
| 3P | 15% | |
| 4P | 3% | |
| Table 1C. Mass spectrometry analysis of tryptic peptides of CENP-F after in vitro phosphorylation with Cdk1/cyclin B | ||
| # of phosphorylation sites | Phosphate load | |
| Tryptic phosphopeptide, | 0P | 57% |
| residues 2995-3016 | 1P | 43% |
| Tryptic phosphopeptide, | 0P | 7% |
| residues 3031-3052 | 1P | 64% |
| 2P | 29% | |
| 3P | 1% |
Table 1: Mass spectrometry analysis of the CENP-F fragment after in vitro phosphorylation with Cdk1/cyclin B. This table was created with data from10.
Discussion
Our in vitro kinase assay is a very powerful method to identify molecular targets for the kinase Cdk1, which is a master controller of the cell cycle and regulates many important cellular processes. The method determines if a purified protein is a substrate for Cdk1 and allows identification of specific phosphorylation sites. This facilitates mechanistic studies for regulation of cellular processes by phosphorylation through Cdk1.
The most critical factor for successful identification of the phosphorylation sites by mass spectrometry is the phosphorylation efficiency of the kinase assay. The phosphorylation efficiency of the individual sites should be as high as possible, preferably 100%. This can be achieved by increasing the amount of Cdk1/cyclin B and increasing the incubation time to 16 h. When calculating the amount of Cdk1/cyclin B needed, the molar concentration of the protein and the number of phosphorylation sites should be considered. This is especially important if multiple phosphorylation sites are present in the target protein. It should also be noted that the activity of commercial Cdk1/cyclin B can vary from batch to batch, and that the kinase can lose activity rapidly. If in vitro phosphorylation is unsuccessful, it is recommended to test the activity of the kinase. As a positive control for troubleshooting, in vitro phosphorylation of CENP-F (residues 2,987-3,065) can be used. As a negative control, a reaction without Cdk1/cyclin B addition should be performed and analyzed. Another useful negative control is the use of a kinase-dead mutant of Cdk1, which is catalytically inactive, due to a simple point mutation (D146N)47,48,49. In addition, to verify identified phosphorylation sites, a phosphonegative version of the target protein can be created, where the phosphorylation sites are replaced by alanines. If the identified phosphorylation sites are correct, the corresponding phosphonegative mutant cannot be phosphorylated in vitro.
A simple tool to detect successful phosphorylation of the target protein is the simple SDS-PAGE gel shift assay described here. Phosphorylated proteins typically migrate slower on SDS-PAGE than their unphosphorylated counterparts, due to the added size and negative charges. Note that for large proteins, optimization of the SDS-PAGE analysis is required to enhance the resolution sufficiently to visualize the gel shift. A useful tool for the specific separation of phosphorylated proteins is phosphate-binding tag (Phos-tag) SDS-PAGE. Phosphorylated proteins in a gel prepared with Phos-tag acrylamide are visualized as slower migrating bands compared with corresponding dephosphorylated proteins50,51.
To obtain high quality mass spectra, it is very important that the protein for the in vitro kinase assay is pure and homogeneous. Our purification protocol achieves this by combining several highly selective chromatography steps and by preventing protein degradation and oxidation through the addition of protease inhibitors and reductants. The purification protocol can be modified to elute the protein by glutathione (i.e., with the GST-tag intact); however, it should be considered that the GST-tag adds another 25 kDa, and large proteins are challenging targets for mass spectrometry. Alternatively, our purification protocol for His6-tag fusion proteins can also be used.
This protocol can also be modified to identify phosphorylation sites that are specific for other kinases. The only requirement is that the purified kinase is available, active, and sufficiently stable. Assay conditions need to be adjusted for each kinase to achieve maximum activity. Parameters to optimize include the amount of the kinase, the reaction buffer (pH, salt concentration), incubation time, reaction temperature, and ATP concentration. In vitro kinase assays have been successfully used to identify phosphorylation sites for many kinases including Plks (polo-like kinases)52,53,54, and MAPK/ERK kinases (mitogen-activated protein kinases /extracellular-signal-regulated kinases)55,56.
While the strength of this method is to identify specific phosphorylation sites in a purified protein, it should be noted that a phosphorylation site that is a target for Cdk1 in vitro may not necessarily be phosphorylated in vivo. In vivo, additional regulatory mechanisms are in place, which could render a phosphorylation site inaccessible for recognition by Cdk1. For example, additional interaction partners may be present in vivo, which could either conceal a phosphorylation site or make it accessible through structural changes. Furthermore, the substrate needs to colocalize with Cdk1/cyclin B in the nucleus in the G2 phase in order to be phosphorylated. In addition, post-translational modifications or other regulatory processes could impact the conformation of the target protein in vivo, which could render a phosphorylation site inaccessible. These limitations can be overcome by designing appropriate experiments in cells or animal models that verify that the identified phosphorylation sites have a physiological function. Here, we use a simple binding assay for functional verification, since phosphorylation diminishes the interaction of CENP-F with karyopherin α. Functional verification should also be performed in the context of cells or an animal model. Therefore, we also transfected fluorescent fusion proteins of CENP-F-fragments that contained the cNLS into HeLa cells10. Phosphomimetic mutations within the cNLS of CENP-F diminished nuclear localization, which confirmed a physiological function of these phosphorylation sites10.
Several tools are available for functional verification of phosphorylation sites in cells or animal models. Phosphomimetic mutations, which mimic the effects of phosphorylation by negative charges, are widely used for this purpose. For these, the phosphorylation site (serine or threonine) is replaced by aspartate or glutamate. The advantage is that only a point mutation of the target protein is required. It should be noted, that while phosphomimetic mutations were successfully used in many cases for functional verification, they especially work in cases where charge changes are the predominant contributor for regulation rather than a structural change. Phosphomimetic mutations fail to mimic effects of phosphorylation in many other cases (e.g., 11). However, other approaches are available for functional verification of phosphorylation sites, including the use of phosphonegative mutations: these mutations prevent phosphorylation by replacing phosphorylation sites with alanine. Another useful approach for functional verification in cellular context is the expression of a kinase-dead mutant of Cdk1, which is inactivated by an easy-to-introduce point mutation (D146N)47,48,49. Notably, several small molecule Cdk1 inhibitors are commercially available such as Flavopiridol and RO-3306, which can be used for functional studies either in cells or animal models7,8,11. These inhibitors are well-characterized, and several are in clinical trials for cancer treatment (for a review see9).
While a large number of protein phosphorylation sites have been identified in proteomic studies, the kinase specificity is in most cases unknown, and the in vitro kinase assay is one of the few methods available to identify substrates of Cdk1. Other approaches have also been used. Cdk1 targets were identified by using an engineered Cdk1 enzyme. It has a larger substrate binding pocket and accepts more bulky versions of ATP as a substrate6. This bulky substrate cannot bind to wild-type kinases. The addition of radiolabeled bulky ATP to a cell extract containing the modified Cdk1 enzyme therefore leads to the specific labeling of the direct substrates of Cdk1. 200 Cdk1 targets were identified in budding yeast with this method; however, it cannot be applied to mammalian cells, which is a crucial limitation. In another study, Cdk1 substrates were identified in human tissue culture cells by performing quantitative phosphoproteomics analysis using two small molecule inhibitors of Cdk1, Flavopiridol, and RO-3306. 1215 phosphopeptides on 551 proteins were significantly reduced upon Cdk1 inhibitor addition7. This method can screen an entire proteome for potential Cdk1 targets, but resulting targets need to be verified, e.g., by an in vitro kinase assay.
In the future, the in vitro kinase assay will likely be combined with high throughput screens for Cdk1 substrates that are currently under development. Due to the large number of Cdk1 substrates in the proteome, many human Cdk1 substrates remain to be identified, and the in vitro kinase assay will be an important tool to identify, map, and verify these sites.
The in vitro kinase assay is a powerful tool to identify targets for the kinase Cdk1 and to identify specific phosphorylation sites. Identification of these phosphorylation sites enables mechanistic studies for the regulation of cellular processes by Cdk1.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank Dr. David King, Howard Hughes Medical Institute, University of California at Berkeley for mass spectrometry analysis and helpful comments. We thank Dr. Xuelian Zhu, Shanghai, Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, China for providing a full-length CENP-F construct. Finally, we thank Dr. Susan Bane, Dr. Brian Callahan and Dr. Christof Grewer at Binghamton University for access to equipment. This research was funded by the Research Foundation for the State University of New York and the Department of Chemistry, State University of New York at Binghamton.
References
- Nurse P. Cyclin dependent kinases and cell cycle control (Nobel Lecture) ChemBioChem. 2002;3(7):596–603. doi: 10.1002/1439-7633(20020703)3:7<596::AID-CBIC596>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Lee MG, Nurse P. Complementation used to clone a human homologue of the fission yeast cell cycle control gene cdc2. Nature. 1987;327:31. doi: 10.1038/327031a0. [DOI] [PubMed] [Google Scholar]
- Lohka MJ, Hayes MK, Maller JL. Purification of maturation-promoting factor, an intracellular regulator of early mitotic events. Proc Natl Acad of Sci U S A. 1988;85(9):3009–3013. doi: 10.1073/pnas.85.9.3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier J, Norbury C, Lohka M, Nurse P, Maller J. Purified maturation-promoting factor contains the product of a Xenopus homolog of the fission yeast cell cycle control gene. Cell. 1988;54(3):433–439. doi: 10.1016/0092-8674(88)90206-1. [DOI] [PubMed] [Google Scholar]
- Gautier J, Minshull J, Lohka M, Glotzer M, Hunt T, Maller JL. Cyclin is a component of maturation-promoting factor from Xenopus. Cell. 1990;60(3):487–494. doi: 10.1016/0092-8674(90)90599-a. [DOI] [PubMed] [Google Scholar]
- Ubersax JA, Woodbury EL, Quang PN, Paraz M, Blethrow JD, Shah K, Shokat KM, Morgan DO. Targets of the cyclin-dependent kinase Cdk1. Nature. 2003;425(6960):859–864. doi: 10.1038/nature02062. [DOI] [PubMed] [Google Scholar]
- Petrone A, Adamo ME, Cheng C, Kettenbach AN. Identification of Candidate Cyclin-dependent kinase 1 (Cdk1) Substrates in Mitosis by Quantitative Phosphoproteomics. Mol Cell Proteomics. 2016;15(7):2448–2461. doi: 10.1074/mcp.M116.059394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev LT, Tovar C, Chen S, Knezevic D, Zhao X, Sun H, Heimbrook DC, Chen L. Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc Natl Acad of Sci U S A. 2006;103(28):10660–10665. doi: 10.1073/pnas.0600447103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan A, Vyas A, Deshpande K, Vyas D. Pharmacological cyclin dependent kinase inhibitors: Implications for colorectal cancer. World J Gastroenterol. 2016;22(7):2159–2164. doi: 10.3748/wjg.v22.i7.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loftus KM, Coutavas E, Cui H, King D, Ceravolo A, Pereiras D, Solmaz S. Mechanism for G2 phase-specific nuclear export of the kinetochore protein CENP-F. Cell Cycle. 2017;16(15):1414–1429. doi: 10.1080/15384101.2017.1338218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baffet AD, Hu DJ, Vallee RB. Cdk1 activates pre-mitotic nuclear envelope dynein recruitment and apical nuclear migration in neural stem cells. Dev Cell. 2015;33(6):703–716. doi: 10.1016/j.devcel.2015.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Songyang Z, Blechner S, Hoagland N, Hoekstra MF, Piwnica-Worms H, Cantley LC. Use of an oriented peptide library to determine the optimal substrates of protein kinases. Curr Biol. 1994;4(11):973–982. doi: 10.1016/s0960-9822(00)00221-9. [DOI] [PubMed] [Google Scholar]
- Malumbres M. Cyclin-dependent kinases. Genome Biol. 2014;15(6):122. doi: 10.1186/gb4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeper DS, Parker LL, Ewen ME, Toebes M, Hall FL, Xu M, Zantema A, van der Eb AJ, Piwnica-Worms H. A- and B-type cyclins differentially modulate substrate specificity of cyclin-cdk complexes. EMBO J. 1993;12(5):1947–1954. doi: 10.1002/j.1460-2075.1993.tb05844.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trembley J, Ebbert J, Kren B, Steer C. Differential regulation of cyclin B1 RNA and protein expression during hepatocyte growth in vivo. Cell Growth Differ. 1996;7(7):903–916. [PubMed] [Google Scholar]
- Pines J, Hunter T. The differential localization of human cyclins A and B is due to a cytoplasmic retention signal in cyclin B. EMBO J. 1994;13(16):3772–3781. doi: 10.1002/j.1460-2075.1994.tb06688.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan DO. Principles of CDK regulation. Nature. 1995;374:131. doi: 10.1038/374131a0. [DOI] [PubMed] [Google Scholar]
- Larochelle S, Merrick KA, Terret M-E, Wohlbold L, Barboza NM, Zhang C, Shokat KM, Jallepalli PV, Fisher RP. Requirements for Cdk7 in the assembly of Cdk1/cyclin B and activation of Cdk2 revealed by chemical genetics in human cells. Mol cell. 2007;25(6):839–850. doi: 10.1016/j.molcel.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker LL, Sylvestre PJ, Byrnes MJ, Liu F, Piwnica-Worms H. Identification of a 95-kDa WEE1-like tyrosine kinase in HeLa cells. Proc Natl Acad of Sci U S A. 1995;92(21):9638–9642. doi: 10.1073/pnas.92.21.9638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atherton-Fessler S, Parker LL, Geahlen RL, Piwnica-Worms H. Mechanisms of p34cdc2 regulation. Mol Cell Biol. 1993;13(3):1675–1685. doi: 10.1128/mcb.13.3.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Stanton JJ, Wu Z, Piwnica-Worms H. The human Myt1 kinase preferentially phosphorylates Cdc2 on threonine 14 and localizes to the endoplasmic reticulum and Golgi complex. Mol Cell Biol. 1997;17(2):571–583. doi: 10.1128/mcb.17.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan CH, Russell P. Human Wee1 kinase inhibits cell division by phosphorylating p34cdc2 exclusively on Tyr15. EMBO J. 1993;12(1):75–85. doi: 10.1002/j.1460-2075.1993.tb05633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strausfeld U, Labbé JC, Fesquet D, Cavadore JC, Picard A, Sadhu K, Russell P, Dorée M. Dephosphorylation and activation of a p34cdc2/cyclin B complex in vitro by human CDC25 protein. Nature. 1991;351:242. doi: 10.1038/351242a0. [DOI] [PubMed] [Google Scholar]
- Leuken R, Clijsters L, Wolthuis R. To cell cycle, swing the APC/C. Biochim Biophys Acta. 2008;1786(1):49–59. doi: 10.1016/j.bbcan.2008.05.002. [DOI] [PubMed] [Google Scholar]
- Acquaviva C, Pines J. The anaphase-promoting complex/cyclosome: APC/C. J Cell Sci. 2006;119(12):2401–2404. doi: 10.1242/jcs.02937. [DOI] [PubMed] [Google Scholar]
- Zhu X, Chang K-H, He D, Mancini MA, Brinkley WR, Lee W-H. The C-terminus of mitosin is essential for its nuclear localization, centromere/kinetochore targeting, and dimerization. J Biol Chem. 1995;270(33):19545–19550. doi: 10.1074/jbc.270.33.19545. [DOI] [PubMed] [Google Scholar]
- Liao H, Winkfein RJ, Mack G, Rattner JB, Yen TJ. CENP-F is a protein of the nuclear matrix that assembles onto kinetochores at late G2 and is rapidly degraded after mitosis. J Cell Biol. 1995;130(3):507–518. doi: 10.1083/jcb.130.3.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattner JB, Rao A, Fritzler MJ, Valencia DW, Yen TJ. CENP-F is a ca 400 kDa kinetochore protein that exhibits a cell-cycle dependent localization. Cell Motil Cytoskeleton. 1993;26(3):214–226. doi: 10.1002/cm.970260305. [DOI] [PubMed] [Google Scholar]
- Christie M, Chang C-W, Rona G, Smith KM, Stewart AG, Takeda AAS, Fontes MRM, Stewart M, Vertessy BG, Forwood JK, Kobe B. Structural biology and regulation of protein import into the nucleus. J Mol Biol. 2016;428(10A):2060–2090. doi: 10.1016/j.jmb.2015.10.023. [DOI] [PubMed] [Google Scholar]
- Zuccolo M, Alves A, Galy V, Bolhy S, Formstecher E, Racine V, Sibarita JB, Fukagawa T, Shiekhattar R, Yen T, Doye V. The human Nup107/160 nuclear pore subcomplex contributes to proper kinetochore functions. EMBO J. 2007;26:1853–1864. doi: 10.1038/sj.emboj.7601642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolhy S, Bouhlel I, Dultz E, Nayak T, Zuccolo M, Gatti X, Vallee R, Ellenberg J, Doye V. A Nup133-dependent NPC-anchored network tethers centrosomes to the nuclear envelope in prophase. J Cell Biol. 2011;192(5):855–871. doi: 10.1083/jcb.201007118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu DJ, Baffet AD, Nayak T, Akhmanova A, Doye V, Vallee RB. Dynein recruitment to nuclear pores activates apical nuclear migration and mitotic entry in brain progenitor cells. Cell. 2013;154(6):1300–1313. doi: 10.1016/j.cell.2013.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergnolle MS, Taylor SS. Cenp-F links kinetochores to Ndel1/Nde1/Lis1/Dynein microtubule motor complexes. Curr Biol. 2007;17(13):1173–1179. doi: 10.1016/j.cub.2007.05.077. [DOI] [PubMed] [Google Scholar]
- Yang ZY, Guo J, Li N, Qian M, Wang SN, Zhu XL. Mitosin/CENP-F is a conserved kinetochore protein subjected to cytoplasmic dynein-mediated poleward transport. Cell Res. 2003;13(4):275–283. doi: 10.1038/sj.cr.7290172. [DOI] [PubMed] [Google Scholar]
- Yang Z, Guo J, Chen Q, Ding C, Du J, Zhu X. Silencing mitosin induces misaligned chromosomes, premature chromosome decondensation before anaphase onset, and mitotic cell death. Mol Cell Biol. 2005;25(10):4062–4074. doi: 10.1128/MCB.25.10.4062-4074.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Ren J, Gao X, Jin C, Wen L, Yao X. GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol Cell Proteomics. 2008;7(9):1598–1608. doi: 10.1074/mcp.M700574-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C, Ye M, Liu Z, Cheng H, Jiang X, Han G, Songyang Z, Tan Y, Wang H, Ren J, Xue Y, Zou H. Systematic analysis of protein phosphorylation networks from phosphoproteomic data. Mol Cell Proteomics. 2012;11(10):1070–1083. doi: 10.1074/mcp.M111.012625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt-Consortium. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017;45(D1):D158–D169. doi: 10.1093/nar/gkw1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen JV, Vermeulen M, Santamaria A, Kumar C, Miller ML, Jensen LJ, Gnad F, Cox J, Jensen TS, Nigg EA, Brunak S, Mann M. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Science Signal. 2010;3(104):ra3. doi: 10.1126/scisignal.2000475. [DOI] [PubMed] [Google Scholar]
- Dephoure N, Zhou C, Villén J, Beausoleil SA, Bakalarski CE, Elledge SJ, Gygi SP. A quantitative atlas of mitotic phosphorylation. Proc Natl Acad of Sci U S A. 2008;105(31):10762–10767. doi: 10.1073/pnas.0805139105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rona G, Marfori M, Borsos M, Scheer I, Takacs E, Toth J, Babos F, Magyar A, Erdei A, Bozoky Z, Buday L, Kobe B, Vertessy BG. Phosphorylation adjacent to the nuclear localization signal of human dUTPase abolishes nuclear import: structural and mechanistic insights. Acta Cryst D. 2013;69(12):2495–2505. doi: 10.1107/S0907444913023354. [DOI] [PubMed] [Google Scholar]
- Harreman MT, Kline TM, Milford HG, Harben MB, Hodel AE, Corbett AH. Regulation of nuclear import by phosphorylation adjacent to nuclear localization signals. J Biol Chem. 2004;279(20):20613–20621. doi: 10.1074/jbc.M401720200. [DOI] [PubMed] [Google Scholar]
- Kosugi S, Hasebe M, Tomita M, Yanagawa H. Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc Natl Acad Sci U S A. 2009;106(25):10171–10176. doi: 10.1073/pnas.0900604106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLachlin DT, Chait BT. Analysis of phosphorylated proteins and peptides by mass spectrometry. Curr Opin Chem Biol. 2001;5(5):591–602. doi: 10.1016/s1367-5931(00)00250-7. [DOI] [PubMed] [Google Scholar]
- Van Berkel GJ, Glish GL, McLuckey SA. Electrospray ionization combined with ion trap mass spectrometry. Anal Chem. 1990;62(13):1284–1295. [Google Scholar]
- Hodel AE, Harreman MT, Pulliam KF, Harben ME, Holmes JS, Hodel MR, Berland KM, Corbett AH. Nuclear localization signal receptor affinity correlates with in vivo localization in Saccharomyces cerevisiae. J Biol Chem. 2006;281(33):23545–23556. doi: 10.1074/jbc.M601718200. [DOI] [PubMed] [Google Scholar]
- Hong KU, Kim H-J, Kim H-S, Seong Y-S, Hong K-M, Bae C-D, Park J. Cdk1-Cyclin B1-mediated Phosphorylation of Tumor-associated Microtubule-associated Protein/Cytoskeleton-associated Protein 2 in Mitosis. J Biol Chem. 2009;284(24):16501–16512. doi: 10.1074/jbc.M900257200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraldi P, Lukas J, Fry AM, Bartek J, Nigg EA. Centrosome duplication in mammalian somatic cells requires E2F and Cdk2–Cyclin A. Nature Cell Biol. 1999;1:88. doi: 10.1038/10054. [DOI] [PubMed] [Google Scholar]
- Heuvel S, Harlow E. Distinct roles for cyclin-dependent kinases in cell cycle control. Science. 1993;262(5142):2050–2054. doi: 10.1126/science.8266103. [DOI] [PubMed] [Google Scholar]
- Kinoshita E, Kinoshita-Kikuta E, Takiyama K, Koike T. Phosphate-binding Tag, a New Tool to Visualize Phosphorylated Proteins. Mol Cell Proteomics. 2006;5(4):749–757. doi: 10.1074/mcp.T500024-MCP200. [DOI] [PubMed] [Google Scholar]
- Takeda H, Kawasaki A, Takahashi M, Yamada A, Koike T. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry of phosphorylated compounds using a novel phosphate capture molecule. Rapid Commun Mass Spectrom. 2003;17(18):2075–2081. doi: 10.1002/rcm.1154. [DOI] [PubMed] [Google Scholar]
- Linder MI, Köhler M, Boersema P, Weberruss M, Wandke C, Marino J, Ashiono C, Picotti P, Antonin W, Kutay U. Mitotic Disassembly of Nuclear Pore Complexes Involves CDK1- and PLK1-Mediated Phosphorylation of Key Interconnecting Nucleoporins. Dev Cell. 2017;43(2) doi: 10.1016/j.devcel.2017.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai T, Haze K, Iimura-Morita Y, Machida T, Iida M, Tanaka K, Komatani H. Identification of β-catenin as a novel substrate of polo-like kinase 1. Cell Cycle. 2008;7(22):3556–3563. doi: 10.4161/cc.7.22.7072. [DOI] [PubMed] [Google Scholar]
- Hansen DV, Tung JJ, Jackson PK. CaMKII and Polo-like kinase 1 sequentially phosphorylate the cytostatic factor Emi2/XErp1 to trigger its destruction and meiotic exit. Proc Natl Acad of Sci U S A. 2006;103(3):608–613. doi: 10.1073/pnas.0509549102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Dong Z, Nomura M, Zhong S, Chen N, Bode AM, Dong Z. Signal Transduction Pathways Involved in Phosphorylation and Activation of p70S6K Following Exposure to UVA Irradiation. J Biol Chem. 2001;276(24):20913–20923. doi: 10.1074/jbc.M009047200. [DOI] [PubMed] [Google Scholar]
- Richard DE, Berra E, Gothié E, Roux D, Pouysségur J. p42/p44 Mitogen-activated Protein Kinases Phosphorylate Hypoxia-inducible Factor 1α (HIF-1α) and Enhance the Transcriptional Activity of HIF-1. J Biol Chem. 1999;274(46):32631–32637. doi: 10.1074/jbc.274.46.32631. [DOI] [PubMed] [Google Scholar]