

Abstract
The ability to study hematopoietic stem cell (HSC) genesis during embryonic development has been limited by the rarity of HSC precursors in the early embryo and the lack of assays that functionally identify the long-term multilineage engraftment potential of individual putative HSC precursors. Here, we describe methodology that enables the isolation and characterization of functionally validated HSC precursors at the single cell level. First, we utilize index sorting to catalog the precise phenotypic parameter of each individually sorted cell, using a combination of phenotypic markers to enrich for HSC precursors with additional markers for experimental analysis. Second, each index-sorted cell is co-cultured with vascular niche stroma from the aorta-gonad-mesonephros (AGM) region, which supports the maturation of non-engrafting HSC precursors to functional HSC with multilineage, long-term engraftment potential in transplantation assays. This methodology enables correlation of phenotypic properties of clonal hemogenic precursors with their functional engraftment potential or other properties such as transcriptional profile, providing a means for the detailed analysis of HSC precursor development at the single cell level.
Keywords: Developmental Biology, Issue 135, Hematopoietic stem cell (HSC), pre-HSC, aorta-gonad-mesonephros region (AGM), embryonic hematopoiesis, index sorting, single cell, clonal analysis, hematopoietic stem cell transplantation
Introduction
Clonal studies have revealed heterogeneity in the long-term engraftment properties of adult HSCs, providing new insight into HSC subtypes and changes in HSC behavior during aging1. However, similar studies of embryonic HSCs and their precursors have been more challenging. During early embryonic development, HSCs arise from a population of precursors known as hemogenic endothelium in a transient process referred to as the endothelial to hematopoietic transition2. The first HSC, defined by their ability to provide robust, long-term multilineage engraftment following transplantation into conditioned adult recipients, are not detected until after embryonic day 10.5 (E10.5) in the murine embryo, at very low frequency3. During their development, precursors to HSC (pre-HSC) arising from hemogenic endothelium must undergo maturation prior to acquiring the properties of adult HSC which allow for efficient engraftment in transplantation assays4,5,6. Obscuring the study of rare HSC origin, a multitude of hematopoietic progenitors with erythroid, myeloid, and lymphoid potential are already detected prior to the emergence of HSC from pre-HSC7,8. Thus, distinguishing pre-HSC from other hematopoietic progenitors requires methods to clonally isolate cells and provide them with the signals sufficient for their maturation to HSC, to detect their engraftment properties in transplantation assays.
A number of approaches have been described which allow for the detection of pre-HSC by either ex vivo or in vivo maturation to HSC. Ex vivo methods have depended on culture of embryonic tissues, such as the AGM region, where the first HSC are detected in development9. Building on these methods, protocols which incorporate the dissociation, sorting, and re-aggregation of AGM tissues have permitted the characterization of sorted populations containing HSC precursors during development from E9.5 to E11.5 in the para-aortic splanchnopleura (P-Sp)/AGM regions4,5,10; however, these approaches are not amenable to high-throughput analysis of precursors at the single cell level required for clonal analysis. Similarly, in vivo maturation by transplantation into newborn mice, where the microenvironment is presumed to be more suitable for the support of earlier stages of HSC precursors, has also enabled studies of sorted populations from the yolk sac and AGM/P-Sp (P-Sp is the precursor region to the AGM) with characteristics of pre-HSC, but these methods also fail to provide a robust platform for single cell analysis11,12.
Studies from Rafii et al. demonstrated that Akt-activated endothelial cell (EC) stroma can provide a niche substrate for the support of adult HSC self-renewal in vitro13,14,15. We recently determined that Akt-activated EC derived from the AGM region (AGM-EC) provides a suitable in vitro niche for the maturation of hemogenic precursors, isolated as early as E9 in development, to adult-engrafting HSC, as well as the subsequent self-renewal of generated HSC16. Given that this system employs a simple 2-dimensional co-culture, it is readily adaptable for clonal analysis of the HSC potential of individually isolated hemogenic precursors.
We have recently reported an approach to assay the HSC potential of clonal hemogenic precursors by combining index sorting of individual hemogenic precursors from murine embryos with AGM-EC co-culture and subsequent functional analysis in transplantation assays17. Index sorting is a mode of fluorescence-activated cell sorting (FACS) that records (indexes) all phenotypic parameters (i.e., forward scatter (FSC-A), side scatter (SSC-A), fluorescence parameters) of each individually sorted cell such that these features can be retrospectively correlated to subsequent functional analysis following sorting. FACS software records both phenotypic information for each cell and the position/well of the 96-well plate into which it was placed. This technique has previously been elegantly used to identify heterogeneity in adult HSC, determine phenotypic parameters that further enrich for the long-term engrafting subset of HSC, and correlate phenotypic parameters of HSC with transcriptional properties at the single cell level18,19. Here, we provide detailed methodology of this approach that enables identification of unique phenotypic parameters and lineage contributions of pre-HSC during early stages of embryonic development.
Protocol
All methods described here have been approved by the Institutional Animal Care and Use Committee (IACUC) of the Fred Hutchinson Cancer Research Center.
1. Preparation of AGM-EC Monolayers for Co-culture
24 h prior to the AGM dissection and sort, make AGM-EC culture media and filter sterilize.
Add 0.1% gelatin in water (100 µL/well) to each well of a 96-well plate and incubate at room temperature for 15 min. Aspirate the gelatin and leave the plate in a tissue culture hood with the lid removed until the wells are dry.
- Dissociate the AGM-EC monolayers and plate to 96-well plates. NOTE: Maintain the AGM-EC, prepared as previously described16, in AGM-EC culture media. For consistency, AGM-EC cell lines should be frozen as soon as sufficient numbers are obtained (generally passage 1-3) and used at a defined passage number (generally passage 5-12) for co-culture experiments. AGM-EC cell lines are derived from C57BL/6J (CD45.2) AGM. See the Table of Materials for all media compositions.
- Aspirate the media from the AGM-EC culture in a 75 cm2 flask and add 5 mL PBS. Aspirate PBS, and add 3 mL trypsin solution (see Table of Materials).
- Incubate at 37 °C for 2-4 min until most cells are lifting off the plate. Add 7 mL of AGM-EC culture media and pipet 8-10 times with a 10 mL pipette to prepare single cells suspension, and transfer to a 15 mL tube.
- Spin at 300 x g for 5 min, remove the supernatant, re-suspend in 10 mL AGM-EC culture media, and estimate the cell number with a hemocytometer. Dilute the cells to a concentration of 1 x 105 cells/mL in AGM-EC culture media.
Add 100 µL AGM-EC cell suspension (1 x 104 cells) to each well of a gelatinized 96-well plate. Place in a 37 °C, 5% CO2 tissue culture incubator overnight to allow the AGM-EC to attach and form a confluent monolayer. NOTE: Evenly distribute the AGM-EC stromal cells so there is limited overgrowth or cell clumping for optimal co-culture.
- Prepare the AGM-EC monolayer for AGM co-culture.
- The following morning, prepare the AGM serum-free media.
- Using a 12-well multichannel pipette, remove the AGM-EC culture media from the AGM-EC and add 200 µL/well of AGM serum-free media without cytokines to wash out any residual serum-containing media.
- Remove the media and add 200 µL/well of AGM serum-free media with cytokines. Work quickly when removing and adding the media so that the endothelial layer is not compromised; e.g., remove and re-add media one row at a time. Place the 96-well plates in a tissue culture incubator at 37 °C until the embryonic cells are ready for sorting.
2. Preparation of Single Cell Suspension from Murine Embryonic Tissues
- Dissect the AGM from timed matings.
- Set up timed matings for generating embryonic tissues of the desired age.
- Harvest embryos from pregnant females at 9.5 to 11.5 days post coitum (dpc), depending on the stage of pre-HSC to be analyzed. NOTE: Embryonic tissues are harvested from C57BL/6J (CD45.2) mice as previously described and precisely staged based on somite count16. We have focused on analysis of populations from the embryonic AGM region and its precursor, the P-Sp, between E9.5 to E11.5, based on somite staging.
- Dissociate the dissected embryonic tissues.
- Collect the dissected tissues in a 15 mL conical tube containing 10 mL PBS with 10% FBS on ice. Gravity settle tissues, aspirate the PBS/FBS, and then immediately add 1 mL of 0.25% collagenase and place in a 37 °C water bath for 25 min.
- Add 1 mL of PBS/10% FBS and pipet about 20-30 times with a 1 mL pipette tip to obtain a single cell suspension. Add an additional 8 mL of PBS/10% FBS and centrifuge cells at 300 x g for 5 min. Discard the supernatant.
3. Antibody Staining of Murine Embryonic Cells
- Prepare the blocking buffer for antibody staining.
- Add 10 µg/mL anti-mouse CD16/CD32 (Fc receptor (FcR) block) and 1 µg/mL DAPI (1 mg/mL stock in H2O) to 1 mL PBS with 10% FBS.
- Draw up in a 3 mL syringe and pass through a 0.22 µm syringe filter to sterilize. Re-suspend the cell pellet from the dissociated murine embryonic tissues (from step 2.2.2) in 500 µL blocking buffer and incubate on ice for 5 min.
- Prepare the antibody mix17.
- Add 10 µg/mL FcR block to 1 mL PBS with 10% FBS and 10 µL of each fluorochrome-conjugated anti-VE-Cadherin antibody (CD144, see the Table of Materials) and fluorochrome-conjugated anti-EPCR antibody (CD201, see the Table of Materials). Use a 1:100 final dilution of antibodies unless otherwise indicated based on the manufacturer's recommendations or according to titrations. NOTE: This antibody combination is used for defining gates for sorting, as described below (step 4.2). Sorting based on the co-expression of VE-Cadherin and EPCR enables significant enrichment for the portion of AGM-derived cells containing HSC precursor activity, as previously described17.
- Add additional fluorochrome-conjugated antibodies for further phenotypic analysis of individual index-sorted precursors. NOTE: These antibodies are not necessarily used for defining gates for sorting. In this sample protocol, we have included PE-conjugated anti-CD41 antibody and FITC-conjugated anti-CD45 antibody to analyze the relative expression of these hematopoietic markers, which evolve during the emergence of pre-HSC from hemogenic endothelium4,5,16,17. Other markers of interest can be included as desired. Samples stained with fluorochrome-conjugated isotype antibody controls are used to define negative and positive gates for analysis.
- Draw up the antibody mix in a 3 mL syringe and pass through a 0.22 µm syringe filter to sterilize. Add 500 µL of the antibody mix to the cell suspension in blocking buffer, pipette to mix, and incubate on ice for at least 15-20 min.
- Wash the stained cells.
- Add 8 mL PBS/10% FBS. Centrifuge the cells at 300 x g for 5 min, aspirate, and re-suspend the cell pellet with 1 mL PBS/10% FBS.
- Remove cell clumps by pipetting the cell suspension through a 35 μm cell strainer cap on a 5 mL tube. Wash the cell strainer with an additional 3 mL PBS/10% FBS. Centrifuge the cells at 300 x g for 5 min, aspirate, re-suspend in 500 µL PBS/10% FBS, and place on ice until FACS.
4. Index Sorting of Single Hemogenic Precursors to 96-wells with AGM-EC Stroma for Co-culture
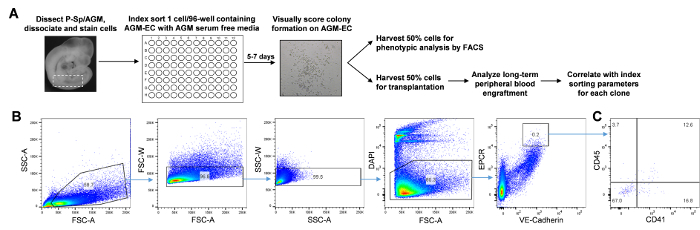
Prepare the FACS machine for plate mode and align for sorting to 96-well plates according to the manufacturer's instructions. Select single cell mode and index sorting mode in the software for maximal purity mask settings and to enable acquisition of index parameters for each sorted cell.
- Acquire a sample of cells from the antibody stained samples to set gates for sorting.
- Load the stained cell sample on the FACS machine and acquire cells at the lowest flow rate. Plot FSC-A verses SSC-A and select a gate that includes cells of varying size (Figure 1B) (which is based on the observation that pre-HSC activity is identified in cells of different size profiles in the AGM17). Plot FSC-A verses FSC-W and SSC-A verses SSC-W, and select gates to exclude doublets. Then plot FSC-A verses DAPI to gate live cells (negative for DAPI) (Figure 1B).
- Plot PECy7 (or the relevant fluorochrome for the VE-Cadherin stain) versus PerCP (or the relevant fluorochrome for the EPCR stain). Gate cells that are positive for VE-Cadherin (which includes a mixed population of endothelial cells as well as hematopoietic progenitors and pre-HSC from the AGM) and that have high EPCR expression (which enriches for pre-HSC from P-Sp/AGM17). This is the final gate that will be used to index sort single cells in step 4.3 (Figure 1B).
- Plot additional fluorochromes used for staining. NOTE: Depending on the subpopulation to be analyzed, additional gates can be set based on the detection of other markers included for staining cells. However, index sorting enables the recording of the fluorescent parameters for each sorted cell such that these parameters can be retrospectively analyzed. Thus, no additional gating is necessary at this point. For all FACS setups, samples stained with single antibodies should be used to adjust compensation, to account for spillover in the emission of overlapping fluorochromes, and with isotype control antibodies, to account for background staining and determine negative/positive gates.
- Index sort the AGM cells.
- Place a 96-well plate prepared with the AGM-EC stroma in AGM serum-free media with cytokines (from step 1.5.3) in the plate block of the FACs sorting machine. Load the sample and begin sample acquisition. Select index sorting/single cell mode and begin sortingsingle cells to individual 96-wells. Use the lowest flow rate to minimize shear stress on embryonic cells.
- Ensure that all events are recorded during the index sorting. This will enable enumeration of the total number of cells analyzed in the sorting gate relative to the actual number sorted to individual 96-wells, as not all events recorded will be sorted to wells.
- Once the cells have been sorted to an entire 96-well plate, place the plate in a tissue culture incubator at 37 °C set at 5% CO2. Repeat for additional plates required per experiment.
- Co-culture individually sorted cells in 96-wells containing AGM-EC (from step 4.3) for up to 7 days (5 days for cells sorted from E11-11.5 embryos, 6 days from E10-10.5 embryos, 7 days from E9-9.5 embryos). Visualize hematopoietic colonies of various sizes and morphologies with an inverted microscope at 100X magnification (Figure 2B) after the co-culture period. NOTE: The media does not need to be changed during the co-culture period. Sorted hemogenic precursors may initially integrate into the AGM-EC layer with an EC-like morphology and cannot be initially distinguished from AGM-EC stroma (Figure 2A). However, following 24-48 h in co-culture, small, rounded hematopoietic cells or clusters of cells may be detected. At the end of co-culture (5-7 days), individual colonies of hematopoietic cells can be visualized in the subset of 96-wells that received a sorted cell with clonal hematopoietic potential. If visually inspected wells contain more than one distinct colony, then this sample is excluded from downstream analysis to exclude samples that may have received more than one cell sorted per well.

5. Analysis of Clonal Hematopoietic Progeny Following Co-culture
- Harvest clones from each 96-well for FACS analysis.
- Vigorously pipet to dissociate hematopoietic cells from endothelial layers using a multichannel pipette with 200 µL pipette tips. Try not to dissociate the endothelial layer. Remove 100 µL (~50% of the sample) for phenotypic analysis of hematopoietic progeny by FACS.
- Transfer to a 96-well V-bottom plate and centrifuge at 300 x g for 2 min to pellet the cells. Remove the media by flicking media from the plate with a single rapid motion while the plate is inverted (flick plate).
- Place the remaining 100 µL cells in the original 96-well plate in a 37 °C incubatorfor use in the subsequent engraftment assay (step 6).
- Incubate the cells with the appropriate antibodies for hematopoietic analysis.
- Re-suspend the cell pellets in each well of a 96-well V-bottom with 50 µL of PBS/2% FBS containing 10 µg/mL FcR block and 1 µg/mL DAPI. Incubate the plate at 4 °C for 5 min.
- Add 50 µL of PBS/2% FBS containing 10 µg/mL FcR block and an antibody mix for HSC analysis containing PE-Cyanine7-conjugated anti-VE-Cadherin, PerCP-conjugated anti-CD45, PE-conjugated anti-EPCR, APC-conjugated anti-Sca1, and FITC-conjugated anti-Gr1 and anti-F4/80 (each antibody at 1:100 final dilution unless otherwise indicated based on the manufacturer's recommendations or according to titrations). Incubate the plate at 4 °C for at least 20 min.
- Remove unbound antibody with sequential dilutions and analyze the cells.
- Add 200 µL/well PBS/2% FBS and centrifuge at 300 x g for 2 min to pellet the cells. Then, flick plate to discard the supernatant and add 200 µL PBS/2% FBS to each cell pellet and centrifuge at 300 x g for 2 min to pellet cells.
- Flick plate to discard the supernatant and add 50 µL/well PBS/2% FBS for analysis. Proceed to analyze cells by FACS using a flow cytometer equipped with a plate reader. NOTE: Acquire a fixed volume of cells (e.g., 35 µL of the total 50 µL used for re-suspension) to enumerate the subsets of hematopoietic progeny.
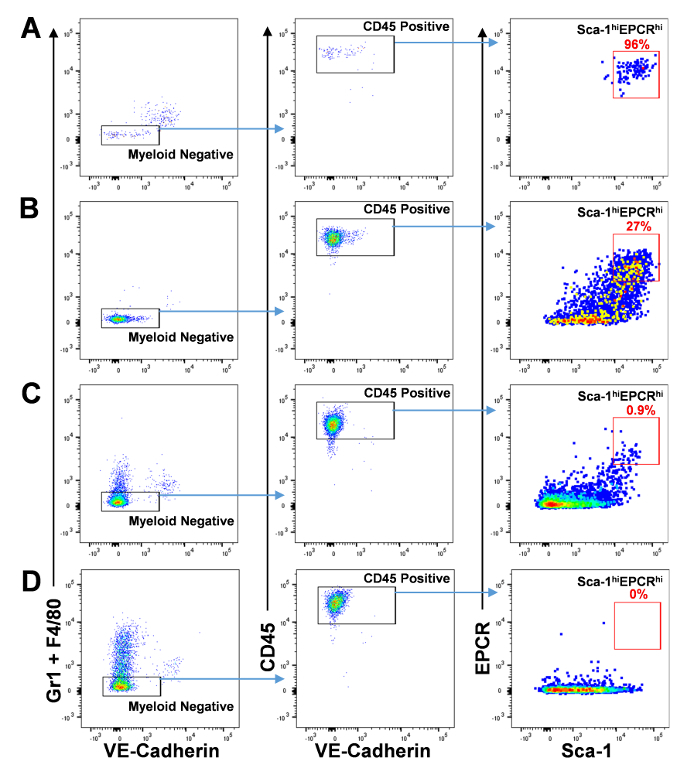
- Analyze the FACS data for each well containing hematopoietic progeny to screen for colonies that contain HSC potential (Figure 3).
- Gate first the CD45 positive population that is negative for myeloid markers Gr1 and F480. Do not gate out cells that still express low levels of VE-Cadherin. Cells from the AGM-EC layer that are disrupted during harvest of hematopoietic colonies are VE-Cadherin positive and CD45 negative.
- Gate CD45+VE-Cad-/lowGr1-F4/80- cells further on the subset that express high levels of EPCR and Sca1 (Figure 3A - C). NOTE: In prior studies, colonies lacking cells with CD45+VE-Cad-/lowGr1-F4/80-Sca1hiEPCRhi phenotype (Figure 3D) reproducibly failed to provide detectable multilineage peripheral blood engraftment in irradiated recipient mice17 (data not shown); thus, these colonies need not be included in the subsequent transplantation analysis in step 6.
6. Analysis of Engraftment Properties of Individual Clones and Correlation with Phenotypic Properties Elucidated by Index Sorting
The day of (if possible) or the morning following the phenotypic analysis in step 5, re-suspended the remaining 100 µL cells from each 96-well containing hematopoietic colonies following co-culture (from step 5.1) by vigorous pipetting using a 200 µL pipette tip.
- Prepare and add rescue cells from whole bone marrow of congenic strain B6.SJL-PtprcaPepcb/BoyJ (B6 CD45.1) adult mice16,17.
- Count nucleated bone marrow cells in Turks solution under a hemocytometer. Dilute to a concentration of 5 x 105 nucleated cells/mL in PBS with 2% FBS.
- Add 100 µL of rescue bone marrow cells (5 x 104) to each well containing hematopoietic progeny for transplantation analysis and mix by pipetting.
- Transplant progeny of individual clones into a single lethally irradiated recipient congenic strain B6.SJL-PtprcaPepcb/BoyJ (B6 CD45.1) adult mouse.
- Lethally irradiate the same number of B6 CD45.1 mice as the number of clones to individually inject progeny of a single clone (using 1,000 cGy from a Cesium source).
- Draw 200 µL total cell suspension (this includes cells from the clone as well as 5 x 104 B6 CD45.1 rescue bone marrow cells) into a ½ mL insulin syringe with a 29G ½ inch needle.
- Lethally irradiate B6 CD45.1 adult recipients 2-24 h prior to injection.
- Using a plastic mouse restrainer that allows tail access, inject via the tail vein 200 µL volume containing both cells from each clone and 5 x 104 B6 CD45.1 adult bone marrow rescue cells to aid in recipient survival.
- Ear tag and weigh the recipient mice, and check their weight/health at regular intervals (e.g., 3-4 times/week post irradiation/transplant, or per individual institutional standard operating procedures) to ensure good health.
- Perform analysis of peripheral blood engraftment at regular intervals (e.g., 2, 16, 24 weeks) following transplantation.
- Remove 50-100 µL of blood via retro-orbital bleed or another ethically approved blood letting method.
- Add 3 mL lysis buffer (16.6 g NH4Cl, 2 g NaHCO3 and 74.4 mg EDTA to 2 L H2O) to blood and incubate at room temperature for 5 min. Centrifuge at 300 x g for 5 min. Aspirate and re-suspend the pellet with 2 mL PBS/2% FBS. Centrifuge at 300 x g for 5 min.
- Re-suspend the pellet with 150 µL PBS/2% FBS containing 10 µg/mL FcR block and 1 µg/mL DAPI, and dispense 1/3 of the volume to a well containing 50 µL isotype controls for donor and lineage, 1/3 to a well containing 50 µL antibody for donor and isotype controls for lineage, and 1/3 to a well containing 50 µL antibodies to detect both donor and lineage. NOTE: Antibodies for detection of multilineage engraftment: APCeFluor780-conjugated anti-CD45.2, Pe-Cyanine7-conjugated anti-CD45.1, FITC-conjugated anti-CD3, PE-conjugated anti-F4/80, PerCP-conjugated anti-Gr1, and APC-conjugated anti-CD19, or relevant isotype controls.
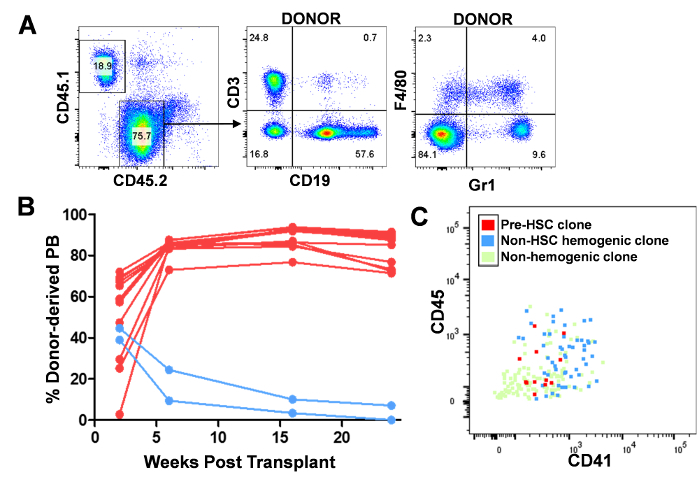
- Identify clones with long-term engraftment by assessing peripheral blood contribution by FACS over time of CD45.2 (donor)-derived hematopoietic cells to myeloid (Gr1 and/or F4/80 positive), B-cell (CD19 positive), and T-cell (CD3 positive) (Figure 4A-B). NOTE: Hematopoietic engraftment can also be analyzed by CD45.2 (donor)-derived contribution to hematopoietic populations from tissues harvested from bone marrow, spleen, thymus, and peritoneum, or following secondary transplantation of bone marrow cells into B6 CD45.1 mice to analyze serial engraftment properties17.
Correlate each clone's phenotypic properties as determined by initial single cell index sorting with its engraftment properties. Using flow analysis software, upload sorting parameters for each index-sorted cell (correlated to the 96-well to which it was sorted). Map functional data onto flow plots to evaluate the phenotypic properties of single cells as they relate to their functional output (for example, long-term, multilineage engraftment) (Figure 4C).
Representative Results
Figure 1A shows a schematic of the experimental design. Once P-Sp/AGM tissues are dissected, pooled, and dissociated in collagenase, they are stained with antibodies to VE-Cadherin and EPCR for index sorting. Pre-HSC are enriched in cells sorted at VE-Cadherin+EPCRhigh (Figure 1B). Other fluorochrome-conjugated antibodies can be included to retrospectively analyze additional phenotypic parameters, which are recorded for each cell during index sorting. In this representative experiment from E11 AGM, the cells are also stained with FITC-conjugated anti-CD45 and PE-conjugated anti-CD41 antibodies. Heterogeneity within the VE-Cadherin+EPCRhigh population for these hematopoietic-specific markers is observed (Figure 1C), consistent with the known asynchrony in hematopoietic development at this stage5.
Following index sorting of single VE-Cadherin+EPCRhigh AGM cells to individual 96-wells containing AGM-EC in AGM serum-free media and cytokines, colony formation occurs. Initially, sorted VE-Cadherin+EPCRhigh AGM cells integrate into the AGM-EC layer before they begin to round up and form hematopoietic clusters (Figure 2A, shown here from cells sorted from a transgenic murine embryo expressing GFP under the Ly6a promoter21, to distinguish them from the AGM-EC stroma). After 5-7 days of co-culture, colonies of various sizes and morphologies can be detected by inspection with an inverted microscope (Figure 2B), shown here from a representative experiment, 6 days following sorting from E11 AGM.
Next, phenotypic analysis by FACS is performed on half of the progeny of each VE-Cadherin+EPCRhigh cell which has formed a hematopoietic colony. Colonies containing cells with HSC phenotype are identified as VE-Cadherin-/lowGr1-F4/80-CD45+Sca1hiEPCRhi (Figure 3). Here, we show four different colonies obtained following co-culture of index-sorted E11 AGM VE-Cadherin+EPCRhigh cells, three containing different proportions of phenotypic HSC with other more differentiated cell types (Figure 3A-C), and the fourth lacking cells with HSC phenotype (Figure 3D). The number of colonies observed per number of cells plated and the percentage that resulted in engraftable stem cell colonies varies depending on the embryonic age and sort performed. VE-Cadherin-/lowGr1-F4/80-CD45+Sca1hiEPCRhi E11 AGM clones generated 53 ± 15 colonies out of 192 cells plated with 5% ± 1.3% of the 192 cells plated generating clones that engrafted long term (mean ± SD for 3 experiments).
To correlate the phenotype with engraftment potential, the remaining half of the progeny of each VE-Cadherin+EPCRhigh cell that has formed a hematopoietic colony containing cells with HSC phenotype detected by FACS (e.g., the colonies represented in Figure 3A-C) is transplanted to irradiated (1,000 cGy) adult congenic strain (B6 CD45.1) mice along with a dose of rescue cells from B6 CD45.1 marrow. Long-term multilineage engraftment is confirmed for each clone by following donor (CD45.2) contribution to the myeloid (Gr1 and F4/80), B-cell (CD19), and T-cell (CD3) compartment of the peripheral blood over time (Figure 4A). Although most colonies containing cells with HSC phenotype detected by FACS provide long-term engraftment, transplantation is necessary to confirm functional multilineage engraftment as some clones lose engraftment over time (Figure 4B), or display unique engraftment properties such as lymphoid-biased engraftment (data not shown). Once the functional HSC potential of each VE-Cadherin+EPCRhigh clone is determined, this is correlated back to that clone's index sorting parameters to identify its precise phenotypic characteristics. In this example, we see that functionally confirmed pre-HSC clones (red dots) are heterogeneous in their expression of CD41 and CD45, consistent with established dynamic expression of these markers during pre-HSC maturation to HSC (Figure 4C). Clones forming hematopoietic colonies but either lacking HSC potential by phenotypic screening or by absence of long-term, multilineage engraftment following transplantation (non-HSC hemogenic clones, blue dots) are also heterogeneous for CD41 and CD45, although a subset express higher levels of CD41 compared with pre-HSC clones, which are primarily CD41low. Clones that did not form hematopoietic colonies (non-hemogenic clones, light green dots) are predominantly CD41-/lowCD45-, which likely reflect non-hemogenic VE-Cadherin+EPCRhigh endothelial cells.
Figure 1: Protocol overview and representative flow cytometry analysis for index sorting of single cells. (A) Schematic representation of the protocol. (B) Flow cytometry analysis of dissociated E11 AGM cells showing gating strategy for index sorting of VE-Cadherin+EPCRhigh cells enriched for pre-HSC (analysis with FACS analysis software). Numbers in each gate represent the percent of cells. (C) Flow cytometry analysis showing CD41 and CD45 staining within the VE-Cadherin+EPCRhigh population. Please click here to view a larger version of this figure.
Figure 2: Formation of hematopoietic colonies during AGM-EC co-culture. (A) Time-lapse imaging of VE-Cadherin+EPCRhighGFP+ sorted cells from Ly6a-GFP transgenic embryos21 co-cultured on AGM-EC. Scale bars in µm are shown in bottom left of each image. (B) Representative colonies formed from VE-Cadherin+EPCRhigh index-sorted single cells after 6 days co-culture on AGM-EC. Please click here to view a larger version of this figure.
Figure 3: Flow cytometry analysis of the progeny of a single E11 AGM-derived VE-Cadherin+EPCRhigh cells following co-culture on AGM-EC. Gating for cells with HSC potential is shown as the subset that is VE-Cadherin-/lowGr1-F4/80- (left panel), CD45+ (middle panel), and Sca1hiEPCRhi (right panel). (A) Representative colony consisting of a homogeneous population of cells with the HSC phenotype. (B-C) Representative colonies containing a mix of cells with the HSC phenotype and more differentiated cell types. (D) Representative colony consisting of cells lacking the HSC phenotype. Please click here to view a larger version of this figure.
Figure 4: Analysis of peripheral blood engraftment of progeny of individual clones following AGM-EC co-culture and correlation with index sorting parameters. (A) Donor (CD45.2)-derived peripheral blood engraftment of myeloid (Gr1 and F4/80), B-cell (CD19), and T-cell (CD3) subsets from a representative recipient (B6 CD45.1) transplanted with progeny of individual E11 VE-Cadherin+EPCRhigh clone following AGM-EC co-culture. (B) Donor (CD45.2)-derived peripheral blood engraftment over time after transplantation of progeny of individual E11 VE-Cadherin+EPCRhigh clones following AGM-EC co-culture. (C) Correlation of CD41 and CD45 expression for each index sorted VE-Cadherin+EPCRhigh clone with its HSC potential following AGM-EC co-culture based on long-term, multilineage engraftment in transplantation assay. Please click here to view a larger version of this figure.
Discussion
The study of HSC genesis during embryonic development necessitates means to detect HSC potential in hemogenic precursors yet lacking the competence to provide long-term multilineage hematopoietic reconstitution in transplanted adult recipients. In this protocol, we present a clonal assay of embryonic hemogenic precursors by stromal co-culture on vascular niche ECs from the AGM, which supports the maturation of precursors to HSC, with subsequent functional analysis in transplantation assays. Incorporation of index sorting permits the retrospective analysis of phenotypic parameters to characterize the properties of pre-HSC as defined by surface markers included in the assay. This approach thus provides an advantage over other methods that rely on bulk sorting of precursor populations with re-aggregation-based HSC assays4,5,10, enabling efficient screening for potential markers of pre-HSC while at the same time providing information about the relative expression level of surface markers for each individual cell. Using this methodology, for example, we previously reported that, in addition to the use of EPCR to enrich for pre-HSC populations at different stages of development, intermediate expression of the Notch ligand Dll4 further defined a subpopulation of clonal VE-Cadherin+EPCR+ precursors with HSC potential, whereas Dll4 negative VE-Cadherin+EPCR+ hemogenic precursors mostly lacked HSC potential17.
Another advantage of this method is the ability to screen for HSC potential immediately following co-culture based on the detection of hematopoietic progeny with a phenotype (VE-Cadherin-/lowGr1-F4/80-CD45+Sca1hiEPCRhi) that is correlated with long-term, multilineage engraftment. This eliminates the need to transplant colonies lacking hematopoietic progeny with this HSC phenotype, as these colonies do not provide long-term engraftment in transplantation assays. Furthermore, initial information distinguishing potential pre-HSC from hemogenic precursors lacking HSC potential immediately following co-culture, without the need to await results from long-term engraftment assays, is useful, for example, in rapidly screening candidate markers of HSC-precursors. However, the correlation of phenotypic data with long-term engraftment following transplantation provides additional valuable insights into variations of the precise engraftment properties of pre-HSC clones. We found, for example, that clonal pre-HSC from the P-Sp/AGM region between E9.5 and E11.5 have the unique potential to give rise to both B1a and B2-lymphocytes, whereas B1a-lymphocyte potential is deficient in adult HSC17. Furthermore, we detected an evolution in the engraftment properties of pre-HSC clones between E9.5 and E11.5 with earlier pre-HSC demonstrating a relative skew toward peritoneal B1a verses B2-lymphocyte engraftment and less robust self-renewal potential based on secondary transplantation assays17.
While this protocol provides a valuable approach to elucidate the unique properties of clonal HSC precursors, there are some limitations that must be acknowledged. Although detection of progeny with HSC phenotypic by FACS following AGM-EC co-culture is generally correlated to long-term multilineage engraftment, some colonies with this phenotype provide only short-term engraftment or engraftment deficient in one or more hematopoietic lineages (Figure 4B; data not shown). Thus, transplantation remains the gold standard in validating functional HSC. Furthermore, it cannot be ruled out that some of the differences in engraftment properties of pre-HSC clones, rather than reflecting cell-intrinsic differences in hemogenic precursor potential, may result from stochastic variability during AGM-EC co-culture in self-renewal verses differentiation decisions, as pre-HSC are simultaneously maturing to HSC and undergoing cell divisions. Indeed, this may in part account for the observed heterogeneity of colony morphology and phenotype of cells generated from individual pre-HSC clones (e.g., Figure 3A-C), and may result in an underestimation of pre-HSC by this assay if not all potential pre-HSC clones are detected by generation of functional HSC following AGM-EC co-culture. However, the ability to distinguish functionally distinct hemogenic clones by their differential expression of unique phenotypic markers (such as Dll417) enabled by this method provides a means to identify subpopulations of hemogenic precursors with inherent cell-intrinsic differences.
Building on this basic protocol, a number of extended applications can be included to further study hematopoietic potential of clonal hemogenic precursors during development. Beyond the use of antibodies for detection of surface markers, expression of fluorescence markers in transgenic embryos (e.g., Ly6a-GFP21 or Runx1-GFP22 expressed in hemogenic endothelium/pre-HSC) can be incorporated for index sorting of hemogenic precursors. In addition to transplantation assays following AGM-EC co-culture to detect putative pre-HSC, progeny can also be assessed for hematopoietic lineage potential based on secondary in vitro assays, such as culture on OP9 or OP9-Dl1 to detect B- and T-cell potential23, or clonogenic colony forming assays in methylcellulose to detect erythromyeloid potential. Indeed, some of the non-HSC hemogenic clones (e.g., Figure 3D) likely represent multipotent progenitors, erythromyeloid progenitors, or T- and B-cell progenitors previously described that precede and are independent of HSC development8,24,25,26,27,28, and this assay may further facilitate clonal studies of these distinct waves of hematopoietic precursors. Finally, index sorting of pre-HSC can be combined with other downstream analyses such as single cell RNA-sequencing, to correlate phenotypic properties with the global transcriptional profiles of developing HSC precursors. Altogether, this protocol provides a method for the robust analysis of clonal hematopoiesis from embryonic hemogenic precursors that should provide novel insights into HSC development.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We would like to thank Andrew Berger, Stacey Dozono, and Brian Raden in the Fred Hutchinson Flow Cytometry Core for assistance with FACS. This work was supported by the National Institutes of Health NHLBI UO1 grant #HL100395, Ancillary Collaborative Grant #HL099997, and NIDDK grant #RC2DK114777. Brandon Hadland is supported by the Alex’s Lemonade Stand Foundation and Hyundai Hope on Wheels Foundation.
References
- Copley MR, Beer PA, Eaves CJ. Hematopoietic stem cell heterogeneity takes center stage. Cell Stem Cell. 2012;10(6):690–697. doi: 10.1016/j.stem.2012.05.006. [DOI] [PubMed] [Google Scholar]
- Medvinsky A, Rybtsov S, Taoudi S. Embryonic origin of the adult hematopoietic system: advances and questions. Development. 2011;138(6):1017–1031. doi: 10.1242/dev.040998. [DOI] [PubMed] [Google Scholar]
- Kumaravelu P, et al. Quantitative developmental anatomy of definitive haematopoietic stem cells/long-term repopulating units (HSC/RUs): role of the aorta-gonad-mesonephros (AGM) region and the yolk sac in colonisation of the mouse embryonic liver. Development. 2002;129(21):4891–4899. doi: 10.1242/dev.129.21.4891. [DOI] [PubMed] [Google Scholar]
- Taoudi S, et al. Extensive hematopoietic stem cell generation in the AGM region via maturation of VE-cadherin+CD45+ pre-definitive HSCs. Cell Stem Cell. 2008;3(1):99–108. doi: 10.1016/j.stem.2008.06.004. [DOI] [PubMed] [Google Scholar]
- Rybtsov S, et al. Hierarchical organization and early hematopoietic specification of the developing HSC lineage in the AGM region. J Exp Med. 2011;208(6):1305–1315. doi: 10.1084/jem.20102419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybtsov S, et al. Tracing the origin of the HSC hierarchy reveals an SCF-dependent, IL-3-independent CD43(-) embryonic precursor. Stem Cell Reports. 2014;3(3):489–501. doi: 10.1016/j.stemcr.2014.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath KE, et al. Distinct Sources of Hematopoietic Progenitors Emerge before HSCs and Provide Functional Blood Cells in the Mammalian Embryo. Cell Rep. 2015;11(12):1892–1904. doi: 10.1016/j.celrep.2015.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Yoder MC, Yoshimoto M. Lymphoid progenitor emergence in the murine embryo and yolk sac precedes stem cell detection. Stem Cells Dev. 2014;23(11):1168–1177. doi: 10.1089/scd.2013.0536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvinsky A, Dzierzak E. Definitive hematopoiesis is autonomously initiated by the AGM region. Cell. 1996;86(6):897–906. doi: 10.1016/s0092-8674(00)80165-8. [DOI] [PubMed] [Google Scholar]
- Sheridan JM, Taoudi S, Medvinsky A, Blackburn CC. A novel method for the generation of reaggregated organotypic cultures that permits juxtaposition of defined cell populations. Genesis. 2009;47(5):346–351. doi: 10.1002/dvg.20505. [DOI] [PubMed] [Google Scholar]
- Yoder MC, Hiatt K, Mukherjee P. In vivo repopulating hematopoietic stem cells are present in the murine yolk sac at day 9.0 postcoitus. Proc Natl Acad Sci U S A. 1997;94(13):6776–6780. doi: 10.1073/pnas.94.13.6776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora N, et al. Effect of developmental stage of HSC and recipient on transplant outcomes. Dev Cell. 2014;29(5):621–628. doi: 10.1016/j.devcel.2014.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler JM, Rafii S. Generation of a vascular niche for studying stem cell homeostasis. Methods Mol Biol. 2012;904:221–233. doi: 10.1007/978-1-61779-943-3_18. [DOI] [PubMed] [Google Scholar]
- Butler JM, et al. Endothelial cells are essential for the self-renewal and repopulation of Notch-dependent hematopoietic stem cells. Cell Stem Cell. 2010;6(3):251–264. doi: 10.1016/j.stem.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H, et al. Angiocrine factors from Akt-activated endothelial cells balance self-renewal and differentiation of haematopoietic stem cells. Nat Cell Biol. 2010;12(11):1046–1056. doi: 10.1038/ncb2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadland BK, et al. Endothelium and NOTCH specify and amplify aorta-gonad-mesonephros-derived hematopoietic stem cells. J Clin Invest. 2015;125(5):2032–2045. doi: 10.1172/JCI80137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadland BK, et al. A Common Origin for B-1a and B-2 Lymphocytes in Clonal Pre- Hematopoietic Stem Cells. Stem Cell Reports. 2017;8(6):1563–1572. doi: 10.1016/j.stemcr.2017.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson NK, et al. Combined Single-Cell Functional and Gene Expression Analysis Resolves Heterogeneity within Stem Cell Populations. Cell Stem Cell. 2015;16(6):712–724. doi: 10.1016/j.stem.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte R, et al. Index sorting resolves heterogeneous murine hematopoietic stem cell populations. Exp Hematol. 2015;43(9):803–811. doi: 10.1016/j.exphem.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan K, Kharas M, Dzierzak E, Gilliland DG. Isolation of early hematopoietic stem cells from murine yolk sac and AGM. J Vis Exp. 2008. [DOI] [PMC free article] [PubMed]
- deBruijn MF, et al. Hematopoietic stem cells localize to the endothelial cell layer in the midgestation mouse aorta. Immunity. 2002;16(5):673–683. doi: 10.1016/s1074-7613(02)00313-8. [DOI] [PubMed] [Google Scholar]
- Lorsbach RB, et al. Role of RUNX1 in adult hematopoiesis: analysis of RUNX1-IRES-GFP knock-in mice reveals differential lineage expression. Blood. 2004;103(7):2522–2529. doi: 10.1182/blood-2003-07-2439. [DOI] [PubMed] [Google Scholar]
- Holmes R, Zúñiga-Pflücker JC. The OP9-DL1 system: generation of T-lymphocytes from embryonic or hematopoietic stem cells in vitro. Cold Spring Harb Protoc. 2009;2009(2):pdb.prot5156. doi: 10.1101/pdb.prot5156. [DOI] [PubMed] [Google Scholar]
- Yoshimoto M. The first wave of B lymphopoiesis develops independently of stem cells in the murine embryo. Ann N Y Acad Sci. 2015;1362:16–22. doi: 10.1111/nyas.12612. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, et al. Functional B-1 progenitor cells are present in the hematopoietic stem cell-deficient embryo and depend on Cbfβ for their development. Proc Natl Acad Sci U S A. 2014;111(33):12151–12156. doi: 10.1073/pnas.1407370111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimoto M, et al. Autonomous murine T-cell progenitor production in the extra-embryonic yolk sac before HSC emergence. Blood. 2012;119(24):5706–5714. doi: 10.1182/blood-2011-12-397489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inlay MA, et al. Identification of multipotent progenitors that emerge prior to hematopoietic stem cells in embryonic development. Stem Cell Reports. 2014;2(4):457–472. doi: 10.1016/j.stemcr.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palis J. Hematopoietic stem cell-independent hematopoiesis: emergence of erythroid, megakaryocyte, and myeloid potential in the mammalian embryo. FEBS Lett. 2016;590(22):3965–3974. doi: 10.1002/1873-3468.12459. [DOI] [PubMed] [Google Scholar]