

Abstract
Virus-like particles (VLPs) have been used as nanocarriers to display foreign epitopes and/or deliver small molecules in the detection and treatment of various diseases. This application relies on genetic modification, self-assembly, and cysteine conjugation to fulfill the tumor-targeting application of recombinant VLPs. Compared with genetic modification alone, chemical conjugation of foreign peptides to VLPs offers a significant advantage because it allows a variety of entities, such as synthetic peptides or oligosaccharides, to be conjugated to the surface of VLPs in a modulated and flexible manner without alteration of the VLP assembly.
Here, we demonstrate how to use the hepatitis E virus nanoparticle (HEVNP), a modularized theranostic capsule, as a multifunctional delivery carrier. Functions of HEVNPs include tissue-targeting, imaging, and therapeutic delivery. Based on the well-established structural research of HEVNP, the structurally independent and surface-exposed residues were selected for cysteine replacement as conjugation sites for maleimide-linked chemical groups via thiol-selective linkages. One particular cysteine-modified HEVNP (a Cys replacement of the asparagine at 573 aa (HEVNP-573C)) was conjugated to a breast cancer cell-specific ligand, LXY30 and labeled with near-infrared (NIR) fluorescence dye (Cy5.5), rendering the tumor-targeted HEVNPs as effective diagnostic capsules (LXY30-HEVNP-Cy5.5). Similar engineering strategies can be employed with other macromolecular complexes with well-known atomic structures to explore potential applications in theranostic delivery.
Keywords: Bioengineering, Issue 135, Cysteine replacement, chemical conjugation, hepatitis E, virus-like particles, multivalent ligand display, targeting ligand
Introduction
The development of nano-sized vectors in therapeutic and diagnostic delivery, known as nanotheranostics, has shifted much of the biomedical field away from generalized treatments towards targeted delivery1. Targeted nanotheranostic delivery integrates nano-sized vectors (nanoparticles) with theranostic molecules to stably direct theranostic molecules to a specific diseased tissue or biochemical pathway2,3,4. Nanomedicine has come to the forefront of targeted delivery because optimally sized nanoparticles have the capacity to stabilize circulation of theranostic molecules and selectively target cell surface molecules presented on diseased tissues. Many nanotheranostic platforms still suffer from passive cell uptake, pre-mature degradation, toxicity, and insufficient association with theranostic molecules. VLPs overcome many of these obstacles in targeted delivery. They have been used as nanocarriers to display foreign epitopes and/or deliver small molecules: a regimen that can be used to combat many diseases1. This application relies mainly on the property of self-assembly as well as the ease of genetic modifications, to fulfill the designed application for the given VLP. Compared to genetic engineering, chemical conjugation of foreign peptides to VLP displays a significant advantage because it allows a great variety of entities, such as peptides or oligosaccharides, to be conjugated to the surface of VLPs in a modulated and flexible manner without alteration of VLP assembly.
HEVNPs, derived from the recombinant HEV capsid protein, 2nd open reading frame (ORF2), are non-infectious, self-assembling capsids capable of cell-binding and entry. Because HEV evolved for mucosal transmission, the assembled capsid protein is similarly stable in proteolytic and acidic mucosal conditions5. HEVNPs form a hollow, T = 1 icosahedral capsid, composed of 60 identical units6,7 of ORF2, rendering it highly stable both in storage and in harsh physiological conditions. Lacking any viral genetic elements, the efficient, high yield production is achieved through baculovirus expression system in insect cells. Because of their proteolytic stability, self-assembled HEVNPs are extracted and purified from cell supernatant, substantially reducing necessary purification steps. Additionally, HEVNPs possess a surface exposed protrusion domain (P domain) connected through a flexible hinge to a stable icosahedral base. The P domain forms surface-exposed spikes atop the icosahedral base while the flexible hinge makes it possible to significantly modify the P domain without compromising the base icosahedral structure. With 60 repeated units, single site-specific modification results in 60 symmetric sites for chemical modulation. Recently, we proposed a nano-platform using HEVNP that can chemically conjugate ligands or small molecules for theranostic applications. This was achieved by replacing a single amino acid with cysteine on the protrusion domain of HEV-VLP as a reaction site with maleimide-linked peptides or molecules. Based on previous structural analysis of HEV-VLP and well-studied immunogenic epitopes8,9, the following five HEV-VLP amino acids were replaced with cysteine as potential candidates: Y485C, T489C, S533C, N573C, and T586C (Figure 1). After expression and purification from insect cells, their VLP formations were confirmed by transmission electron microscopy (TEM) observation (Figure 2), and the exposed cysteine sites were analyzed by Western blot after maleimide-linked biotin conjugation (Figure 2). Among the five mutants, HEVNP-573C displayed the strongest signal of maleimide-biotin conjugation (Figure 2) and was used for follow up demonstration as the nanocarrier for breast cancer cell targeting4 (Figure 3).
This protocol depicts chemical conjugation methods to attach tumor-targeting molecules to HEVNPs through surface cysteine conjugation. We detail the conjugation of tumor targeting and detection molecules for tumor delivery with recombinant HEVNPs containing a cysteine at N573 (HEVNP-573C). We focused on a two-step click chemistry conjugation process to bind a breast cancer tumor targeting peptide, LXY3010 to HEVNPs to form LXY30-HEVNP (Figure 4). Subsequently, N-hydroxysuccimide (NHS)-Cy5.5 were conjugated to the separate Lys site on HEVNPs to build LXY30-HEVNP-Cy5.5 for fluorescent detection both in vitro (Figure 5) and in vivo4.
Protocol
1. HEVNP Production in Insect Cells
NOTE: All following steps should be performed in a cell culture hood. Refer to our previous publication for more detailed HEVNP production procedures11.
Culture Sf9 cells in insect cell media (see Table of Materials) to 50 - 75% confluence in 6-well plates.
Using insect cell transfection reagents according to the manufacturer's protocols, transfect Bacmids containing HEVNP-573C ORF2 into Sf99 cells to produce recombinant baculovirus. Incubate the transfected cells at 27 °C for 3 - 6 days, depending on the viability of cells.
Collect supernatant at 3 - 6 days post-infection (after the transfected cells are lysed due to baculovirus infection) as P0 baculovirus stock.
Remove culture medium before applying 200 µL of P0 stock to 50 - 75% confluent Sf9 cells in a 25 cm2 monolayer flask. Rock the flask every 15 min to ensure the full coverage of the inoculum. Repeat 4 times, for a total of 60 min.
Add 2 mL of insect cell culture medium to the flask and keep at 27 °C for 3 - 6 days, depending on viability of cells, to amplify baculovirus to a higher titer.
Carry out plaque assays to obtain the baculovirus titer reading12.
Culture the suspended Tn5 insect cells (Table of Materials) with 100 mL of insect cell medium in 250 mL flask and shake in 27 °C at 150 rpm to a titer of 0.5 x 105 - 1 x 106 for inoculation.
Add baculovirus at a multiplicity of infection (MOI) of 5 - 10 to 100 mL Tn5 cells in a 250 mL flask. After inoculation, shake in 27 °C at 150 rpm for 5 - 7 days.
Once most cells appear to have vesicles and 70 - 90% of cells are dead under optical microscope observation, collect the Tn5 cells and transfer into 33 mL ultracentrifuge tubes. Place the ultracentrifuge tubes in swinging bucket rotors, balance, and spin down the cell debris and recombinant baculoviruses at 10,000 x g for 90 min at 25 °C.
Keep the supernatant that contains the released HEVNPs at 4 °C for further purification. For extended storage of baculovirus supernatant, add protease inhibitors.
2. HEVNP Purification
- Pellet and isolate the HEVNPs using cesium chloride (CsCl) gradient separation:
- Transfer the collected supernatant (from step 1.10) and add 20% NP-40 into each tube to make a final concentration of 0.5% NP-40 to dissolve any remaining cellular membrane. Gently mix by pipetting and incubate for at least 30 min at 25 °C.
- Ultracentrifuge the HEVNPs at 112,400 x g in swinging bucket rotors for 2 h at 4 °C to pellet down the HEVNPs from the supernatant. Discard the supernatant after centrifugation and gently re-suspend the crude pellet in 200 µL of 10 mM MES buffer pH 6.2 in each tube overnight (O/N) at 4 °C. Run SDS-PAGE (step 3.1) to confirm the presence of HEVNP ORF2 in the crude pellet as a 52 kDa band.
- Prepare a 38.5% (w/v) CsCl gradient by mixing 1.96 g CsCl, the re-suspended crude pellet, and ~4 mL of 0.01 M MES pH 6.2 in a 5 mL ultracentrifuge tube. Balance the tubes and place in a swinging bucket rotor. Ultracentrifuge at 147,000 x g for 16 h at 4 °C.
- Collect the fractions after CsCl gradient:
- Discard the top 500 µL fraction, which is primarily light-weight cell membrane debris. Collect 500 µL fractions, starting from the top of the tube, and change tips in between each fraction. Place each fraction into numbered/labeled 1.5 mL tubes. NOTE: The presence of HEVNP ORF2 in fractions separated from CsCl gradient cannot be detected by running SDS PAGE gels due to the high concentration of CsCl. The CsCl in each fraction can be removed or diluted by following a CsCl clean-up procedure. Alternatively, the CsCl gradient can be replaced by a 10 - 40% sucrose gradient13 to avoid CsCl residual.
- Transfer each fraction to a 5 mL ultracentrifuge tube and dilute the CsCl with 4.5 mL of 10 mM MES, pH 6.2. Balance the tubes and place in a swinging bucket rotor. Ultracentrifuge at 147,000 x g for 2 h at 4 °C to pellet down the HEVNPs.
- Discard the supernatant and gently re-suspend the HEVNPs in 100 µL of 10 mM MES pH 6.2 in each tube. Cover the tubes to avoid evaporation and incubate O/N at 4 °C.
- Record the A280 reading and A260/A280 nm ratio using a spectrophotometer. Determine the approximate concentration of ORF2 as:
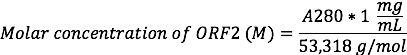 Each ORF2 will contain 1 Cys site and 1 Lys site for chemical conjugation. NOTE: The molar extinction coefficient of HEVNP ORF2 is 60,280, which is equivalent to 1.019 mg/mL × A280. This is so close to 1:1 that the concentration of HEVNPs (in mg/mL) can be approximated by the A280, and therefore, the concentration of ORF2 by the above equation. For example, an HEVNP with an A280 reading of 1 will have a concentration of 1 mg/mL, which is equivalent to 18.8 µM ORF2.
Each ORF2 will contain 1 Cys site and 1 Lys site for chemical conjugation. NOTE: The molar extinction coefficient of HEVNP ORF2 is 60,280, which is equivalent to 1.019 mg/mL × A280. This is so close to 1:1 that the concentration of HEVNPs (in mg/mL) can be approximated by the A280, and therefore, the concentration of ORF2 by the above equation. For example, an HEVNP with an A280 reading of 1 will have a concentration of 1 mg/mL, which is equivalent to 18.8 µM ORF2. - Prepare an SDS-PAGE. Use a 6 µL sample from each fraction to determine the fractions containing 53.3 kDa HEVNP ORF2 protein (step 3.1). The HEVNPs should be found in fractions 3 - 5, with a density of ~1.25 g/mL.
- Confirm the presence and the purity of HEVNPs by TEM observation. Prepare or dilute the HEVNP samples to 0.5 - 2.0 mg/mL for TEM. The HEVNPs appear in TEM as empty icosahedral proteins, ~27 nm in diameter (Figure 2). Some protein contaminants may remain in the fractions and will be observed under TEM (step 3.2).
In the case that impurities are present under TEM, repeat step 2.1.3 - 2.2.6 for better purity of the HEVNPs. NOTE: Extra purification through CsCl gradient may cause yield loss of HEVNPs. Alternatively, the CsCl gradient purification can be replaced by a 10 - 40% sucrose gradient to avoid residual CsCl13.
3. HEVNP Characterization
- Prepare SDS PAGE 4-12% Bis-Tris Protein Gels, 1.0 mm, 17-wells (see Table of Materials) according to the user's manual14:
- Add 2 µL of 4x loading buffer to 6 µL of protein sample. Incubate the sample mixture in a heat block for 10 min at 100 °C to denature the protein. Load the protein samples onto the gel.
- Run the SDS-PAGE by setting the DC power supply at 100 V for 10 min, then 150 V for 45 min until the samples run to about 1 cm above the bottom of the gel.
- Stain the SDS PAGE gel with Coomassie blue (0.25% (w/v) Coomassie Brilliant Blue R250, 30% (v/v) methanol, 10% (v/v) acetic acid), for 1 h.
- After the staining procedure, remove the Coomassie blue stain and apply de-staining buffer (30% (v/v) methanol, 10% (v/v) acetic acid) onto the protein gel for >12 h at room temperature.
- Document the gel under white light to confirm the presence of HEVNP ORF2 at the 52 kDa band.
- Observe the HEVNPs using TEM.
- Prepare or dilute the HEVNP samples to 0.5 - 2 mg/mL with 10 mM MES pH 6.2 for TEM imaging.
- Ionize the carbon-coated grids with 40 mA glow discharge for 30 s to produce a hydrophilic carbon surface. The glow discharge equipment is described in the Table of Materials. NOTE: The hydrophilic carbon surface of the grids can only last for 30 min after glow discharge treatment.
- Hold in tweezers and add 2 µL of HEVNP sample to the grid, wait for 15 - 30 s, and blot with filter paper.
- Immediately wash the grid with ddH20 and blot with filter paper.
- Immediately add 2 µL of 2% uranyl acetate to the grid, wait 15 s, then blot with filter paper. Dry the sample grids by putting them in an electronic dehumidify dry cabinet for O/N.
- Transfer the grid into a TEM and image at 10-80k magnification. HEVNPs appear in TEM as empty icosahedral proteins that are ~27 nm in diameter, due to the absence of viral RNA.
4. Chemical Conjugation of HEVNPs with Biotin, Cancer Targeting Ligand, and Fluorophores
- Perform one step conjugation of HEVNPs and maleimide linked biotin.
- Buffer change: Apply the HEVNPs in mini dialysis units and dialyze against 0.01 M PBS pH 7.4 at room temperature for 1 h according to the manufacturer's protocol (Table of Materials). Transfer the HEVNPs to 1.5 mL tubes and measure the protein concentration at 280 nm using a spectrophotometer.
- Mix the HEVNP to 1 mg/mL, which is equivalent to 18.8 µM of Cys reaction sites (see details in step 2.2.4), with an equal amount of maleimide-biotin (100 µM) in 0.01 M PBS, pH 7.4, to make a 1:5 molar ratio; react O/N at 4 °C. Remove unbound maleimide-biotin with a 40 K MWCO Spin Desalting column procedure according to the manufacturer's protocol (Table of Materials).
- Analyze the samples through a standard reducing SDS-PAGE (step 3.1).
- Using standard procedures, prepare a chemiluminescent Western Blot using HRP-linked Streptavidin. Capture the chemiluminescent signal by X ray film (Figure 2).
- Perform two step LXY30 conjugation to surface-exposed cysteine on HEV NPs (Figure 5).
- Buffer exchange: Apply HEVNPs in mini dialysis units and dialyze against 0.01 M PBS pH 7.4 at room temperature for 1 h. Transfer the HEVNPs to 1.5 mL tubes and measure protein concentration at 280 nm using a spectrophotometer.
- Add 650 µM maleimide-azide and 650 µM alkyne-LXY3010 in 0.01 M PBS pH 7.4 with 200 µM CuSO4 and 1 mM ascorbic acid to form maleimide-linked LXY30 (Mal-LXY30) at 650 µM. Incubate the mixture at 4 °C for O/N.
- Mix the HEVNP to 1 mg/mL, which is equivalent to 18.8 µM of Cys reaction site (see details in step 2.2.4), with about 10% volume of Mal-LXY30 (650 µM) in 0.01 M PBS pH 7.4, to make a 1:3 molar ratio; react O/N at 4 °C. NOTE: Due to the relatively high concentration of Mal-LXY30, the final concentrations of the reactants, such as CuSO4, are reduced about 10 times after mixing, to avoid their damage to HEVNPs. Another option is the Cu-free conjugation method15.
- Remove unbound maleimide-click-LXY30 with a 40 K MWCO Spin Desalting column according to the manufacturer's protocol (see Table of Materials). Keep the LXY30-linked HEVNPs (LXY30-HEVNPs) at 4 °C.
- Perform one step conjugation of the LXY30-HEVNPs and Cy5.5 NHS ester (NHS-Cy5.5)
- Mix the LXY30-linked HEVNPs (LXY30-HEVNPs) to 1 mg/mL, which is equivalent to 18.8 µM of the Cys reaction site (see details in step 2.2.4), with equal volume of NHS-Cy5.5 (100 µM) in 0.01 M PBS pH 7.4 to make a 1:5 molar ratio; react O/N at 4 °C.
- Remove unbound Cy5.5-NHS with a 40K MWCO Spin Desalting column procedure according to the manufacturer's protocol (see Table of Materials). Keep the LXY30, Cy5.5-linked HEVNPs (LXY30-HEVNP-Cy5.5) at 4 °C.
5. HEVNP Binding to and Internalization in MDA-MB231 Breast Cancer Cells
Seed MDA-MB231 breast cancer cells into a 35-mm glass bottom dishes (5 x 104 per dish) O/N in a mammalian cell culture cabinet.
- For the cell binding experiment, prepare LXY30-HEVNP-Cy5.5 by following steps 4.2 - 4.3. Dilute the LXY30-HEVNP-Cy5.5 to 0.01 mg/mL, which is equivalent to 0.188 µM of HEVNP ORF2 (refer to step 2.2.4), in 250 µL of 0 - 1% FBS/DMEM.
- Prepare the negative control sample at 0.01 mg/mL HEVNP-Cy5.5 by following step 4.3 but conjugated with NHS-Cy5.5 dye only, (HEV-Cy5.5). Dilute the HEVNP-Cy5.5 to 0.01 mg/mL, which is equivalent to 0.188 µM of HEVNP ORF2 (refer to step 2.2.4), in 250 µL of 10% FBS supplemented with DMEM.
Wash the cells once by applying 250 µL of 1 M PBS buffer, pH 7.4. Remove the PBS buffer after the wash, while keeping some buffer in the cell culture dish.
Apply 250 µL of LXY30-HEVNP-Cy5.5 in 10% FBS supplemented with DMEM or HEVNP-Cy5.5 in 10% FBS supplemented with DMEM to the cultured MDA-MB231 breast cancer cells. Shield the cell cultured dishes from light with aluminum foil.
Keep the cell cultured dishes in a 37 °C cell culture cabinet for 1 h for internalization.
Wash the cultured cells on ice 3 times, 5 min per wash, with 250 µL of 1 M PBS, pH 7.4.
Fix the cells in 4% PFA in 1 M PBS, pH 7.4 for 20 min and then wash once with 250 µL of 1 M PBS, pH 7.4. NOTE: The cells are now ready to be imaged by confocal microscope. Representative data are shown in Figure 5.
Representative Results
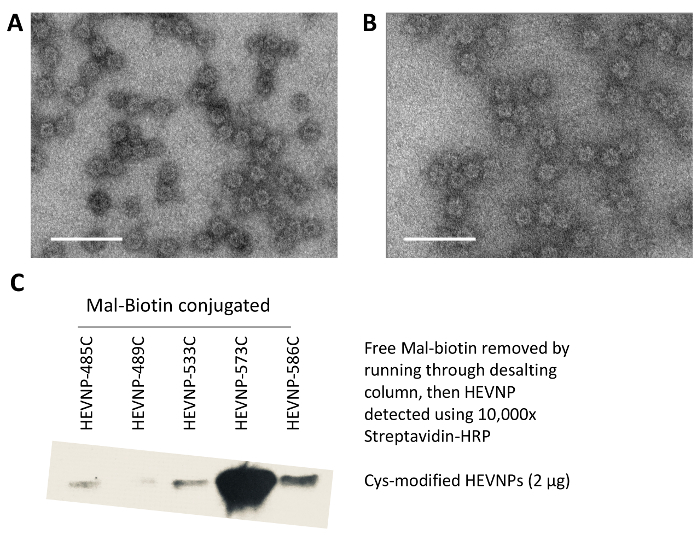
Akin to HEV-VLPs, all Cys modified HEVNPs formed soluble icosahedral capsids and did not aggregate in solution during production or purification. Before and after single-step maleimide-biotin conjugation, each of the Cys modified HEVNPs were indistinguishable from HEV-VLPs in the negative stain EM (Figure 2). Maleimide-biotin conjugation efficiency to Cys modified HEVNPs was first tested with Western blotting via chemiluminescent streptavidin binding. Following maleimide-biotin conjugation, Cys modified HEVNPs displayed streptavidin binding signals (Figure 2).
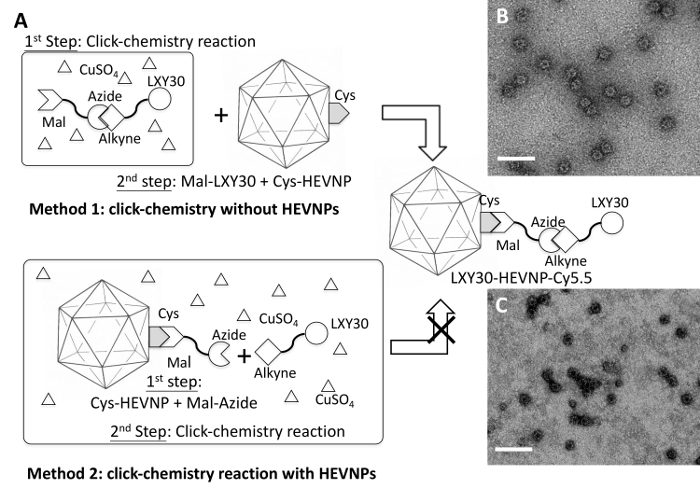
Effective two-step cysteine conjugation was influenced by the order in which conjugation was carried out. Because α3β1 integrin is be overexpressed in breast cancer tumor cells, a synthetic ligand, LXY30, with specific affinity to α3β110, was selected as a tumor-targeting conjugate molecule. Direct maleimide (MaI) functionalization of LXY30 was not feasible because LXY30 is cyclized through an intermolecular disulfide bond; hence, there was a concern for self-reactivity. Instead, LXY30 was functionalized with an alkyne group and bound to maleimide-azide through a click chemistry reaction. When HEV-573C NPs were bound to maleimide-azide first, followed by a click chemistry reaction to LXY30-alkyne, the HEV-573C NPs appeared to disassemble under TEM observation (Figure 4C). However, an initial click chemistry reaction between LXY30-alkyne and maleimide-azide to form MaI-LXY30, followed by MaI-LXY30 conjugation to Cys-modified HEVNPs (LXY30-Mal-Cy5.5) did not affect its structure (Figure 4B). Figure 4 depicts a schematic of the LXY30-functionalization of HEVNPs for tumor cell targeting.
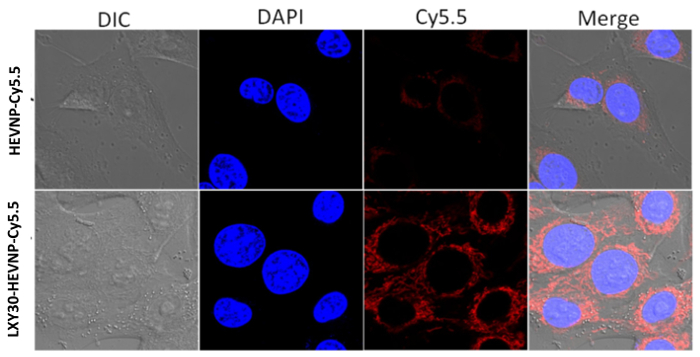
HEVNPs conjugated to the breast cancer targeting ligand, LXY30 (LXY30-HEVNPs) bound and were internalized into cultured breast cancer cells. For fluorescent detection, LXY30-HEVNPs and unbound HEVNP-573Cs were labeled with NHS functionalized Cy5.5 fluorescent dye (NHS-Cy5.5) through primary amine coupling of lysine on HEVNP-573Cs. LXY30-HEVNP-Cy5.5 and HEVNP-Cy5.5 were applied to cultured breast cancer cells (MDA-MB-231), and observed by confocal microscopy. The NIR fluorescence images by confocal microscope indicate LXY30-HEVNP-Cy5.5 had higher binding affinity for and increased internalization in MDA-MB-231 breast cancer cells compared with HEVNP-Cy5.5 (Figure 5).
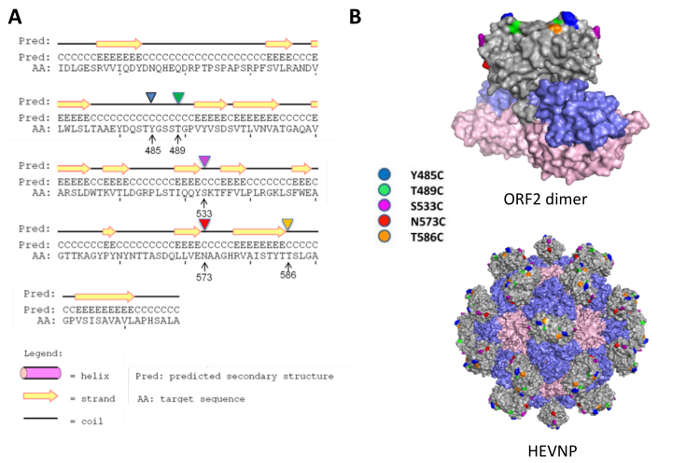
Figure 1: Cysteine replacement on the surface protrusion domain of hepatitis E virus nanoparticle. Cysteine mutations were proposed for residues Y485, T489, S533, N573, and T586 at the flexible coil region to minimize the disturbance to the secondary structure of HEV ORF2 (A). All the selected residues for cysteine mutation are located at the surface of the protrusion (P) domain (grey; the other domains are the shell (S)-domain in blue and middle (M)-domain in pink), with residue Y485 (cyan), T489 (yellow), T586 (orange) on the outmost surface, and residue S533 (magenta) and N573 (red) on the side facing the five-fold axis of HEVNP (B). The figure has been modified from Chen et al. 20164 with permission. Please click here to view a larger version of this figure.
Figure 2: Characterization of HEVNP carrying N573C as an example for all Cys modified HEVNPs. The HEVNP-573C appeared as spherical projections on electron micrographs (A) and remained as intact particles after Mal-biotinylating conjugation (B). Representative results of the conjugation and epitope exposure of Cys-modified HEVNPs. They were bound to maleimide-biotin via cysteine-maleimide coupling and subsequently assayed through Western blot for streptavidin binding. The cysteine mutation at N573 allowed efficient biotinylation to the HEVNP compared to the other mutation sites, i.e., Y485, T489, S533, and T586 (C). Bar equals 100 nm. Please click here to view a larger version of this figure.
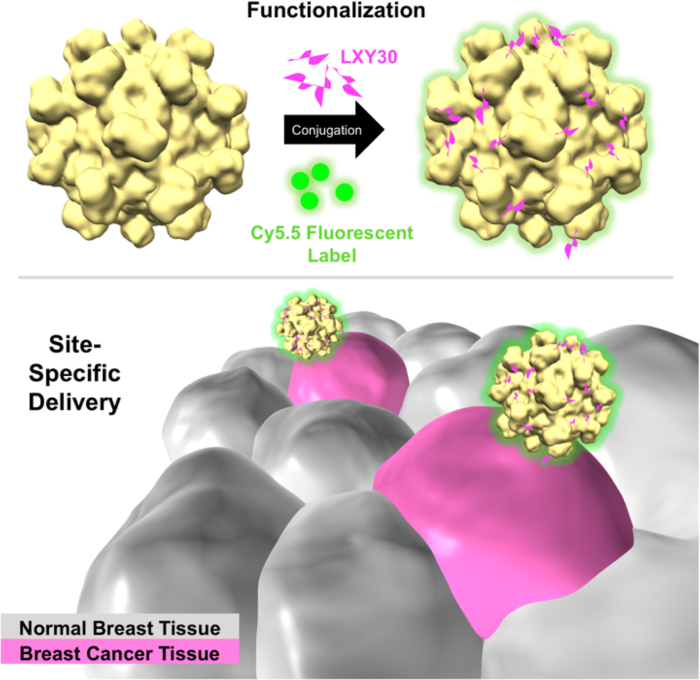
Figure 3: Schematic of breast cancer cells that are specifically targeted with Cy5.5-labeled LXY30-HEVNPs (LXY30-HEVNP-Cy5.5). LXY30-HEVNP-Cy5.5 selectively bind to breast cancer cells (magenta) due to LXY30's specific affinity to α3β1, which is an over-expressed integrin on the breast cancer cell membrane. Please click here to view a larger version of this figure.
Figure 4: A schematic of the two-step chemical conjugation processes, including a thiol-selective reaction and a copper catalyzed azide-alkyne cycloaddition or 'click chemistry' reaction to build LXY30-HEVNPs. Two methods have been tested. Method 1: the copper catalyzed azide-alkyne cycloaddition reaction between Mal-azide and alkyne-LXY30 to form Mal-linked LXY30 (Mal-LXY30) was performed first. Then the Mal-LXY30 was added to react with the Cys site of HEV-573C NPs (A). The LXY30 and Cy5.5 decorated HEV-573C NPs (LXY30-HEVNP-Cy5.5) remained intact after all these chemical conjugation processes (B). Method 2: the conjugation process begins by labeling Mal-linked azide (Mal-Azide) at the Cys site of HEV-573C NPs through a thiol-selective reaction to build azide-linked HEVNPs (Azide-HEVNPs), followed by adding LXY30-alkyne, ascorbic acid, and CuSO4 to form LXY30-linked HEVNPs (LXY30-HEVNPs) (A). However, most LXY30-HEVNPs were damaged or disassembled from the conjugation processes, which may be caused by the reactive reagents, including azide, ascorbic acid, and CuSO4 (C). Mal: Maleimide. This figure has been modified from Chen et al. 20164 with permission. Please click here to view a larger version of this figure.
Figure 5: Representative results of cell binding and internalization of fluorescently labeled LXY30-HEVNPs, which binds to MDA-MB-231 breast cancer cells. NIR fluorescence images of LXY30-HEVNP-Cy5.5 targeting to MDA-MB-231 cells. The signal of the nuclei, which were stained by DAPI (blue), and the signal of Cy5.5 (red) were acquired using a confocal fluorescence microscope. This figure has been modified from Chen et al. 20164 with permission. Please click here to view a larger version of this figure.
Discussion
In contrast to the time consuming genetic engineering procedure, which usually takes weeks, here we demonstrate simple two-step and one-step chemical conjugation procedures, which can be completed within 3 days, of adding the cancer targeting ligand and/or fluorescence detection dye to the Cys/Lys sites of HEVNPs. The technique can be used to screen for the best ligand target from a pool of candidates, and thus takes advantage of the available peptide/small molecule synthesis services at a reasonable cost and delivery time.
Unlike traditional genetic engineering, which can only insert the polypeptides into proteins of interest, the chemical conjugation can connect various materials including polypeptides, small chemical molecules, and nano-particles onto the surface of the protein of interest, as long as there is a Cys or Lys residue to be used as the reactive site. If there is no such residue at the chemical conjugation site, simple Cys/Lys replacement can be genetically modified on the protein of interest to make it chemically activatable as demonstrated in previous research4.
To control that the chemical conjugation only occurs on the selective reaction sites, the structure of both the conjugate molecule and protein to be conjugated need to be thoroughly studied to avoid intramolecular reactivity. As demonstrated herein with the breast cancer targeting peptide LXY3010, a critical disulfide bond stabilizes cyclic ligand conformation. Given the thiol-reactivity of maleimide, direct maleimide-functionalization of LXY30 ligands will likely result in intramolecular reactivity. Instead, two step conjugation (step 4.2) was performed by reacting the maleimide-linked azide with alkyne-linked LXY30 through click chemistry reaction. However, the sequence of the click chemistry reaction and thiol-selected reaction is critical for the conformational intactness of the LXY30-HEVNPs (Figure 4). Through a CuSO4 catalyzed click-chemistry reaction, LXY30 was indirectly bound to maleimide to build MaI-LXY30. Subsequently, the MaI-LXY30 are conjugated to HEVNP-573C such that the formed LXY30-HEVNPs can retain their native icosahedral structure (Figure 4).
Disclosures
The authors declare that they have no competing interests.
Acknowledgments
The authors acknowledge the sponsorship of the funding to RHC by NIH grant #'s: AI095382, EB021230, CA198880, National Institute of Food and Agriculture, as well as the Finland Distinguished Professor program.
References
- Ludwig C, Wagner R. Virus-like particles-universal molecular toolboxes. Curr Opin Biotechnol. 2007;18(6):537–545. doi: 10.1016/j.copbio.2007.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galaway FA, Stockley PG. MS2 viruslike particles: a robust, semisynthetic targeted drug delivery platform. Mol Pharm. 2013;10(1):59–68. doi: 10.1021/mp3003368. [DOI] [PubMed] [Google Scholar]
- Ma Y, Nolte RJ, Cornelissen JJ. Virus-based nanocarriers for drug delivery. Adv Drug Deliv Rev. 2012;64(9):811–825. doi: 10.1016/j.addr.2012.01.005. [DOI] [PubMed] [Google Scholar]
- Chen CC, et al. Chemically activatable viral capsid functionalized for cancer targeting. Nanomedicine (Lond) 2016;11(4):377–390. doi: 10.2217/nnm.15.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jariyapong P, et al. Chimeric hepatitis E virus-like particle as a carrier for oral-delivery. Vaccine. 2013;31(2):417–424. doi: 10.1016/j.vaccine.2012.10.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing L, et al. Recombinant hepatitis E capsid protein self-assembles into a dual-domain T = 1 particle presenting native virus epitopes. Virology. 1999;265(1):35–45. doi: 10.1006/viro.1999.0005. [DOI] [PubMed] [Google Scholar]
- Li TC, et al. Essential elements of the capsid protein for self-assembly into empty virus-like particles of hepatitis E virus. J Virol. 2005;79(20):12999–13006. doi: 10.1128/JVI.79.20.12999-13006.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing L, et al. Structure of hepatitis E virion-sized particle reveals an RNA-dependent viral assembly pathway. J Biol Chem. 2010;285(43):33175–33183. doi: 10.1074/jbc.M110.106336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing L, et al. Spatial configuration of hepatitis E virus antigenic domain. J Virol. 2011;85(2):1117–1124. doi: 10.1128/JVI.00657-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W, et al. Discovery and characterization of a high-affinity and high-specificity peptide ligand LXY30 for in vivo targeting of α3 integrin-expressing human tumors. EJNMMI research. 2016;6(1) doi: 10.1186/s13550-016-0165-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li TC, et al. Expression and self-assembly of empty virus-like particles of hepatitis E virus. J Virol. 1997;71(10):7207–7213. doi: 10.1128/jvi.71.10.7207-7213.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Technologies IbL. Bac-to-Bac Baculovirus Expression System: User Manual. 2015. Available from: http://tools.thermofisher.com/content/sfs/manuals/bactobac_man.pdf.
- Peyret H. A protocol for the gentle purification of virus-like particles produced in plants. J Virol Methods. 2015;225:59–63. doi: 10.1016/j.jviromet.2015.09.005. [DOI] [PubMed] [Google Scholar]
- Technologies NbL. Vol. MAN0007891 1-2. Life Technologies; 2013. [Google Scholar]
- Baskin JM, et al. Copper-free click chemistry for dynamic in vivo imaging. Proc Natl Acad Sci U S A. 2007;104(43):16793–16797. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]