

Abstract
Live-cell imaging is a powerful technique that can be used to directly visualize biological phenomena in single cells over extended periods of time. Over the past decade, new and innovative technologies have greatly enhanced the practicality of live-cell imaging. Cells can now be kept in focus and continuously imaged over several days while maintained under 37 °C and 5% CO2 cell culture conditions. Moreover, multiple fields of view representing different experimental conditions can be acquired simultaneously, thus providing high-throughput experimental data. Live-cell imaging provides a significant advantage over fixed-cell imaging by allowing for the direct visualization and temporal quantitation of dynamic cellular events. Live-cell imaging can also identify variation in the behavior of single cells that would otherwise have been missed using population-based assays. Here, we describe live-cell imaging protocols to assess cell fate decisions following treatment with the anti-mitotic drug paclitaxel. We demonstrate methods to visualize whether mitotically arrested cells die directly from mitosis or slip back into interphase. We also describe how the fluorescent ubiquitination-based cell cycle indicator (FUCCI) system can be used to assess the fraction of interphase cells born from mitotic slippage that are capable of re-entering the cell cycle. Finally, we describe a live-cell imaging method to identify nuclear envelope rupture events.
Keywords: Cancer Research, Issue 135, FUCCI, G1/S, mitotic slippage, phase-contrast microscopy, mitotic catastrophe, tetraploidy, cell cycle arrest, Taxol, nuclear rupture
Introduction
Anti-mitotic drugs have long been used in the chemotherapeutic regimens of various types of solid tumors and often show great efficacy1,2,3. Mechanistically, these drugs disrupt normal mitotic progression and promote mitotic arrest in rapidly proliferating cancer cells. However, cell fate in response to mitotic arrest is highly variable: while a fraction of cells undergoes cell death directly from mitosis, others exit out of mitosis and return to interphase as tetraploid cells (a process termed mitotic slippage)4,5,6,7,8. These interphase cells can execute apoptosis, undergo permanent cell cycle arrest, or even re-enter the cell cycle4,5,6,7,8,9,10,11. Cells that evade mitotic cell death by slipping into interphase, only to re-enter the cell cycle following drug removal, may therefore contribute to the re-emergence of cancer cell populations. Moreover, cells that slip from mitosis are tetraploid, and tetraploidy is known to promote chromosome instability that drives tumor relapse12,13,14,15. Defining the factors that control cell fate in response to anti-mitotic drug treatments is therefore critical to optimize current therapeutics.
In this protocol, we describe methods to directly observe and study the fate of cells that undergo prolonged mitotic arrest in response to the anti-mitotic drug paclitaxel. Paclitaxel is an established therapeutic in the clinic and has proven highly efficacious in many tumors types, including those of the breast, ovaries, and lungs16,17,18,19,20. Paclitaxel, which is a plant alkaloid derived from the bark of the Yew tree, stabilizes microtubules and thus prevents their dynamicity21,22. While dampening of microtubule dynamics by paclitaxel does not affect cell cycle progression from G1 through G2, the drug does lead to sustained activation of the spindle assembly checkpoint during mitosis by hindering kinetochore-microtubule attachment (reviewed in depth here23,24)25. As a consequence, anaphase onset is prevented in paclitaxel-treated cells and results in a prolonged mitotic arrest.
This protocol will first describe approaches to identify mitotic cells in live-cell imaging experiments. Mitosis can be visualized in adherent tissue culture cells due to two noticeable cell biological changes. First, chromosomes become highly condensed immediately prior to nuclear envelope breakdown. While often detectable by standard phase-contrast microscopy, chromosome condensation can be more clearly detected using fluorescent tags that label chromosomes (e.g. fluorescently-labeled histone proteins). Second, mitotic cells can also be identified by the dramatic morphological changes that result from cell rounding.
This protocol will then demonstrate how to use live-cell imaging approaches to track the fates of cells experiencing prolonged mitotic arrest. Cells arrested in mitosis undergo one of three distinct fates. First, cells can undergo cell death during mitosis. This phenomenon is readily visualized by light microscopy, as dying cells are observed to shrink, bleb, and/or rupture. Second, cells can exit from mitosis and return back to interphase without chromosome segregation or cytokinesis, a process termed mitotic slippage. The decondensation of chromosomes and/or the flattening of the mitotic cell readily identifies this process. Cells that slip from mitosis also often display irregular, multi-lobed nuclei and frequently harbor several micronuclei5. Third, cells arrested in mitosis can initiate anaphase and proceed through mitosis after a long delay. While uncommon at higher drug concentrations, this behavior suggests that the arrested cells may have satisfied the spindle assembly checkpoint, or that the spindle assembly checkpoint is partially weakened or defective. Anaphase onset can be visualized by chromosome segregation and subsequent cytokinesis using live-cell imaging.
Live-cell imaging methods to track the fate of cells that evade mitotic cell death by undergoing mitotic slippage will also be described. Cells that undergo mitotic slippage either die in the subsequent interphase, trigger a durable G1 cell cycle arrest, or re-enter the cell cycle to initiate a new round of cell division4. An approach using the FUCCI (fluorescent ubiquitination-based cell cycle indicator) system to determine the fraction of cells that re-enter the cell cycle following mitotic slippage will be described. FUCCI allows for the direct visualization of the G1/S transition and can be used in conjunction with long-term live-cell imaging both in vitro and in vivo26,27. The FUCCI system takes advantage of two fluorescently labeled proteins, truncated forms of hCdt1 (chromatin licensing and DNA replication factor 1) and hGeminin, whose levels oscillate based on cell cycle position. hCdt1 (fused to a red fluorescent protein) is present at high levels during G1 phase where it acts to license DNA for replication, but is ubiquitinated by the E3 ubiquitin ligase SCFSkp2 and degraded during S/G2/M phases to prevent re-replication of DNA26. By contrast, hGeminin (fused to a green fluorescent protein), is an inhibitor of hCdt1 whose levels peak during S/G2/M, but is ubiquitinated by the E3 ubiquitin ligase APCCdh1 and degraded at the end of mitosis and throughout G126. Consequently, FUCCI delivers a straightforward fluorescence readout of cell cycle phase, as cells exhibit red fluorescence during G1, and green fluorescence during S/G2/M. The FUCCI system is a significant advance over other approaches (such as bromodeoxyuridine staining) to identify proliferative cells, because it does not require cell fixation and allows for single cell imaging without the need for additional pharmacological treatments to synchronize cell populations. Though not discussed in this protocol, additional live-cell sensors have also been developed to visualize cell cycle progression, including a helicase B sensor for G128, DNA ligase-RFP29 and PCDNA-GFP30 sensors for S-phase, and the recent FUCCI-4 sensor, which detects all stages of the cell cycle31.
Finally, a live-cell imaging method to detect nuclear envelope rupture will be described. Recent studies have revealed that the nuclear envelopes of cancer cells are unstable and prone to bursting, thereby allowing the contents of the nucleoplasm and cytoplasm to intermix. This phenomenon, termed nuclear rupture, can promote DNA damage and stimulation of the innate immune response32,33,34,35,36,37,38,39,40,41,42,43,44. While the underlying causes of nuclear rupture remain incompletely characterized, it is known that deformations in nuclear structure correlate with an increased incidence of nuclear rupture42. One well-known effect of paclitaxel treatment is the generation of strikingly abnormal nuclear structures following mitosis; as such, a method using live-cell imaging to quantify nuclear rupture will be described, while also exploring if paclitaxel treatment increases the frequency of nuclear rupture events. Nuclear rupture can be detected by the observed leakage of a nuclear-targeted fluorescent protein into the cytoplasm (e.g. a tandem dimer repeat of RFP fused to a nuclear localization signal, TDRFP-NLS). This leakage is distinctly visible by eye, which enables simple quantitation of rupture events.
This protocol requires a widefield epifluorescence microscope that is equipped with an encoded stage and autofocusing software. The encoded stage allows for precise automated movement to defined X-Y coordinates, while autofocus software maintains cells in focus for the duration of the imaging period. In addition, this protocol requires equipment to maintain cells at 37 °C with humidified 5% CO2 atmosphere. This can be achieved by enclosing the entire microscope within a temperature and atmosphere controlled enclosure, or by using stage-top devices that locally maintains temperature and environment. The phase-contrast objective used in this protocol is a plan fluor 10x with a numerical aperture of 0.30. However, 20X objectives are also sufficient to identify both rounded mitotic and flattened interphase cells in a single focal plane. If performing phase-contrast imaging (as described in this method), the cover can be either glass or plastic. If differential interference contrast (DIC) microscopy is used, it is imperative to use a glass cover to prevent depolarization of light.
Protocol
This protocol focuses on using the non-transformed and chromosomally stable retinal pigmented epithelial (RPE-1) cell line for live-cell imaging experiments. However, this protocol can be adapted to any adherent cell line so long as cell culture conditions are adjusted as necessary. All procedures must adhere to institutional biosafety and ethical guidelines and regulations.
1. Preparing Cells for Live-cell Imaging
- Use freshly thawed and early passage RPE-1 cells expressing either human histone H2B fused to a fluorescent protein (e.g. H2B-GFP), or the FUCCI system (a detailed protocol on how to generate FUCCI-expressing cells can be found in reference45) to assess mitotic cell fate.
- To measure the frequency of nuclear rupture, use RPE-1 cells expressing both H2B-GFP and a tandem dimer of red fluorescent protein fused to a single nuclear localization signal (TDRFP-NLS). NOTE: Constructs of three green fluorescent proteins fused in tandem to a single nuclear localization signal (GFP3-NLS) have also been used to demonstrate rupture (as in reference42).
- Maintain cells on 10 cm tissue culture plates in the appropriate growth medium. RPE-1 cells are maintained in phenol red-free, Dulbecco's Modified Eagle Medium/Nutrient Mixture F-12 (DMEM:F12) supplemented with 10% fetal bovine serum (FBS), 100 IU/mL penicillin, and 100 μg/mL streptomycin.
- Aspirate medium from the cells, wash the tissue culture dish with 10 mL of sterile, room temperature phosphate buffered saline (PBS) to remove remaining medium, and then aspirate the PBS.
- Add 2 mL of 0.25% Trypsin with ethylenediaminetetraacetic acid (EDTA) to the cells and incubate at 37 °C for 3 min or until the majority of cells have detached from the plate. NOTE: Do not keep cells in trypsin longer than needed.
- Add 10 mL of complete medium to collect the trypsinized cells with a 10 mL serological stripette and dispense in a 15 mL conical tube.
- Pellet the cells by centrifugation at 180 x g for 3 min at room temperature.
Aspirate the supernatant, being careful not to disrupt the pellet, and then thoroughly resuspend the pellet in 10 mL of fresh medium by gently pipetting the medium up and down in the serological stripette.
- Use a hemocytometer or automated cell counting machine to count cells46.
- Dilute cells in a new conical tube to 30,000 cells/mL of complete medium.
- Plate 30,000 RPE-1 cells (1 mL volume) per well of a 12-well glass bottomed (#1.5 thickness) imaging dish to achieve the required cellular density the next day (30–50% confluent). NOTE: Always handle the plate with gloves and be careful not to touch the glass-bottom.
Allow cells to grow in a 37 °C tissue culture incubator until they fully attach and flatten on the glass-bottomed imaging plate. While cells can attach in as little as 4 h, it is recommended that cells are not treated with drugs and imaged until the following day.
- Add paclitaxel (dissolved in dimethyl sulfoxide (DMSO)) to the desired final concentration in complete medium and mix thoroughly.
- Prepare complete medium with an equal volume of DMSO alone to use as a control.
- Warm the medium containing drug or DMSO to 37 °C before adding to cells. This will prevent focal drift due to a sudden temperature change.
- Add 1 mL of medium containing either paclitaxel or DMSO alone to individual wells of the 12-well imaging dish.
2. Setting Up the Microscope for Live-cell Imaging
Clean the glass-bottom of the imaging dish with optical cleaner to remove any fingerprints or dust that may interfere with imaging. Use sufficient optical cleaner to wet the entire glass surface.
Place the glass-bottom dish on the microscope stage in the imaging dish adaptor, remove the plastic cover from the dish, and cover the dish with a glass-topped chamber. Ensure the chamber has a valve to allow a steady flow of air consisting of 5% CO2. NOTE: To humidify the 5% CO2, the gas is flowed through tubing inserted into a sterile water bath housing. This allows for the gas to become equilibrated to 95% humidity.
Initiate/calibrate the encoded stage using the software program controlling the microscope. This will ensure accurate X-Y coordinates and prevent focal drift.
Focus on the cells using phase-contrast optics and perform Kohler illumination (described in detail in reference47) to focus the light and provide optimal contrast.
- Use acquisition software to determine the optimal exposure times for white light and all fluorescent channels being used (exposures that give 75% pixel saturation on the camera are ideal, provided this amount of light is not toxic to cells).
- Use acquisition software to select several, non-overlapping fields of view from each well for imaging. Select imaging regions where cells have adhered well to the glass-bottom and are between 50% and 70% confluent. Avoid areas of clumped cells, as this will make subsequent analysis difficult. NOTE: It is important to have non-overlapping fields of view from each well to avoid tracking the same cells twice. If cells are highly motile, it may be necessary to acquire several images radiating out from a central point and stitch them back together to make one large field of view.
Activate the microscope's autofocusing feature to ensure all points are maintained in focus for the duration of the experiment.
To assess mitotic cell fate, set the imaging software to collect images from each selected field of view every 10 min for up to 96 h. Unperturbed mitosis lasts 20–40 min, and thus 10 min intervals will provide enough sampling to identify when cells divide. To identify nuclear rupture, which is a transient event, acquire images every 5 min. NOTE: Make sure that the computer driving the image acquisition software has auto-updating, screensavers, and energy-savings modes disabled, as these can often interfere with image acquisition over long experiments.
Initiate the imaging experiment. Periodically confirm that image acquisition is running smoothly over the course of the video.
3. Video Analysis to Identify Cell Fate in Response to Paclitaxel
Confirm that imaged cells are healthy throughout the course of the imaging experiment. Control cells treated with DMSO should be viable and actively proliferating. NOTE: If cells exhibit signs of stress, do not quantitate the video and instead focus on optimizing imaging conditions48. Signs of imaging stress include blebbing/dying cells, cells that fail to attach/spread on the plate, and cells that show a low mitotic index and/or prolonged mitosis. Possible sources of stress include fluctuations in temperature or CO2 levels inside the imaging chamber or phototoxicity48.
When imaging an entire field of view with a 10X or 20X objective, hundreds of individual cells may be present. Therefore, to assist with quantitation, divide the field of view into smaller quadrants using available software tools. Score cells within each quadrant separately (as described in steps 3.3.1–3.6.2).
- When analyzing the video, track each cell in a merged view using phase-contrast and/or fluorescent images. Use software analysis tools to create the merged view.
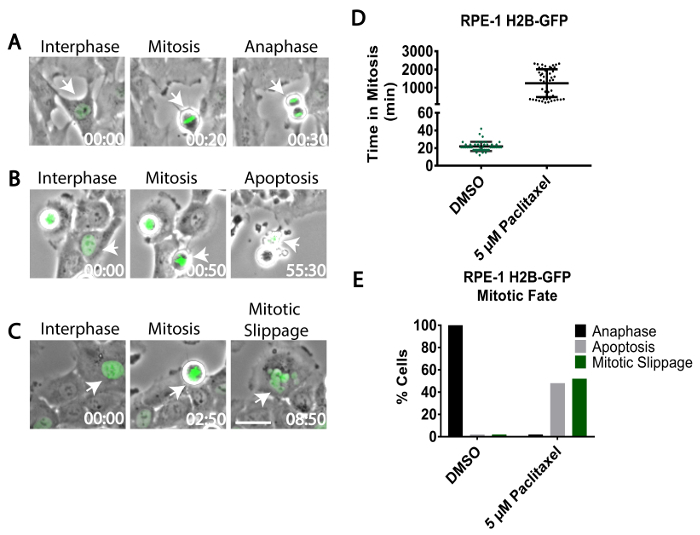
- Starting at the beginning of the video, identify a single interphase cell and track its progress through the cell cycle using phase optics. To track a single cell, observe the cell from frame to frame by eye. Identify interphase cells due to their flattened morphology (as assessed by phase-contrast) and their lack of DNA condensation (as assessed by H2B-GFP) (Figure 1A).
- Identify cells that enter mitosis by observation of cell rounding (using phase-contrast optics) and/or chromosome condensation (using H2B-GFP) (Figure 1A-C). Both cell rounding and chromosome condensation are readily visualized by eye.
- Annotate the time when the cell enters mitosis. Continue tracking the cell until it reaches its fate (anaphase as in step 3.5, cell death from mitosis as in step 3.6.1, or mitotic slippage as in step 3.6.2).
Control cells should efficiently align their chromosomes and enter anaphase within 1 h (Figure 1D). Visualize anaphase by phase-contrast optics as the cell begins to pinch into two or through visualization of poleward-moving chromosomes labeled with H2B-GFP (Figure 1A). Annotate the time when the cell undergoes anaphase.
- By contrast, cells treated with paclitaxel will remain rounded with condensed chromosomes for several hours (3-40 h) (Figure 1B-D).
- Identify mitotically-arrested cells that undergo cell death. NOTE: Cells that die during mitosis are visualized by phase-contrast microscopy, as cells will bleb, shrink, and/or rupture (Figure 1B). If imaging H2B-GFP, the chromosomes will also fragment during cell death.
- Identify mitotically-arrested cells that undergo cell slippage. NOTE: Cells that undergo mitotic slippage are observed by phase-contrast microscopy, as they flatten back out into interphase and decondense chromosomes without undergoing anaphase (Figure 1C). Cells that undergo mitotic slippage give rise to large tetraploid cells that are often multinucleated (Figure 1C).
Continue tracking cells from the original field of view. Once a whole field of view is tracked, move to a separate field of view acquired from the same well and continue tracking cells.
4. Video Analysis to Identify Cell Fate Following Mitotic Slippage
- Assess cell fate following mitotic slippage (as described in step 3.6.2) using RPE-1 cells expressing the FUCCI system. In addition to phase-contrast imaging, it is necessary to acquire both red fluorescence (indicative of G1 phase) and green fluorescence (indicative of S/G2/M) images).
- Track FUCCI RPE-1 cells as described in step 3.3 through 3.7.
- To confirm that the FUCCI system is working properly, validate that control cells alternate expression of the red and green fluorescent proteins appropriately from analysis of the live-cell imaging data. Control cells should transition from exhibiting entirely nuclear red fluorescence to exhibiting entirely nuclear green fluorescence during interphase as cells progress from G1 to S phase. Cells should continue exhibiting green fluorescence throughout the completion of mitosis. Immediately following mitosis, cells should once again exhibit entirely red fluorescence during interphase. NOTE: While methods exist to quantify both RFP and GFP fluorescence intensities from the FUCCI system in single cells using fluorescent traces49, this is often not necessary as the fluorescence color change is robust and visible by eye.
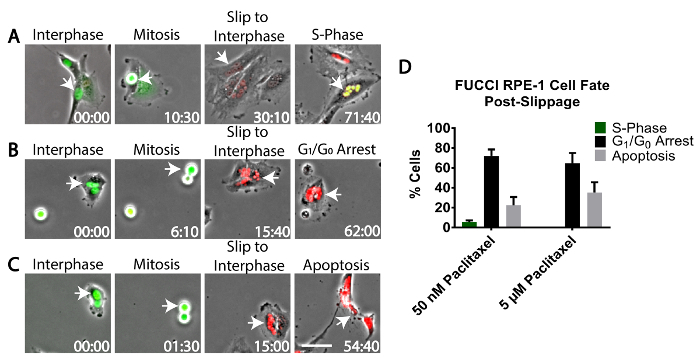
- Identify cells arrested in mitosis using phase-contrast microscopy and track them until they undergo mitotic slippage, as described in 3.4 and 3.6.2. Cells that slip out of mitosis and back into interphase will change from exhibiting green fluorescence during mitosis to red fluorescence during G1 phase (Figure 2A-C).
- Track these slipped cells using phase-contrast and epifluorescent imaging by eye to assess their cell fate (Figure 2D).
- Identify cells that re-enter the cell cycle. These cells are identified by the red-to-green change in fluorescence expression using the FUCCI system that indicates G1/S transition (Figure 2A).
- Identify cells that undergo G1 cell cycle arrest. These cells are identified by expression of red fluorescence that persists for >24 h (Figure 2B).
- Identify cells that die in interphase. These cells are identified by cell rupture/blebbing/shrinking using phase-contrast imaging (Figure 2C).
5. Video Analysis to Identify Frequency of Nuclear Envelope Rupture
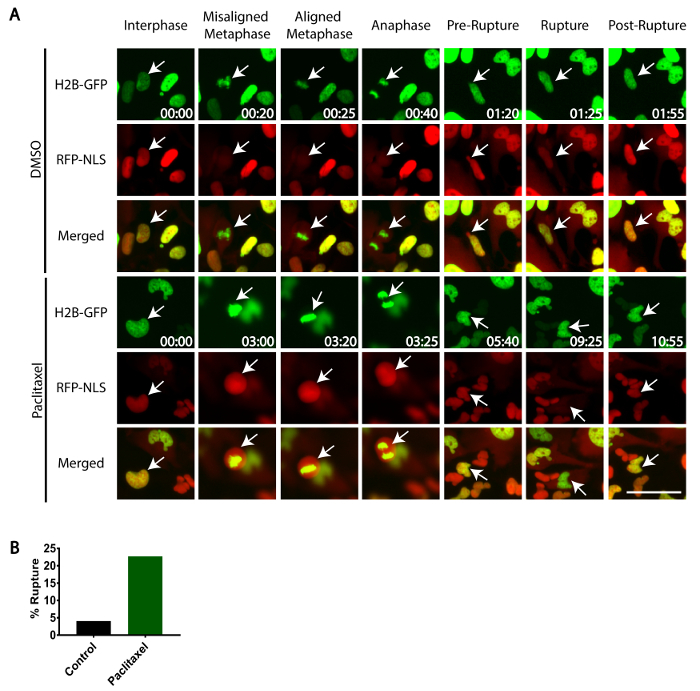
Use RPE-1 cells expressing H2B-GFP and TDRFP-NLS to assess nuclear envelope rupture. In addition to phase-contrast imaging, acquire both red fluorescence (TRITC) and green fluorescence (FITC) images. Acquire images every 5 min to visualize rupture. NOTE: It is critical to generate cell lines in which the TDRFP-NLS is efficiently imported into the nucleus with minimal cytoplasmic fluorescence.
Image cells as described in step 3.3 through 3.4. The TDRFP-NLS fluorescence signal and H2B-GFP should co-localize during interphase. Upon mitosis (as visualized by cell rounding using phase optics and/or chromosome condensation by H2B-GFP), the nuclear envelope will break down and the TDRFP-NLS signal will become cytoplasmic (Figure 3A). Following mitosis, the nuclear envelope will reform in the daughter cells and the TDRFP-NLS signal will become nuclear.
Track daughter cells throughout the subsequent interphase by live-cell imaging. Identify nuclear rupture events by observing a transient burst of nuclear-localized TDRFP-NLS into the surrounding cytoplasm. Within minutes, the nuclear envelope will be repaired and the TDRFP-NLS will be relocalized to the nucleus (Figure 3A). NOTE: Often, the nuclear DNA, as visualized by H2B-GFP, will be seen to protrude outside the rupture site as a small bleb.
Score the fraction of nuclei that undergo a rupture event during interphase. To score this fraction, count the number of cells that undergo a rupture event over the total number of cells tracked.
Representative Results
Using the protocol described above, RPE-1 cells expressing H2B-GFP were treated with an anti-mitotic or vehicle control (DMSO) and analyzed by live-cell imaging. Analysis revealed that 100% of control mitotic RPE-1 cells initiated anaphase an average of 22 min after entering mitosis (Figure 1A, 1D). By contrast, RPE-1 cells treated with paclitaxel exhibited a profound mitotic arrest lasting several hours (Figure 1D). Imaging revealed that 48% of these mitotically arrested cells underwent mitotic cell death (Figure 1B, 1E), while the remaining 52% underwent mitotic slippage (Figure 1C, 1E).
To assess the fate of RPE-1 cells that underwent mitotic slippage, RPE-1 cells expressing the FUCCI system were treated with either low-dose 50 nM paclitaxel, high-dose 5 µM paclitaxel, or vehicle control (DMSO) and analyzed by long term live-cell imaging. The data revealed that of the RPE-1 cells that underwent mitotic slippage following treatment with 50 nM paclitaxel, 22% died in the subsequent cell cycle (Figure 2C, 2D), 72% induced cell cycle arrest (Figure 2B, 2D), and only 5% re-entered S-phase (Figure 2A, 2D). By contrast, of the RPE-1 cells treated with high-dose 5 µM paclitaxel that underwent mitotic slippage, 35% died in the subsequent cell cycle, 65% induced cell cycle arrest, and none re-entered S-phase (Figure 2D).
Interestingly, low-dose paclitaxel-treatment promotes nuclear envelope rupture in RPE-1 cells following mitosis. After mitotic completion daughter cells were tracked and scored for any nuclear rupture events, revealing that only ~4% of control (DMSO-treated) RPE-1 daughter cells ruptured, whereas ~23% of daughter cells treated with 10 nM paclitaxel exhibited rupture (Figure 3A, 3B).
Figure 1: Mitotic cell fate in response to anti-mitotic treatment. RPE-1 cells stably expressing H2B-GFP were treated with DMSO vehicle control (A) or 5 µM paclitaxel (B, C) and imaged using time-lapse phase-contrast and widefield epifluorescence microscopy to assess mitotic cell fate. Interphase cells (left panels) were tracked as they entered mitosis (middle panels) and until they either initiated anaphase (A), underwent mitotic cell death (B), or slipped from mitosis back to interphase (C). White arrows indicate tracked cells. (D) The amount of time that control cells (DMSO-treated) and anti-mitotic-treated cells spent in mitosis, as quantitated by live-cell imaging (n =100 cells for each condition). (E) The fraction of cells (n = 100) that underwent anaphase, mitotic cell death, or mitotic slippage in DMSO-treated cells or cells treated with 5 µM paclitaxel. Time, h:min. Scale Bar = 50 µm. Please click here to view a larger version of this figure.
Figure 2: Fate of cells that undergo mitotic slippage. RPE-1 cells stably expressing the FUCCI system were treated with 50 nM paclitaxel or 5 µM paclitaxel and imaged using time-lapse phase-contrast and widefield fluorescence microscopy to quantitate cell fate following mitotic slippage. Cells were scored as either re-entering the cell cycle, as judged by a red-to-green fluorescence change in the FUCCI system (A); arresting in G1 phase, as judged by persistent red fluorescence for >24 h (B); or dying during the subsequent mitosis, as judged by cellular blebbing/rupture (C). White arrows indicate tracked cells. (D) The fraction of cells (n = 100) that underwent each fate. Error bars represent the standard deviation from the mean from two independent experiments. Time, h:min. Scale Bar = 50 µm. Please click here to view a larger version of this figure.
Figure 3: Frequency of nuclear envelope rupture in paclitaxel-treated cells. RPE-1 cells stably expressing TDRFP-NLS and H2B-GFP were treated with 10 nM paclitaxel or vehicle control (DMSO) and imaged using time-lapse phase-contrast and widefield epifluorescence microscopy (A). During mitosis (Metaphase/Anaphase) the nuclear envelope is broken down and TDRFP-NLS becomes cytoplasmic. Following mitosis, the TDRFP-NLS relocalizes to the nucleus of interphase cells (Pre-Rupture). Nuclear rupture events are identified by the delocalization of TDRFP-NLS from the nucleus to the cytoplasm in interphase cells (Rupture), followed by the relocalization of TDRFP-NLS to the nucleus following nuclear envelope repair (Post-Rupture). White arrows indicate tracked cells. (B) The fraction of cells (n >100 per condition) that show nuclear envelope rupture following mitosis in the presence of paclitaxel or vehicle control. Time, h:min. Scale Bar = 50 µm. Please click here to view a larger version of this figure.
Discussion
The most critical aspect of long-term live-cell imaging is ensuring the health of the cells being imaged. It is essential that cells be exposed to minimal extraneous environmental stressors, such as substandard conditions regarding temperature, humidity, and/or CO2 levels. It is also important to limit photodamage from fluorescent excitation illumination, as imaging stress is known to affect cell behavior50. Limiting photodamage can be achieved in multiple ways as detailed elsewhere48. In general, it is best to use the shortest exposure time necessary to produce a sufficiently bright enough fluorescence signal to efficiently track cells, thus limiting phototoxicity. However, if cells are imaged only once every 10–20 min, longer exposures (that approach pixel saturation) are typically well tolerated (though this must be determined empirically for each fluorophore and cell type). Because imaged cells are sensitive to both environmental and imaging stresses, it is imperative that control cells are always included in live-cell imaging experiments and monitored to confirm normal proliferation.
One limitation to long-term live-cell imaging is that it requires equipping an existing microscope with either an environmental chamber that maintains both temperature and 5% humidified CO2 atmosphere, or a stage-top chamber that is also capable of supporting the appropriate temperature and atmosphere. While flowing CO2 is optimal, cells can also be imaged for up to 48 h using CO2-independent medium if an environmental chamber with 5% humidified CO2 is not available. However, many cell types are intolerant of such medium, and this must be determined empirically by measuring cell growth and viability. Overlaying the cells with mineral oil can also be used to prevent medium evaporation.
A second major limitation to this method is that manually tracking cell fates is highly laborious and time-intensive. However, cell-tracking programs are available and may be optimized to automate image analysis51,52,53. A further limitation with this method is that highly motile cells are often difficult to track over extended periods of time. If this poses a significant problem, the entire well can be imaged and the images stitched together to form one large field of view. Many software programs support this method.
Autofluorescence is another constraint that must be addressed with this protocol. Phenol red free medium reduces background autofluorescence during live cell imaging. It has also been demonstrated that two vitamins commonly found in growth medium, riboflavin and pyridoxal, can decrease photostability of fluorescent proteins. Thus, medium lacking these vitamins can also be used during fluorescence imaging to enhance signal to noise rations.While plastic bottom dishes are less expensive and may provide adequate imaging results, they also produce a greater amount of autofluorescence that negatively affects signal-to-noise ratios. In addition, plastic bottom dishes have greater variation in thickness, which can disrupt the autofocus feature of many microscopes. The thickness of the glass bottomed imaging dish in this protocol is 0.17 mm, which is the ideal glass for use with modern microscopes.
Lastly, long-term movies encompassing multiple fields of view and acquiring several fluorescence channels produces massive data files (tens of gigabytes to even terabytes each), which pose a problem for data storage and back-up. To mitigate this issue, it is recommended that all images be acquired using 2 x 2 or 4 x 4 binning, provided the resulting loss in resolution is acceptable. Binning not only limits exposure times (i.e. cells stably expressing H2B-GFP or the FUCCI system should have exposures less than 500 ms), but also drastically reduces the size of data files (an image that is binned 2 x 2 is one-fourth the file size of a non-binned image).
Despite these limitations, long-term live-cell imaging represents the best method to track individual cell fates from whole populations, especially when cell fates are highly heterogeneous. As more live-cell fluorescent markers and sensors become available, live-cell imaging approaches will be expanded to visualize and quantitate several additional aspects of cell biology. For example, live-cell sensors currently exist to quantitate DNA damage foci, p53 levels, cell cycle position, and apoptosis26,54,55,56,57,58. Thus, imaging experiments have the capacity to reveal the underlying cellular properties that dictate how and why single cells respond variably to chemotherapeutic agents.
Disclosures
The authors have nothing to disclose.
Acknowledgments
AFB, MAV and NJG would like to thank Ryan Quinton for comments on the manuscript and Adrian Salic for the TDRFP-NLS construct. AFB and MAV are funded by the NIGMS Biomolecular Pharmacology Training Grant 5T32GM008541. NJG is a member of the Shamim and Ashraf Dahod Breast Cancer Research Laboratories and is supported by NIH grants GM117150 and CA-154531, the Karin Grunebaum Foundation, the Smith Family Awards Program, the Searle Scholars Program, and the Melanoma Research Alliance.
References
- van Vuuren RJ, Visagie MH, Theron AE, Joubert AM. Antimitotic drugs in the treatment of cancer. Cancer Chemother Pharmacol. 2015;76(6):1101–1112. doi: 10.1007/s00280-015-2903-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KS, Koh CG, Li HY. Mitosis-targeted anti-cancer therapies: where they stand. Cell Death Dis. 2012;3:e411. doi: 10.1038/cddis.2012.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson JR, Patrick DR, Dar MM, Huang PS. Targeted anti-mitotic therapies: can we improve on tubulin agents? Nat Rev Cancer. 2007;7(2):107–117. doi: 10.1038/nrc2049. [DOI] [PubMed] [Google Scholar]
- Gascoigne KE, Taylor SS. How do anti-mitotic drugs kill cancer cells? J Cell Sci. 2009;122(Pt 15):2579–2585. doi: 10.1242/jcs.039719. [DOI] [PubMed] [Google Scholar]
- Rieder CL, Maiato H. Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Dev Cell. 2004;7(5):637–651. doi: 10.1016/j.devcel.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Gascoigne KE, Taylor SS. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell. 2008;14(2):111–122. doi: 10.1016/j.ccr.2008.07.002. [DOI] [PubMed] [Google Scholar]
- Huang HC, Mitchison TJ, Shi J. Stochastic competition between mechanistically independent slippage and death pathways determines cell fate during mitotic arrest. PLoS One. 2010;5(12):e15724. doi: 10.1371/journal.pone.0015724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Orth JD, Mitchison T. Cell type variation in responses to antimitotic drugs that target microtubules and kinesin-5. Cancer Res. 2008;68(9):3269–3276. doi: 10.1158/0008-5472.CAN-07-6699. [DOI] [PubMed] [Google Scholar]
- Ganem NJ, Pellman D. Limiting the proliferation of polyploid cells. Cell. 2007;131(3):437–440. doi: 10.1016/j.cell.2007.10.024. [DOI] [PubMed] [Google Scholar]
- Ganem NJ, Storchova Z, Pellman D. Tetraploidy, aneuploidy and cancer. Curr Opin Genet Dev. 2007;17(2):157–162. doi: 10.1016/j.gde.2007.02.011. [DOI] [PubMed] [Google Scholar]
- Ganem NJ, Pellman D. Linking abnormal mitosis to the acquisition of DNA damage. J Cell Biol. 2012;199(6):871–881. doi: 10.1083/jcb.201210040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotillo R, Schvartzman JM, Socci ND, Benezra R. Mad2-induced chromosome instability leads to lung tumour relapse after oncogene withdrawal. Nature. 2010;464(7287):436–440. doi: 10.1038/nature08803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotillo R, et al. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell. 2007;11(1):9–23. doi: 10.1016/j.ccr.2006.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem NJ, Godinho SA, Pellman D. A mechanism linking extra centrosomes to chromosomal instability. Nature. 2009;460(7252):278–282. doi: 10.1038/nature08136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silkworth WT, Nardi IK, Scholl LM, Cimini D. Multipolar spindle pole coalescence is a major source of kinetochore mis-attachment and chromosome mis-segregation in cancer cells. PLoS One. 2009;4(8):e6564. doi: 10.1371/journal.pone.0006564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire WP, et al. Cyclophosphamide and Cisplatin Compared with Paclitaxel and Cisplatin in Patients with Stage III and Stage IV Ovarian Cancer. New England Journal of Medicine. 1996;334(1):1–6. doi: 10.1056/NEJM199601043340101. [DOI] [PubMed] [Google Scholar]
- McGuire WP, et al. Taxol: a unique antineoplastic agent with significant activity in advanced ovarian epithelial neoplasms. Ann Intern Med. 1989;111(4):273–279. doi: 10.7326/0003-4819-111-4-273. [DOI] [PubMed] [Google Scholar]
- Ettinger DS. Taxol in the treatment of lung cancer. J Natl Cancer Inst Monogr. 1993. pp. 177–179. [PubMed]
- Weaver BA. How Taxol/paclitaxel kills cancer cells. Mol Biol Cell. 2014;25(18):2677–2681. doi: 10.1091/mbc.E14-04-0916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4(4):253–265. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- Wall ME, Wani MC. Camptothecin and Taxol: Discovery to Clinic-Thirteenth Bruce F. Cain Memorial Award Lecture. Cancer Research. 1995;55(4):753–760. [PubMed] [Google Scholar]
- Schiff PB, Horwitz SB. Taxol stabilizes microtubules in mouse fibroblast cells. Proc Natl Acad Sci U S A. 1980;77(3):1561–1565. doi: 10.1073/pnas.77.3.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara-Gonzalez P, Westhorpe FG, Taylor SS. The spindle assembly checkpoint. Curr Biol. 2012;22(22):R966–R980. doi: 10.1016/j.cub.2012.10.006. [DOI] [PubMed] [Google Scholar]
- Musacchio A. The Molecular Biology of Spindle Assembly Checkpoint Signaling Dynamics. Curr Biol. 2015;25(20):R1002–R1018. doi: 10.1016/j.cub.2015.08.051. [DOI] [PubMed] [Google Scholar]
- Uetake Y, et al. Cell cycle progression and de novo centriole assembly after centrosomal removal in untransformed human cells. J Cell Biol. 2007;176(2):173–182. doi: 10.1083/jcb.200607073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaue-Sawano A, et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 2008;132(3):487–498. doi: 10.1016/j.cell.2007.12.033. [DOI] [PubMed] [Google Scholar]
- Chittajallu DR, et al. In vivo cell-cycle profiling in xenograft tumors by quantitative intravital microscopy. Nat Methods. 2015;12(6):577–585. doi: 10.1038/nmeth.3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, et al. Cell cycle-dependent regulation of a human DNA helicase that localizes in DNA damage foci. Mol Biol Cell. 2004;15(7):3320–3332. doi: 10.1091/mbc.E04-03-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easwaran HP, Leonhardt H, Cardoso MC. Cell cycle markers for live cell analyses. Cell Cycle. 2005;4(3):453–455. doi: 10.4161/cc.4.3.1525. [DOI] [PubMed] [Google Scholar]
- Hahn AT, Jones JT, Meyer T. Quantitative analysis of cell cycle phase durations and PC12 differentiation using fluorescent biosensors. Cell Cycle. 2009;8(7):1044–1052. doi: 10.4161/cc.8.7.8042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajar BT, et al. Fluorescent indicators for simultaneous reporting of all four cell cycle phases. Nat Methods. 2016;13(12):993–996. doi: 10.1038/nmeth.4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gekara NO. DNA damage-induced immune response: Micronuclei provide key platform. J Cell Biol. 2017;216(10):2999–3001. doi: 10.1083/jcb.201708069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow KH, Factor RE, Ullman KS. The nuclear envelope environment and its cancer connections. Nat Rev Cancer. 2012;12(3):196–209. doi: 10.1038/nrc3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crasta K, et al. DNA breaks and chromosome pulverization from errors in mitosis. Nature. 2012;482(7383):53–58. doi: 10.1038/nature10802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch EM, Fischer AH, Deerinck TJ, Hetzer MW. Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell. 2013;154(1):47–60. doi: 10.1016/j.cell.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciejowski J, Li Y, Bosco N, Campbell PJ, de Lange T. Chromothripsis and Kataegis Induced by Telomere Crisis. Cell. 2015;163(7):1641–1654. doi: 10.1016/j.cell.2015.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CZ, et al. Chromothripsis from DNA damage in micronuclei. Nature. 2015;522(7555):179–184. doi: 10.1038/nature14493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denais CM, et al. Nuclear envelope rupture and repair during cancer cell migration. Science. 2016;352(6283):353–358. doi: 10.1126/science.aad7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raab M, et al. ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science. 2016;352(6283):359–362. doi: 10.1126/science.aad7611. [DOI] [PubMed] [Google Scholar]
- Bernhard W, Granboulan N. THE FINE STRUCTURE OF THE CANCER CELL NUCLEUS. Exp Cell Res. 1963;24(Suppl 9):19–53. doi: 10.1016/0014-4827(63)90243-x. [DOI] [PubMed] [Google Scholar]
- de Noronha CM, et al. Dynamic disruptions in nuclear envelope architecture and integrity induced by HIV-1 Vpr. Science. 2001;294(5544):1105–1108. doi: 10.1126/science.1063957. [DOI] [PubMed] [Google Scholar]
- Vargas JD, Hatch EM, Anderson DJ, Hetzer MW. Transient nuclear envelope rupturing during interphase in human cancer cells. Nucleus. 2012;3(1):88–100. doi: 10.4161/nucl.18954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieprath T, Darwiche R, De Vos WH. Lamins as mediators of oxidative stress. Biochem Biophys Res Commun. 2012;421(4):635–639. doi: 10.1016/j.bbrc.2012.04.058. [DOI] [PubMed] [Google Scholar]
- Mitchison TJ, Pineda J, Shi J, Florian S. Is inflammatory micronucleation the key to a successful anti-mitotic cancer drug? Open Biol. 2017;7(11) doi: 10.1098/rsob.170182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenk EM, Ganem NJ. Generation and Purification of Tetraploid Cells. Methods Mol Biol. 2016;1413:393–401. doi: 10.1007/978-1-4939-3542-0_24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morten BC, Scott RJ, Avery-Kiejda KA. Comparison of Three Different Methods for Determining Cell Proliferation in Breast Cancer Cell Lines. J Vis Exp. 2016. [DOI] [PMC free article] [PubMed]
- Salmon ED, Canman JC. Proper alignment and adjustment of the light microscope. Curr Protoc Cell Biol. 2001. Chapter 4 Unit 4 1. [DOI] [PubMed]
- Cole R. Live-cell imaging. Cell Adh Migr. 2014;8(5):452–459. doi: 10.4161/cam.28348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke RT, Orth JD. Through the Looking Glass: Time-lapse Microscopy and Longitudinal Tracking of Single Cells to Study Anti-cancer Therapeutics. J Vis Exp. 2016. [DOI] [PMC free article] [PubMed]
- Douthwright S, Sluder G. Live Cell Imaging: Assessing the Phototoxicity of 488 and 546 nm Light and Methods to Alleviate it. J Cell Physiol. 2017;232(9):2461–2468. doi: 10.1002/jcp.25588. [DOI] [PubMed] [Google Scholar]
- Hilsenbeck O, et al. Software tools for single-cell tracking and quantification of cellular and molecular properties. Nat Biotechnol. 2016;34(7):703–706. doi: 10.1038/nbt.3626. [DOI] [PubMed] [Google Scholar]
- Skylaki S, Hilsenbeck O, Schroeder T. Challenges in long-term imaging and quantification of single-cell dynamics. Nat Biotechnol. 2016;34(11):1137–1144. doi: 10.1038/nbt.3713. [DOI] [PubMed] [Google Scholar]
- Jones TR, et al. Scoring diverse cellular morphologies in image-based screens with iterative feedback and machine learning. Proc Natl Acad Sci U S A. 2009;106(6):1826–1831. doi: 10.1073/pnas.0808843106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas C, Falck J, Bartkova J, Bartek J, Lukas J. Distinct spatiotemporal dynamics of mammalian checkpoint regulators induced by DNA damage. Nat Cell Biol. 2003;5(3):255–260. doi: 10.1038/ncb945. [DOI] [PubMed] [Google Scholar]
- Bekker-Jensen S, Lukas C, Melander F, Bartek J, Lukas J. Dynamic assembly and sustained retention of 53BP1 at the sites of DNA damage are controlled by Mdc1/NFBD1. J Cell Biol. 2005;170(2):201–211. doi: 10.1083/jcb.200503043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewer A, Batchelor E, Gaglia G, Lahav G. Basal dynamics of p53 reveal transcriptionally attenuated pulses in cycling cells. Cell. 2010;142(1):89–100. doi: 10.1016/j.cell.2010.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lekshmi A, et al. A quantitative real-time approach for discriminating apoptosis and necrosis. Cell Death Discovery. 2017;3:16101. doi: 10.1038/cddiscovery.2016.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jullien D, Vagnarelli P, Earnshaw WC, Adachi Y. Kinetochore localisation of the DNA damage response component 53BP1 during mitosis. J Cell Sci. 2002;115(Pt 1):71–79. doi: 10.1242/jcs.115.1.71. [DOI] [PubMed] [Google Scholar]