

Summary
Acot7, encoding acyl-CoA thioesterase-7, is one of ~60 genes expressed ubiquitously across tissues but relatively silenced, or “disallowed”, in pancreatic β-cells. The capacity of ACOT7 to hydrolyse long-chain acyl-CoA esters suggests potential roles in β-oxidation, lipid biosynthesis, signal transduction or insulin exocytosis. Here, we explored the physiological relevance of β-cell-specific Acot7 silencing by re-expressing ACOT7 in these cells. ACOT7 overexpression in clonal MIN6 and INS1(832/13) β-cells impaired insulin secretion in response to glucose plus fatty acids. Furthermore, examined in a panel of transgenic mouse lines, we demonstrate that overexpression of mitochondrial ACOT7 selectively in the adult β-cell reduced glucose tolerance dose-dependently and impaired glucose-stimulated insulin secretion. By contrast, depolarisation-induced secretion was unaffected, arguing against a direct action on the exocytotic machinery. Acyl-CoA levels, ATP/ADP increases, membrane depolarization and Ca2+ fluxes were all markedly reduced in transgenic mouse islets, whereas glucose-induced O2-consumption was unchanged. Whilst glucose-induced increases in ATP/ADP ratio were similarly lowered after ACOT7 over-expression in INS1(832/13) cells, changes in mitochondrial membrane potential (ΔΨ) were unaffected, consistent with an action of Acot7 to increase cellular ATP consumption. Since Acot7 mRNA levels are increased in human islets in type 2 diabetes, inhibition of the enzyme might provide a novel therapeutic strategy.
Introduction
Maintained secretion of insulin is essential for normal blood glucose homeostasis and both the loss and dysfunction of pancreatic β-cells, the sole source of the circulating hormone in man, are implicated in Type 2 diabetes (T2D) (1).
Glucose sensing by β-cells involves a number of gene products such as GLUT2 and glucokinase whose expression is restricted to these, and only a few other, cell types and that ensure that elevated blood glucose concentrations are converted into enhanced glycolytic, and then citrate cycle flux, stimulating respiratory chain activity and, ultimately, ATP production by mitochondria. The resulting rise in cytosolic ATP/ADP ratio closes ATP-sensitive K+ (KATP) channels and this in turn leads to plasma membrane depolarisation and Ca2+ influx through voltage-gated calcium channels, triggering dense core secretory granule exocytosis (1).
In addition to β-cell “signature” genes, a small group of “housekeeping” genes is also relatively repressed in β- compared to other cell types (2–4). Of these, the monocarboxylate (lactate/pyruvate) transporter MCT-1 (SLC16A1) and lactate dehydrogenase (LDH) are particularly weakly expressed in the β-cell, but strongly expressed elsewhere in the body(5). This configuration appears to prevent inappropriate insulin secretion in response to circulating pyruvate derived from muscle during exercise(5–7). Thus, patients with activating mutations in the SLC16A1 promoter display exercise-induced hyperinsulinism (6), a situation mimicked in mice by over-expression of MCT-1 selectively in the adult β-cell(7). Systematic comparisons of the transcriptome of mouse islets versus other tissues (2; 3) revealed a further 64 genes similarly suppressed (or “disallowed”) in β-cells, of which a core of 11 genes (4) were common to two independent studies. Whilst evidence exists for a role for the suppression of some of these genes in the function or survival of β-cells (notably MCT-1, as described above, as well as LDHA (8) and PDGFRA (9)), for the remainder, the biological rationale for β-cell-selective repression is obscure (4).
Acyl-CoA thioesterase 7 (Acot7) has been recently identified by our group as a member of the β-cell “disallowed” genes group (3). ACOT7 is a member of a family of 13 enzymes responsible for the hydrolysis of acyl-CoA (10; 11). The murine Acot7 gene comprises 13 exons and undergoes differential splicing to generate cytosolic and mitochondrial variants (12). ACOT7 acts upon acyl-CoAs with a range of chain lengths (10) and is particularly highly expressed in the brain and testis (12; 13). Additionally, ACOT7 is implicated in the hydrolysis of arachidonoyl-CoA (AA-CoA) (14), which furnishes free arachidonic acid (AA) for the synthesis of prostaglandins. Others (13; 15) have suggested a role for ACOT7 in the brain in the maintenance of low, non-toxic, acyl-CoA levels.
Suggesting a role in β-cell decompensation in T2D, levels of Acot7 are increased in Zucker diabetic fatty rat islets (16) and in micro-dissected β-cell enriched tissue from T2D patients (17). Given the importance of intracellular lipids in the control of many β-cell functions, including membrane trafficking, ion channel activity and insulin exocytosis (18), we have explored here the impact of Acot7 overexpression in these cells both in vitro and in vivo.
Experimental Design and Materials
Generation of constructs over-expressing ACOT7 isoforms
The murine Acot7 coding sequences (CDS) were amplified by RT-PCR from liver and kidney RNA and ligated into P3XFLAG-CMV-14 in-frame with the C-terminal 3xFLAG epitope tag. The CDS of each isoform, complete with epitope tag, were then amplified and ligated into the pBI-L vector generating two plasmids with a bidirectional tetracycline-regulated promoter that simultaneously drives the expression of both firefly luciferase and Flag:Acot7_mit (pBI-LTet FLAG::Acot7_mit) or Flag:Acot7_cyt (pBI-LTet FLAG::Acot7_cyt).
Generation and maintenance of Acot7 transgenic (Acot7 Tg) mice
The expression cassette was excised from pBI-LTet FLAG::Acot7_mitand used for pronuclear microinjection into C57BL/6J oocytes at the Imperial College London/MRC transgenics unit. Successful integrants (F9, F15 and F26) were identified by PCR and backcrossed with C57BL/6 wild-type mice for at least 3 generations. The resulting heterozygous Acot7_mit mice were crossed with homozygous RIP7-rtTA mice (C57BL/6 background) to produce littermates Acot7 Tg (Acot7_mit+/-, Rip7rtTA positive, heterozygous, 1:2 ratio) and controls (Acot7_mit-/-, Rip7rtTA positive, heterozygous, 1:2 ratio). All the animals were administered doxycycline in the drinking water (0.5 g/l) from the age of 5 weeks. High-fat diet (HFD, DIO Rodent Purified Diet w/60% Energy From Fat, DIO-58Y1) was administered from the age of 5 weeks, when indicated.
All in vivo procedures were performed at the Imperial College Central Biomedical Service and approved by the UK Home Office Animals Scientific Procedures Act, 1986 (PPL 70/7349).
Acot7 expression and protein analysis
RNA-Seq read data were downloaded for a range of mouse tissues (Accession numbers: SRA056174 – beta cells; GSE21860 – islets; GSE36026 – other tissues). Reads were mapped to the mouse genome (Ensembl NCBIM37) using Bowtie:Tophat, and genes quantified with Cufflinks (19). Isolation of total RNA, RT-qPCR and Western (immuno-)blotting were performed as previously described (20).
Human growth hormone (hGH) release assay
hGH secretion assays were performed as previously described (21). To reach a final experimental concentration of 0.5 mM, 2:1 unsaturated:saturated, mix of fatty acids, which in the presence of 1% BSA results in unbound FFA concentrations in the nanomolar range (22), oleate and palmitate (Sigma-Aldrich) were incubated with BSA for 2h at 37C.
Immunocytochemistry and Immunohistochemistry
MIN6 and INS1(832/13) cells, co-transfected with pTet-off and pBI-LAcot7_mit or pBI-L-Acot7_cyt, and isolated pancreata were fixed, stained and visualized as previously described (23). Cells and slides were visualized using a LSM-780 microscope (Zeiss, UK) and an Axiovert 200 M microscope (Zeiss, Welwyn Garden City, UK), respectively. ImageJ software (Wayne Rasband, NIMH) was used to calculate the mean intensity of flag (Acot7 transgene) in the β-cell area surrounded by glucagon-positive (alpha-cell) area. For β-cell mass estimation, we determined the percentage of pancreatic surface which was insulin–positive, as measured in whole pancreas sections separated by 25μm in the z-axis.
Intraperitoneal glucose (IPGTT), insulin (IPITT) tolerance tests and in vivo Insulin secretion
IPGTT and IPITT were performed as described in (20). For IS, animals were fasted overnight and glucose (3g/kg) was administered intraperitoneally. Plasma insulin was measured by ELISA (Crystal Chem).
Isolation and analysis of mouse islets
Islets were isolated by digestion with collagenase as described elsewhere (24). Insulin secretion, Ca2+ (Fluo-2-AM, Invitrogen) and ATP/ADP (Perceval) imaging were performed as previously described (23; 25). Insulin release during perifusion was monitored using a custom-built device, using 50 islets and perifusion rate of 500 µl min-1 at 37 °C. Insulin was quantified using a homogeneous time-resolved fluorescence-based (HTRF)assay (CisBio) in a Pherastar Reader (BMG Labtech).
Electrophysiology was performed as described in (26) in perforated patch-clamp configuration using an EPC9 patch-clamp amplifier controlled by Pulse acquisition software (Heka Elektronik, Pfalz, Germany). Data were filtered at 1 kHz and sampled at 2 kHz.
Seahorse experiments were performed as described in (27). Briefly, 50 islets per well (matched size) were placed in 24-well islet plates in Krebs-Ringer buffer-0.1% BSA containing 3mM glucose and pre-incubated at 37C without CO2 for 1h. Islets were then incubated at 3mM glucose followed by 17mM glucose and oligomycin (5μM) to measure oxygen consumption rate (OCR) and extracellular acidification rate (ECAR).
Transfection, ATP/ADP and Δψm analysis in INS1 (832/13) cells
Cells were transfected for 24 h with pBI-LTet FLAG::Acot7_mit or a control empty vector plus pEGFP-C1 (Addgene) (secretion and TMRE experiments) or GW1-PercevalHR (Addgene, ATP/ADP experiments, ratiometric mode) using Lipofectamine2000, with 50-60% efficiency. Cells were pre-incubated for 1h at 3mM glucose prior to stimulation. ATP/ADP and tetramethylrhodamine-ethyl ester (TMRE) dynamics were monitored using a widefield microscope (Olympus IX70) equipped with a Polychrome IV (Till), a heating stage and 40x/1.35NA objective. ATP/ADP measurements were performed as previously described (28). For TMRE experiments, INS1 (832/13) were loaded with 10nM TMRE in imaging buffer (28) for 45 min. and re-equilibrated with 2nM for 10 min. before recordings. TMRE (2nM) was present throughout, and excited at 550nm (0.25 Hz). Data were analysed using ImageJ with a purpose-designed macro.
Lipidomics
Lipids extraction from isolated islets (150-300 per animal and sample) and analysis was conducted by the Lipidomics Facility at Babraham Institute (29).
Statistical analysis
Statistical analysis was performed with GraphPad Prism 6.0. Statistical significance was evaluated as indicated in the figure legends. All data are shown as mean ± standard error of the mean (S.E.M.). P values < 0.05 were considered statistically significant.
Results
Acot7 expression is weak in mouse islets compared with other tissues
Interrogation of publicly-accessible RNA-Seq databases (Fig. 1A), and qRT-PCR (Fig. 1B), confirmed lower levels of mRNA encoding all Acot7 mRNA variants in mouse islets compared to all other tissues examined. Notably, RNA-Seq data showed lowest Acot7 expression in purified β-cells (Fig 1A). Western blotting analysis confirmed strongest expression at the protein level in brain, lung and testis. ACOT7 immunoreactivity was undetectable in islets (Fig. 1C). As observed by relative RT-qPCR, other members of the Acyl-CoA thioesterase family were also poorly expressed in mouse islets (Fig. 1D).
Figure 1. Acot7 expression is weak in mouse islets in comparison to other tissues.

A) RNA-seq data for Acot7 in different mice tissues and isolated islets and β-cells. FKPM: Fragments per kilobase pero million fragments mapped. B) RT-qPCR expression data of Acot7 mRNA in mice tissues and isolated islets. n=3 C57BL6 mice. C) Western blot for ACOT7 in mouse tissues and islets. A representative experiment is shown. D) RT-qPCR expression of Acot family members (Acot1-13) in isolated mouse islets. Data are presented as relative to Actin. n= 4 mice per genotype.
Overexpression of Acot7 in β-cell lines impairs glucose plus fatty acid-induced insulin secretion
We first determined whether Acot7 overexpression may affect glucose-stimulated insulin secretion (GSIS) or secretion in response to depolarisation of the plasma membrane with KCl (KSIS). We generated two DNA constructs expressing the cytosolic (Acot7_cyt) or the mitochondrially-targeted (Acot7_mit) isoforms. Experiments were performed using either isoform co-overexpressed in β-cell lines alongside human growth hormone as a reporter of regulated secretion from the transfected cell population. The latter peptide is co-stored and released with insulin from dense core granules (21).
Both Acot7_cyt and Acot7_mit were expressed with the appropriate sub-cellular localisation in both murine MIN6 (30) and in INS1(832/13) (31) cells (Fig. 2A). Thus, in each case, the epitope tag displayed a cytosolic localisation (Acot7_cyt) or was closely co-localised with the mitochondrial marker TOM20 (Acot7_mit; Fig. 2A).
Figure 2. Overexpression of Acot7 in β-cell lines impairs glucose plus fatty acid-induced insulin secretion.

A) Immunocytochemistry using an anti-Flag antibody to show the localization of Acot7_mit and Acot_cyt in MIN6 or INS1(832/13) cells. Cells were transfected with vectors encoding an Acot7 transcript containing or not a mitochondrial targeting sequence (Acot7_mit and Acot7_cyt, respectively) and including a 3xFLAG tag. An antibody against TOM20C was used as mitochondrial outer membrane marker. Objective 100x/NA 1.4. B, C) Glucose-stimulated secretion was evaluated in MIN6 (B, C) and INS1(832/13) (C) cells over-expressing cytosolic (Acot7_cyt), mitochondrial (Acot7_mit) Acot7 or an empty vector using over-expressed human growth hormone (hGH) as insulin surrogate. hGH secretion was measured after 1h 17/20mM glucose stimulation in presence or absence of 0.5mM fatty acids as indicated (G: Glucose; PA: Palmitic Acid; OA: Oleic Acid, PA/OA ratio 1:2), following o/n pre-incubation at 3mM glucose. Data are from three to four independent experiments and six to eight technical replicates (means ± S.E.M). ns no significant, *p<0.05, **p<0.01, ****p<0.0001; Matched (by experimental day) two-way ANOVA, Bonferroni test.
Examined in MIN6 cells, overexpression of neither isoform affected hormone release in response to 30 vs 3mM glucose, nor in response to 30mM KCl (Fig. 2B). However, since Acot7 action is likely to lead to the degradation of acyl-CoA derived from exogenous fatty acids (FAs) we next asked whether secretion in response to a mixture of glucose plus FAs may be susceptible to Acot7 overexpression. Confirming this possibility, secretion in response to 20mM glucose plus a mixture of palmitate and oleic acid (0.5mM Oleate:Palmitic acid (2:1) / 1% (w/v) BSA), was significantly reduced in MIN6 and in INS1(832/13) cells upon over-expression of either the cytosolic (16% and 13%, respectively) or mitochondrial isoforms of Acot7 (17% and 13% in respectively) (Fig. 2C).
β-cell-selective over-expression of mitochondrial Acot7 in adult mice impairs glucose tolerance
To explore the potential role in vivo for the suppression of insulin release identified above in clonal β-cells, we generated three mouse lines re-expressing the mitochondrial form of Acot7 using the TetON system (32). Our in vitro results suggested that both isoforms inhibited secretory function to a similar extent but, given the central role of mitochondria in β-cell stimulus-secretion coupling (1) we focussed on mitochondrial ACOT7.
Genomic qPCR analysis demonstrated the presence of 4, 7 and 70 copies of the transgene in three lines originating from three different founders (F) (F9, F26 and F15 respectively; results not shown). After establishing stable integration at a single site by the inheritance of the same number of copies for 3 generations, offspring were subsequently crossed to RIP7rtTA mice, expressing the reverse tetracyclin-regulated transactivator selectively in β-cells (32). Islets isolated from animals bearing or lacking the Acot7 transgene were incubated with doxycycline for 48h to induce the expression of the transgene. Alternatively, animals were administered doxycycline for 2-4 weeks to induce expression before islet isolation. RT-qPCR revealed an increase of ~14, 28 and 100-fold over endogenous Acot7 mRNA levels respectively in the three lines (F15, F9 and F26) when doxycyline was administered in vivo, with comparable levels of induction observed in vitro in cultured islets (Fig. 3A). By contrast, Acot7 mRNA levels remained unchanged in the hypothalamus (a potential site of RIP2 activity (33)) or the liver of the mice with maximum Acot7 overexpression in islets (F26) (Fig. S1A). Additionally, the ratio of Acot7:β-actin mRNA in islets from mice displaying the strongest over-expression of the thioesterase in islets (F26) was in the same range as that observed in hypothalamus (Fig. S1B), indicating that levels of over-expression in β-cells were still within the normal physiological range in the context of other cell types. Expression of the other Acot family members was identical in transgenic versus control islets, eliminating the possibility of compensatory changes in the expression of the latter (Fig S1C).
Figure 3. Generation of mice with β-cell-selective over-expression of mitochondrial Acot7.

A) Expression of Acot7 mRNA in isolated islets from different Acot7 Tg lines (F15, F9, F26) assessed by RT-qPCR. Isolated islets were incubated for 48h in the presence of doxycycline (Dox-In vitro) or administered in the drinking water to animals for 2-4 weeks (Dox- In vivo). n=4-7 mice/genotype. Data are presented as fold-change versus the Dox-In vitro control. *p<0.05, ****p<0.0001; Unpaired Student’s t test. B) Western blot for ACOT7 (anti-Acot7) or exogenous ACOT7 (anti-Flag) in isolated islets from F15, F9 and F26 mouse lines. Tubulin is shown as loading control. A representative experiment is presented. C) Immunocytochemistry using anti-Flag antibody to show the expression of exogenous mitochondrial Acot7 (Acot7_mit) in pancreata from control or Acot7 Tg mice (lines F15, F9, F26). An anti-glucagon antibody was used for alpha cells staining. Representative islets are shown. Objective 63X/NA 1.4. D) ImageJ software was used to quantify maximum fluorescence intensity of Flag staining in C). n=3-5 mice/genotype; 2 pancreas sections per animal were used, separated by 50μm. **p<0.01, ****p<0.0001; Unpaired student t test. E) As in C, including an anti-insulin antibody to show co-localization of Acot7_mit and Insulin.
Measurements of expression by Western blotting confirmed a degree of induction of Acot7 in the three lines essentially proportional to changes at the mRNA level (Fig. 3B). Immunocytochemical analysis of pancreata revealed immunoreactivity corresponding to the incorporated FLAG tag within the islets in lines F9 and F26 (Fig. 3C, D). Immunoreactivity was below the level of detection in pancreata from F15 mice (Fig. 3C, D). Examined in the most strongly-expressing line F26, the pattern of immunoreactivity was punctate, consistent with mitochondrial targeting, and was exclusively restricted to the insulin-positive β-cell population (Fig. 3E). We did notice that whereas there was little inter-animal variation in expression levels in animals from lines F26 and F15, F9 offspring expressed Acot7 at highly variable levels (both for mRNA and protein). The reasons for this variability are unclear.
Animals from line F26 displayed indistinguishable weight gain and random-fed glycaemia compared to wild-type littermates (Fig. S2). However, intraperitoneal glucose tolerance was significantly impaired in the transgenic animals at 17, 21 and 25 weeks, with females showing a stronger phenotype (Fig. 4A, B). Similar, but less marked changes were also obtained for lines F15 and F9 (Fig. S3).
Figure 4. β-cell-selective over-expression of mitochondrial Acot7 impairs glucose tolerance.

Glucose tolerance was measured in male (A, C) and female (B, D) Acot7 Tg (green) and littermate control (black) mice of the indicated age. Mice were maintained on a normal chow diet (A, B) or fed a high fat-diet (C, D) for 4 weeks. The area under the curve (AUC) is shown underneath each graph. N=8-10 mice/genotype. *p<0.05, **p<0.01, ***p<0.001 ****p<0.0001; analysis by two-way ANOVA (repeated measures) Fisher least significance different test (Glucose tolerance test); Student’s t test (AUC).
To determine whether the effects on glucose tolerance in Acot7 transgenic line F26 (Acot7 Tg) may be further exacerbated during β-cell stress, we maintained these mice on a high fat diet (HFD, 60% total calories) for 4 weeks (34), sufficient to engender insulin resistance and significant increases in peak glycemia in control non-transgenic mice of the same age (Fig. 4C, D). Differences in glucose tolerance between non-transgenic animals and transgenic littermates became apparent in younger animals (9 weeks) under these conditions than on normal chow (in which the phenotype wasn’t observed until the age of 17 weeks, 12 for the females), especially in male mice (Fig 4A). Comparable results were obtained for the lines with F15 and F9 (Fig. S3). Whether examined in animals on a HFD or regular chow, the observed defects in glucose tolerance were milder in mice from line F15, with lowest Acot7 expression, indicating a dose-dependent effect of Acot7 over-expression.
To exclude insulin insensitivity as the cause of the glucose intolerance detected in Acot7 Tg mice, we evaluated insulin tolerance in 9-week-old F26 female mice fed a HFD and 25-week-old F15 female mice fed a Chow diet (Fig. S4A). No differences between transgenic and control mice were observed (Fig. S4A). β-cell mass was also similar for control and transgenic mice from line F26 kept on chow or HFD (Fig. S4B, C).
Over-expression of mitochondrial Acot7 in the β-cell impairs glucose-induced increases in cytosolic ATP/ADP ratio, membrane depolarization, free Ca2+ and insulin secretion
To explore the mechanism(s) underlying the glucose intolerance after Acot7 overexpression, we measured plasma insulin levels in fasted F26 mice (previously kept on regular chow or HF diet). This revealed a non-significant tendency towards elevated insulin levels in Acot7 Tg mice versus littermate controls but, strikingly, no further increase in plasma insulin was observed post glucose (3g/kg) challenge (Fig. 5A). This result indicates a reduced capacity of the β-cells to secrete insulin in response to increased glucose concentration. Consistent with this in vivo observation, insulin secretion from transgenic mouse isolated islets revealed a marked inhibition in release in response to 17 mM glucose or 8 mM glucose plus fatty acids versus non-transgenic islets (Fig. 5B). As observed in MIN6 cells (Fig. 2B), no differences in insulin secretion were detected in response to depolarization of the plasma membrane with KCl (Fig. 5C).
Figure 5. Over-expression of mitochondrial Acot7 in the β-cell impairs glucose-induced insulin secretion in vivo and in vitro.

A) Glucose (3g/kg)- induced insulin secretion was assessed in Acot7 Tg (green) and littermate control (black) mice fed a normal diet (left hand panel, 23 week-old, n=7M+6F C, 6F+6M Acot7 Tg) or a high-fat diet (right hand panel, 9 week-old, n=6M C, 10M Acot7 Tg), as indicated. Upper graphs show absolute insulin measurements (ng/ml). Insulin levels are presented as fold change over the basal level (time 0) on the lower panels. Matched two-way ANOVA, Bonferroni test. The statistical test was applied to assess the effect of glucose injection on blood insulin content over the time in control (black lines, **p<0.01, ****p<0.0001) and Acot7 Tg animals (green lines, ns: non-significant) B, C) Insulin secretion was evaluated in islets isolated from Acot7 Tg (green) or control (black) 25-week old female mice (fed a chow diet) in response to 30 min exposure to the indicated concentrations of glucose (G) in the presence or absence of 0.5mM fatty acids (FA; 2:1 palmitate/oleic acid) (B) or KCl (C), following 1h pre-incubation at 3mM glucose. Data are from three independent experiments and nine (B) or six (C) technical replicates (means ± S.E.M.). ns: non-significant, *p<0.05; Matched two-way ANOVA, Bonferroni test. D) Following 1h of pre-incubation at 3mM glucose, 50 Acot7 Tg (green) or control (black) isolated islets were perifused with KHB buffer containing 17mM glucose during 30 min. Secreted insulin was measured in samples taken periodically every 30s. n= 5 animals/genotype (3 male, 2 female/genotype, 25 week-old, fed a chow diet). E) Area under the curve (AUC) 0-30, 0-10 and 10-30 min after addition of 17mM glucose. *p<0.05; Unpaired student’s t test.
In contrast to our observations in β-cell lines, Acot7 overexpression reduced secretion in response to high glucose even in the absence of supplementation with exogenous FA. Suggesting that the requirement for exogenous FAs may reflect a depletion of endogenous lipid stores after 24 h, INS1(832/13) cells expressing mitochondrial Acot7, and then pre-cultured for 1h in 3mM glucose, displayed lower insulin secretion in response to 17 mM glucose than control plasmid-transfected cells (Fig. 6A).
Figure 6. Overexpression of Acot7 in INS1 (832/13) cells impairs glucose-induced insulin secretion and increases in cytosolic ATP/ADP ratio.

A) Glucose-stimulated insulin secretion was evaluated in INS1(832/13) cells over-expressing mitochondrial Acot7 (Acot7) or an empty vector (C). Insulin secretion was measured after a 30min. stimulation at 3 or 17mM glucose. (G: Glucose). Data are from three independent experiments and six technical replicates (means ± S.E.M). *p<0.05. Matched two-way ANOVA, Bonferroni test B, C) INS1(832/13) cells were co-transfected with a construct expressing mitochondrial Acot7 (Acot7) or an empty vector (C) and a PercevalHR-containing plasmid to monitor changes in ATP/ADP ratio in response to high glucose (17mM vs 3mM). Data are from two independent experiments and 113 C and 90 Acot7 individual cells. In (B) traces represent the F480/F440 fluorescence ratio. Mean fluorescence and rate of fluorescence increase (slope) at 3mM glucose (min. 0-3) and 17mM glucose (initial response, min. 3-8 and late response, min. 8-20) were estimated and are presented as mean of the values ± S.E.M. *p<0.05, ****p<0.0001; Unpaired student’s t test. In (C), traces represent normalized (to basal) and mean fluorescence intensity (F/F0) over time. Maximum fold change and area under the curve (AUC) were determined and presented as mean of the values ± S.E.M. ***p<0.001, ****p<0.0001; Unpaired student’s t test. (D) INS1(832/13) cells were co-transfected with a construct expressing mitochondrial Acot7 (Acot7) or an empty vector and a GFP-expressing vector to allow selection of transfected cells. Cells were stained with TMRE (sensitive to changes in Δψm) and perifused with 3mM or 17mM glucose as indicated. Data are from three independent experiments and 160 C and 166 Acot7, GFP-positive cells. Traces represent normalized (to basal) and mean fluorescence intensity (F/F0) over time. Maximum fold change and area under the curve (AUC) were determined and presented as mean of the values ± S.E.M. ns: no significant. Unpaired student’s t test.All the experiments were performed 24h after cell transfection and the cells were pre-incubated at 3mM glucose during 1h before starting the experiments.
Dynamic measurements of insulin secretion in transgenic islets revealed changes in both phases of secretion in response to high glucose, with the second (sustained) phase being particularly strongly inhibited (Fig. 5D, E).
The above observations suggested that signalling by glucose to exocytosis (1) may become defective in Acot7 Tg β-cells. To test this hypothesis, we measured the metabolic response to high glucose by following changes in the cytosolic ATP/ADP ratio with the recombinant fluorescent probe, Perceval (35; 36). The glucose-mediated ATP/ADP rise, largely reflective of changes in β-cells (rather than other cell types), was strongly reduced in Acot7 transgenic islets versus controls (Fig 7A). We also assessed ATP/ADP changes in INS1(832/13) cells using the ratiometric sensor Perceval HR in order to provide a measure of this ratio which was independent of the level of probe expression (37). Over-expression of Acot7 had no effect on basal ATP/ADP ratios but significantly lowered the speed and extent of the ATP/ADP increases in response to 17mM glucose. By contrast, glucose-induced increases in mitochondrial membrane potential (ΔΨ), assessed with TMRE, were identical in each case.
Figure 7. Over-expression of mitochondrial Acot7 in the β-cell impairs glucose-induced increases in cytosolic ATP/ADP ratio, membrane depolarization and free Ca2+.

A) Intact Acot7 Tg (green) and control (black) islets were infected with an adenoviral Perceval sensor construct to monitor changes in ATP/ADP ratio in response to high glucose (17mM vs 3mM)., following 1h preincubation at 3mM glucose. n=34 islets (from 4 Control mice, 2M, 2F)/ 30 islets (from 4 Acot7 Tg mice, 2M, 2F). B) Intact Acot7 Tg (green) and control (black) islets were incubated for 45 min with 10 μM Fluo-2-AM in KHB buffer (3mM glucose) before imaging to monitor Ca2+ entry in response to high (17mM) glucose. n=55 islets (from 8 Control mice (4M, 4F))/ 71 islets (from 9 Acot7 Tg mice (4M, 5F). Mice (F26) were 25 weeks old and kept on a normal chow diet. No differences associated with the sex of the animals were observed. Traces represent normalized and averaged fluorescence intensity (F/F0) over time. Maximum fold change and area under the curve (AUC) were determined and presented on the right hand side as mean of the values ± S.E.M. *p<0.05, **<0.01, ***p<0.001; Unpaired student’s t test. C) Representative current clamp recordings of isolated β-cells from control (black) and Acot7 transgenic mice (green) in response to 3 and 17 mM glucose application. The graph on the right hand side shows mean membrane potential ± S.E.M. n=6-11 single cells per genotype. Mice (F26, male) were 9 weeks old fed a HFD for 4 weeks. D, E) OCR and F) ECAR were measured in the presence of 3mM or 17mM glucose and 5μM oligomycin (OM) over time as indicated. In (E) data are presented as fold induction over the basal OCR shown in (D). Each plot represent the mean of 6 (control, two different animals) and 12 (Acot7 Tg, four animals) wells with 70 islets. Maximum fold change and area under the curve (AUC) were determined and presented as mean of the values ± S.E.M. Unpaired student’s t test. Mice (F15) were 25 weeks old and kept on a normal chow diet.
Correspondingly, glucose-induced changes in intracellular free cytosolic Ca2+, imaged with the intracellular fluorescent dye, Fluo-2-AM (38), were also significantly reduced in Acot7 Tg islets (Fig. 7B) consistent with impaired downstream closure of KATP channels (39) (40). The aforementioned defects were not explained by changes in the expression of genes essential for GSIS, including the glucose transporter GLUT2 (Slc2a2), glucokinase (Gck), the KATP channel subunits Kir6.2 (Kcnj11) and SUR1 (Abcc8) or the ATP synthase ATP5 (Atp5a1) in Acot7 Tg islets (Fig. S5).
To investigate a direct role for defective KATP channel closure we used perforated patch clamp electrophysiology (41) to measure changes in membrane potential in single β-cells. These experiments were performed using animals maintained on a high fat diet for 4 weeks where differences in glucose tolerance between transgenic and wild-type were already apparent (see above and Fig 4). Transgenic β-cells showed significantly weaker membrane depolarization in response to high glucose than those from control mice (Fig. 7C), as expected if a reduction in the closure of KATP channels occurs as a consequence of lowered ATP/ADP increase (Fig. 7A).
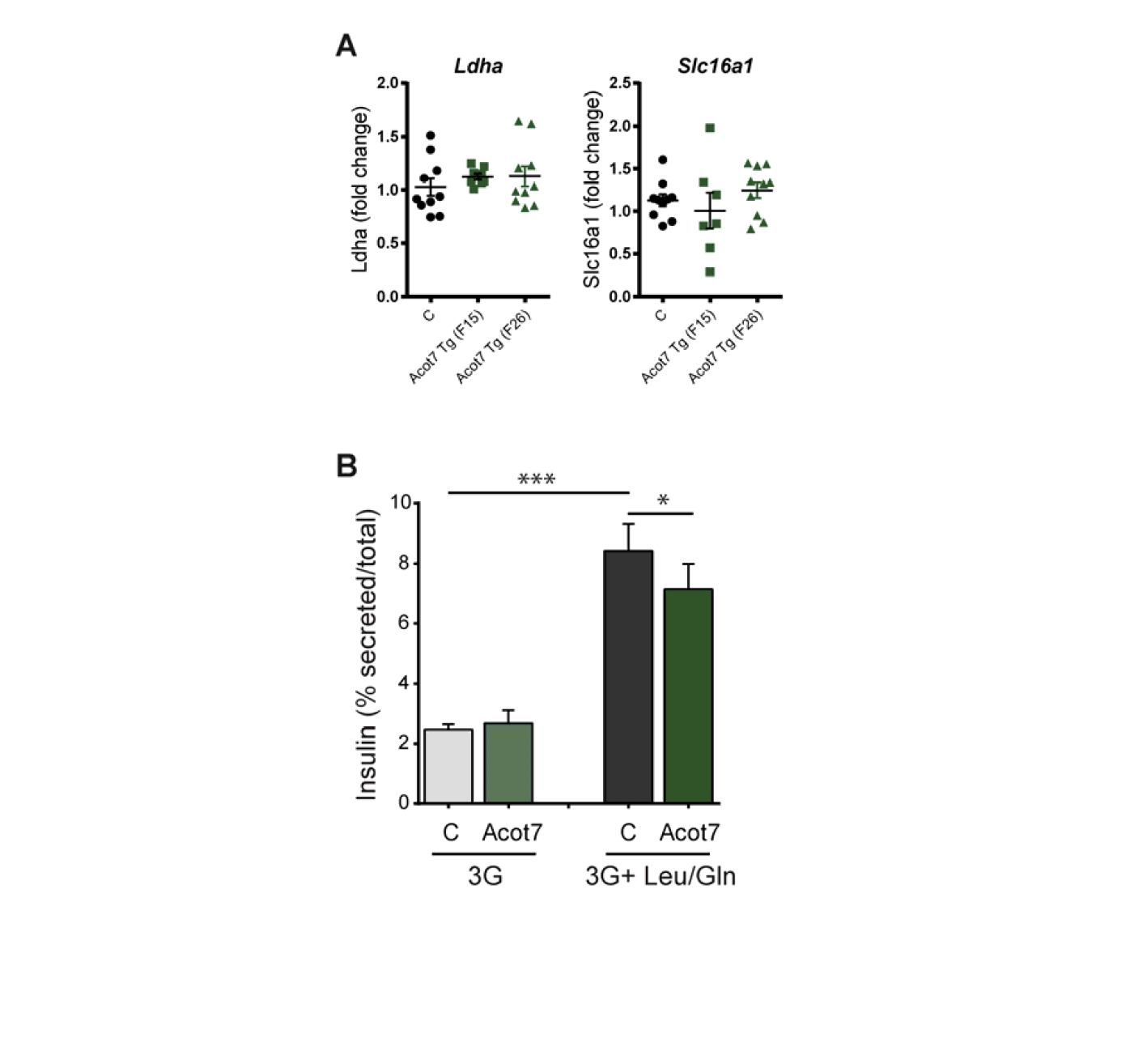
In order to shed more light into the mechanisms underlying the lowered ATP/ADP increases after Acot7 over-expression, we assessed mitochondrial oxidative metabolism upstream of ATP generation. Monitored using a Seahorse XF analyser, no significant differences in oxygen consumption (OCR) were observed between control and transgenic islets (Fig. 7D) though a tendency was observed towards higher basal OCR in transgenic islets. This difference persisted in the presence of oligomycin, an inhibitor of ATP synthase, arguing against increased uncoupling in transgenic islets. Interestingly, the fold increase in OCR in response to 17 mM glucose (% of basal, Fig. 7E) tended to be lower in transgenic islets. Although not significantly different, the rate of extracellular acidification (ECAR) tended to be higher in Acot7 Tg islets at high glucose, possibly reflecting a tendency towards increases in lactate dehydrogenase (Ldha), though not Slc16a1 (MCT-1), expression (Fig S6A).
Over-expression of Acot7 in β-cells results in reduced fatty-acyl-CoA generation but not in significantly altered fatty acid levels
To determine whether an action of ACOT7 to hydrolyse FA-CoA esters into free FA (FFA) and CoA (42) might contribute to impaired metabolic signalling, we performed a complete lipidomic profiling of islets isolated from transgenic or control mice maintained on a HFD for 4 weeks using Electrospray Ionisation Mass Spectrometry (ESI-MS). Islets were incubated overnight at 11mM glucose, starved (3mM glucose) for 1h and subsequently incubated at high glucose (17mM) for 30 min. A clear reduction in FA-CoA was observed in Acot7 Tg islets, as expected (Fig. 8A, C). A tendency towards an increase in the levels of fatty acids of a particular chain length, notably C18:1 (most probably, oleic acid), was also detected (Fig. 8B). No significant changes were observed in the levels of monoacylglycerol (MAG), diacylglycerol (DAG), or other lipids (Fig. 8A). A strong tendency (p=0.06) towards increased fatty acyl carnitine (FaCN) levels was also observed in transgenic islets.
Figure 8. Over-expression of Acot7 in β-cells results in reduced fatty-acyl-CoA generation but not fatty acid levels.

A) Total lyso-phosphatidic acid (LPA), phosphatidic acid (PA), lyso-phosphatidylcholine (LPC), acyl-linked phosphatidycholine (aPC), phosphatidycholine (PC), acyl-lyso-phosphatidylethanolamine (aLPE), lyso-phosphatidylethanolamine (LPE), acyl-phosphatidylethanolamine (aPE), phosphatidylethanolamine (PE), lyso-phosphatidylglycerol (LPG), phosphatidylglycerol (PG), lyso-phosphatidylinositol (LPI), phosphatidylinositol (PI), lyso-phosphatidylserine (LPS), phosphatidylserine (LPS), cardiolipin (CL), free fatty acids (FFA), fatty acyl CoA (FaCoA), fatty acyl carnitine (FaCN), monoacylglycerol (MG), diacylglycerol (DG), triacylglycerol (TG), sphingosine (SG), sphindogsine-1-phosphate (S1P), ceramide (Cer), sphingomyelin (SM), were analysed in isolated islets from 9-week old Acot7 Tg (green) or control littermantes (C, black) fed on a high fat diet for 4 weeks. B, C, D) Detailed free fatty acid (FA, (B)), fatty acyl-CoA (FaCoA, (C)) and fatty acyl-carnitine (FaCN, (D)) content in Acot7 Tg and control islets. n=3-5 mice/genotype, 150-350 islets/mice. Mice (male) were 9 week old and kept on high fat diet for 4 weeks. Isolated islets were pre-incubated at 3mM glucose during 1h and stimulated with 17mM glucose during 30 min prior snap-freezing. Data represent mean values ± S.E.M. *p<0.05; Unpaired Student’s t test.
Discussion
Although at least 11 genes are selectively repressed in the β-cell (4), the physiological relevance, if any, of this tissue-specific suppression has only been examined for Ldha (8), Slc16a1 (MCT1)(7) and Pdgfra (9). Here, we show firstly, using overexpression in β-cell lines, that low ACOT7 levels are required for normal secretory responses to glucose in the presence of adequate FA levels. Extending these data to the in vivo setting, we also demonstrate that β-cell specific over-expression in mice of mitochondrial ACOT7 causes a marked and dose-dependent impairment in glucose tolerance that reflects altered β-cell function rather than reduced β-cell mass (43). No direct evidence was obtained for changes in insulin sensitivity in this model, though clamp studies would be needed to exclude this possibility entirely.
Insulin secretion in response to glucose (and glucose plus FA) was strongly impaired in Acot7 Tg mouse islets, likely explaining the impairment in glucose tolerance and insulin secretion observed in vivo. Although only islets over-expressing the mitochondrial isoform of Acot7 were interrogated, increased levels of fatty acyl-carnitine in transgenic islets suggest increased shuttling of acyl-CoA into the mitochondria in this model expected to result in a decrease of cytosolic FA-CoA.
Consistent with our results, Klett et al. (44) found that siRNA-mediated reduction of Acsl4 (Acyl-CoA synthetase isoform 4), responsible for the conversion of certain FA into FA-CoA, reduced insulin secretion in response to glucose and, more markedly, glucose plus FA in INS1(832/13) cells. In contrast with our findings, and suggesting a different underlying mechanism, the effects occurred without changes in acyl-CoA species. On the contrary, Acsl4 depletion resulted in reduced epoxyeicosatrienoic acids. Although measurement of eicosanoids in transgenic islets would be necessary to discard their contribution to the observed phenotype in Acot7 Tg mice, Acsl4 inhibition also affected KCl-mediated insulin secretion whereas ACOT7 overexpression did not. Furthermore, Ellis and colleagues did not observe changes in prostaglandin D2 or E2 in Acot7-depleted brains (15) nor, more surprisingly, in acyl-CoAs levels. Thus, lipid composition and availability may be critical determinants of Acot7 action in different cell types (45).
FA-CoA has been shown to modulate GSIS through a range of different mechanisms, including the control of secretory granules exocytosis, protein acylation and modulation of the ATP-sensitivity of KATP channels (45). Most recently, Prentki and colleagues (18) have provided evidence that MAGs may play a particularly important role in stimulus-secretion coupling in β-cells, possibly acting via Munc13-1 to enhance a late event in exocytosis downstream of cytosolic Ca2+ increases. In the present study, we did not observe an effect of ACOT7 overexpression on depolarisation-stimulated secretion, nor did we observe significant differences in the levels of MAG arguing against a role for Acot7 in MAG-dependent regulation of GSIS. Of note, ACOT7 overexpression reduced both phases of GSIS, arguing against a preferential effect on a plasma membrane-proximal, readily-releasable granule pool (46).
On the other hand, we did observe a strong reduction in glucose-mediated ATP/ADP and Ca2+ rises in Acot7 Tg animals, suggesting that low ACOT7 levels are necessary to ensure effective KATP channel closure. The exact mechanism(s) underlying the impaired ATP/ADP increases remains unclear. However, preserved OCR rates in islets (Fig. 5D), and mitochondrial ΔΨ glucose-mediated increases (Fig. 6C) in INS1(832/13) cells after Acot7 over-expression lend weight to the view that increased ATP consumption, rather than impaired mitochondrial ATP generation is involved. This may conceivably involve a futile cycle wherein increased acyl-CoA synthesis, occurs in the face of accelerated breakdown of this lipid. Consistent with this view, secretion stimulated by glutamine plus leucine, which allows flux into the TCA cycle downstream of citrate, was slightly reduced in INS1(832/13) cells over-expressing Acot7 (Fig S6B). Further analysis will be needed, however, to determine whether this reflects enhanced acyl-CoA breakdown/resynthesis.
Interestingly, all members of the Acot family were poorly expressed in mouse islets, stressing the importance of maintaining acyl-CoA hydrolysis to a minimum in these cells. Of note, expression of Acot7 is up-regulated in the islet in Zucker diabetic fatty (ZDF) rat (16) and in β-cell-enriched microdissected tissue from T2D patients (17). Pharmacotherapeutic approaches which correct this defect might thus be useful in some forms of T2D.
Supplementary Material
{kind=link}
Acknowledgments
We thank Professor Gerhard Christofori (University of Basel, Basel, Switzerland) for kindly providing the RIP7-rtTA mouse and Professor David Carling (MRC Clinical Sciences Centre, Imperial College London) for permitting us to use their Seahorse XF bioanalyzer. This work was supported by grants to G.A.R. from the Wellcome Trust (WT098424AIA), the MRC, UK (Project GO901521 and Programme MR/J0003042/1), and the BBSRC (BB/J015873/1). G.A.R. is a Royal Society Wolfson Research Merit Award holder and T.J.P. a DRWF post-doctoral fellow. G.A.R. is the guarantor of this work.
Footnotes
Duality of Interest
No conflicts of interest relevant to this article to be reported
Author Contributions
A.M.S. designed and conducted the in vitro and in vivo studies. T.J.P. generated the constructs for Acot7 expression and transgene production and performed analyses of published RNASeq data. P.C. performed and/or assisted with imaging studies and E.H. electrophysiology. M.C.C. performed islet perifusion studies, S.R.S. and A.M.S. performed the Seahorse experiments and M-S. N-T. gave assistance with the in vivo experiments. A.M.S. and Q.Z. performed lipidomic analyses. G.A.R. conceived the study and wrote the paper with A.M.S. G.A.R. is the guarantor of this work.
References
- 1.Rutter GA, Pullen TJ, Hodson DJ, Martinez-Sanchez A. Pancreatic beta cell identity, glucose sensing and the control of insulin secretion. Biochem J. 2015:202–218. doi: 10.1042/BJ20141384. [DOI] [PubMed] [Google Scholar]
- 2.Thorrez L, Laudadio I, Van Deun K, Quintens R, Hendrickx N, Granvik M, Lemaire K, Schraenen A, Van Lommel L, Lehnert S, Aguayo-Mazzucato C, et al. Tissue-specific disallowance of housekeeping genes: the other face of cell differentiation. Genome Res. 2011;21:95–105. doi: 10.1101/gr.109173.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pullen TJ, Khan AM, Barton G, Butcher SA, Sun G, Rutter GA. Identification of genes selectively disallowed in the pancreatic islet. Islets. 2010;2:89–95. doi: 10.4161/isl.2.2.11025. [DOI] [PubMed] [Google Scholar]
- 4.Pullen TJ, Rutter GA. When less is more: the forbidden fruits of gene repression in the adult beta-cell. Diabetes ObesMetab. 2013;15:503–512. doi: 10.1111/dom.12029. [DOI] [PubMed] [Google Scholar]
- 5.Sekine N, Cirulli V, Regazzi R, Brown LJ, Gine E, Tamarit-Rodriguez J, Girotti M, Marie S, MacDonald MJ, Wollheim CB, Rutter GA. Low lactate dehydrogenase and high mitochondrial glycerol phosphate dehydrogease in pancreatic beta-cells. Potential role in nutrient sensing. JBiolChem. 1994;269:4895–4902. [PubMed] [Google Scholar]
- 6.Otonkoski T, Kaminen N, Ustinov J, Lapatto R, Meissner T, Mayatepek E, Kere J, Sipilä I. Physical exercise-induced hyperinsulinemic hypoglycemia is an autosomal-dominant trait characterized by abnormal pyruvate-induced insulin release. Diabetes. 2003;52:199–204. doi: 10.2337/diabetes.52.1.199. [DOI] [PubMed] [Google Scholar]
- 7.Pullen TJ, Sylow L, Sun G, Halestrap AP, Richter EA, Rutter GA. Over-expression of Monocarboxylate Transporter-1 (Slc16a1) in the pancreatic ƒ-cell leads to relative hyperinsulinism during exercise. Diabetes. 2012;61:1725. doi: 10.2337/db11-1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ainscow EK, Zhao C, Rutter GA. Acute overexpression of lactate dehydrogenase-A perturbs beta-cell mitochondrial metabolism and insulin secretion. Diabetes. 2000;49:1149–1155. doi: 10.2337/diabetes.49.7.1149. [DOI] [PubMed] [Google Scholar]
- 9.Chen H, Gu X, Liu Y, Wang J, Wirt SE, Bottino R, Schorle H, Sage J, Kim SK. PDGF signalling controls age-dependent proliferation in pancreatic β-cells. Nature. 2011;478:349–355. doi: 10.1038/nature10502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamada J, Matsumoto I, Furihata T, Sakuma M, Suga T. Purification and properties of long-chain acyl-CoA hydrolases from the liver cytosol of rats treated with peroxisome proliferator. Arch Biochem Biophys. 1994;308:118–125. doi: 10.1006/abbi.1994.1017. [DOI] [PubMed] [Google Scholar]
- 11.Hunt MC, Yamada J, Maltais LJ, Wright MW, Podesta EJ, Alexson SE. A revised nomenclature for mammalian acyl-CoA thioesterases/hydrolases. J Lipid Res. 2005;46:2029–2032. doi: 10.1194/jlr.E500003-JLR200. [DOI] [PubMed] [Google Scholar]
- 12.Hunt MC, Greene S, Hultenby K, Svensson LT, Engberg S, Alexson SE. Alternative exon usage selectively determines both tissue distribution and subcellular localization of the acyl-CoA thioesterase 7 gene products. Cell Mol Life Sci. 2007;64:1558–1570. doi: 10.1007/s00018-007-7062-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamada J. Long-chain acyl-CoA hydrolase in the brain. Amino Acids. 2005;28:273–278. doi: 10.1007/s00726-005-0181-1. [DOI] [PubMed] [Google Scholar]
- 14.Sakuma S, Fujimoto Y, Sawada T, Saeki K, Akimoto M, Fujita T. Existence of acyl-CoA hydrolase-mediated pathway supplying arachidonic acid for prostaglandin synthesis in microsomes from rabbit kidney medulla. Prostaglandins Other Lipid Mediat. 1999;57:63–72. doi: 10.1016/s0090-6980(98)00072-0. [DOI] [PubMed] [Google Scholar]
- 15.Ellis JM, Wong GW, Wolfgang MJ. Acyl coenzyme A thioesterase 7 regulates neuronal fatty acid metabolism to prevent neurotoxicity. Mol Cell Biol. 2013;33:1869–1882. doi: 10.1128/MCB.01548-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parton LE, McMillen PJ, Shen Y, Docherty E, Sharpe E, Diraison F, Briscoe CP, Rutter GA. Limited role for SREBP-1c in defective glucose-induced insulin secretion from Zucker diabetic fatty rat islets: a functional and gene profiling analysis. Am J Physiol Endocrinol Metab. 2006;291:E982–994. doi: 10.1152/ajpendo.00067.2006. [DOI] [PubMed] [Google Scholar]
- 17.Marselli L, Thorne J, Dahiya S, Sgroi DC, Sharma A, Bonner-Weir S, Marchetti P, Weir GC. Gene expression profiles of Beta-cell enriched tissue obtained by laser capture microdissection from subjects with type 2 diabetes. PLoS One. 2010;5:e11499. doi: 10.1371/journal.pone.0011499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Prentki M, Matschinsky FM, Madiraju SR. Metabolic signaling in fuel-induced insulin secretion. Cell Metab. 2013;18:162–185. doi: 10.1016/j.cmet.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 19.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martinez-Sanchez A, Nguyen-Tu MS, Rutter GA. DICER Inactivation Identifies Pancreatic β-Cell "Disallowed" Genes Targeted by MicroRNAs. Mol Endocrinol. 2015;29:1067–1079. doi: 10.1210/me.2015-1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Varadi A, Ainscow EK, Allan VJ, Rutter GA. Involvement of conventional kinesin in glucose-stimulated secretory granule movements and exocytosis in clonal pancreatic beta-cells. J Cell Sci. 2002;115:4177–4189. doi: 10.1242/jcs.00083. [DOI] [PubMed] [Google Scholar]
- 22.Cnop M, Hannaert JC, Hoorens A, Eizirik DL, Pipeleers DG. Inverse relationship between cytotoxicity of free fatty acids in pancreatic islet cells and cellular triglyceride accumulation. Diabetes. 2001;50:1771–1777. doi: 10.2337/diabetes.50.8.1771. [DOI] [PubMed] [Google Scholar]
- 23.Sun G, Tarasov AI, McGinty JA, French PM, McDonald A, Leclerc I, Rutter GA. LKB1 deletion with the RIP2.Cre transgene modifies pancreatic beta-cell morphology and enhances insulin secretion in vivo. Am J Physiol Endocrinol Metab. 2010;298:E1261–1273. doi: 10.1152/ajpendo.00100.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ravier MA, Rutter GA. Isolation and culture of mouse pancreatic islets for ex vivo imaging studies with trappable or recombinant fluorescent probes. Methods Mol Biol. 2010;633:171–184. doi: 10.1007/978-1-59745-019-5_12. [DOI] [PubMed] [Google Scholar]
- 25.Hodson DJ, Tarasov AI, Gimeno Brias S, Mitchell RK, Johnston NR, Haghollahi S, Cane MC, Bugliani M, Marchetti P, Bosco D, Johnson PR, et al. Incretin-modulated beta cell energetics in intact islets of Langerhans. Mol Endocrinol. 2014;28:860–871. doi: 10.1210/me.2014-1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tarasov AI, Semplici F, Ravier MA, Bellomo EA, Pullen TJ, Gilon P, Sekler I, Rizzuto R, Rutter GA. The mitochondrial Ca2+ uniporter MCU is essential for glucose-induced ATP increases in pancreatic β-cells. PLoS One. 2012;7:e39722. doi: 10.1371/journal.pone.0039722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wikstrom JD, Sereda SB, Stiles L, Elorza A, Allister EM, Neilson A, Ferrick DA, Wheeler MB, Shirihai OS. A novel high-throughput assay for islet respiration reveals uncoupling of rodent and human islets. PLoS One. 2012;7:e33023. doi: 10.1371/journal.pone.0033023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tantama M, Martinez-Francois JR, Mongeon R, Yellen G. Imaging energy status in live cells with a fluorescent biosensor of the intracellular ATP-to-ADP ratio. Nature communications. 2013;4 doi: 10.1038/ncomms3550. 2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schug ZT, Peck B, Jones DT, Zhang Q, Grosskurth S, Alam IS, Goodwin LM, Smethurst E, Mason S, Blyth K, McGarry L, et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell. 2015;27:57–71. doi: 10.1016/j.ccell.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miyazaki J, Araki K, Yamato E, Ikegami H, Asano T, Shibasaki Y, Oka Y, Yamamura K. Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology. 1990;127:126–132. doi: 10.1210/endo-127-1-126. [DOI] [PubMed] [Google Scholar]
- 31.Hohmeier HE, Mulder H, Chen G, Henkel-Rieger R, Prentki M, Newgard CB. Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes. 2000;49:424–430. doi: 10.2337/diabetes.49.3.424. [DOI] [PubMed] [Google Scholar]
- 32.Milo-Landesman D, Surana M, Berkovich I, Compagni A, Christofori G, Fleischer N, Efrat S. Correction of hyperglycemia in diabetic mice transplanted with reversibly immortalized pancreatic beta cells controlled by the tet-on regulatory system. Cell Transplant. 2001;10:645–650. [PubMed] [Google Scholar]
- 33.Wicksteed B, Brissova M, Yan W, Opland DM, Plank JL, Reinert RB, Dickson LM, Tamarina NA, Philipson LH, Shostak A, Bernal-Mizrachi E, et al. Conditional gene targeting in mouse pancreatic {beta}-cells: Analysis of ectopic Cre transgene expression in the brain. Diabetes. 2010;59:3090–3098. doi: 10.2337/db10-0624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Heek M, Compton DS, France CF, Tedesco RP, Fawzi AB, Graziano MP, Sybertz EJ, Strader CD, Davis HR. Diet-induced obese mice develop peripheral, but not central, resistance to leptin. J Clin Invest. 1997;99:385–390. doi: 10.1172/JCI119171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tarasov AI, Griffiths EJ, Rutter GA. Regulation of ATP production by mitochondrial Ca(2+) Cell calcium. 2012;52:28–35. doi: 10.1016/j.ceca.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Piccand J, Strasser P, Hodson DJ, Meunier A, Ye T, Keime C, Birling MC, Rutter GA, Gradwohl G. Rfx6 maintains the functional identity of adult pancreatic á-cells. Cell Reports. 2014 doi: 10.1016/j.celrep.2014.11.033. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tantama M, Martínez-François JR, Mongeon R, Yellen G. Imaging energy status in live cells with a fluorescent biosensor of the intracellular ATP-to-ADP ratio. Nat Commun. 2013;4 doi: 10.1038/ncomms3550. 2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hodson DJ, Schaeffer M, Romanò N, Fontanaud P, Lafont C, Birkenstock J, Molino F, Christian H, Lockey J, Carmignac D, Fernandez-Fuente M, et al. Existence of long-lasting experience-dependent plasticity in endocrine cell networks. Nat Commun. 2012;3:605. doi: 10.1038/ncomms1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McTaggart LR, Richardson SE, Witkowska M, Zhang SX. Phylogeny and identification of Nocardia species on the basis of multilocus sequence analysis. J Clin Microbiol. 2010;48:4525–4533. doi: 10.1128/JCM.00883-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rutter GA. Visualising insulin secretion. The Minkowski Lecture 2004. Diabetologia. 2004;47:1861–1872. doi: 10.1007/s00125-004-1541-1. [DOI] [PubMed] [Google Scholar]
- 41.Tarasov AI, Nicolson TJ, Riveline JP, Taneja TK, Baldwin SA, Baldwin JM, Charpentier G, Gautier JF, Froguel P, Vaxillaire M, Rutter GA. A rare mutation in ABCC8/SUR1 leading to altered ATP-sensitive K+ channel activity and beta-cell glucose sensing is associated with type 2 diabetes in adults. Diabetes. 2008;57:1595–1604. doi: 10.2337/db07-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hunt MC, Alexson SE. The role Acyl-CoA thioesterases play in mediating intracellular lipid metabolism. Prog Lipid Res. 2002;41:99–130. doi: 10.1016/s0163-7827(01)00017-0. [DOI] [PubMed] [Google Scholar]
- 43.Donath MY, Ehses JA, Maedler K, Schumann DM, Ellingsgaard H, Eppler E, Reinecke M. Mechanisms of beta-cell death in type 2 diabetes. Diabetes. 2005;54(Suppl 2):S108–113. doi: 10.2337/diabetes.54.suppl_2.s108. [DOI] [PubMed] [Google Scholar]
- 44.Klett EL, Chen S, Edin ML, Li LO, Ilkayeva O, Zeldin DC, Newgard CB, Coleman RA. Diminished acyl-CoA synthetase isoform 4 activity in INS 832/13 cells reduces cellular epoxyeicosatrienoic acid levels and results in impaired glucose-stimulated insulin secretion. J Biol Chem. 2013;288:21618–21629. doi: 10.1074/jbc.M113.481077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Corkey BE, Deeney JT, Yaney GC, Tornheim K, Prentki M. The role of long-chain fatty acyl-CoA esters in beta-cell signal transduction. J Nutr. 2000;130:299S–304S. doi: 10.1093/jn/130.2.299S. [DOI] [PubMed] [Google Scholar]
- 46.Rorsman P, Renström E. Insulin granule dynamics in pancreatic beta cells. Diabetologia. 2003;46:1029–1045. doi: 10.1007/s00125-003-1153-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.