

Abstract
BACKGROUND
Considerable uncertainty exists as to whether lowering low-density lipoprotein cholesterol (LDL-C) by inhibiting the Niemann-Pick C1-Like 1 (NPC1L1) receptor with ezetimibe, either alone or in combination with a 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR) inhibitor (statin), will reduce the risk of coronary heart disease (CHD).
OBJECTIVES
This study evaluated the effect of naturally random allocation to lower LDL-C mediated by polymorphisms in the NPC1L1 gene (target of ezetimibe), the HMGCR gene (target of statins), or both (target of combination therapy) on the risk of CHD.
METHODS
We constructed NPC1L1 and HMGCR genetic LDL-C scores to naturally randomize participants into 4 groups: reference, lower LDL-C mediated by NPC1L1 polymorphisms, lower LDL-C mediated by HMGCR polymorphisms, or lower LDL-C mediated by polymorphisms in both NPC1L1 and HMGCR. We compared the risk of CHD (fatal or nonfatal myocardial infarction) among each group using a 2 × 2 factorial mendelian randomization study design.
RESULTS
A total of 108,376 persons (10,464 CHD events) from 14 studies were included. There were no significant differences in baseline characteristics among the 4 groups, thus confirming that allocation was random. Compared to the reference group, the NPC1L1 group had 2.4 mg/dl lower LDL-C and 4.8% lower risk of CHD (odds ratio [OR]: 0.952, 95% confidence interval [CI]: 0.920 to 0.985); whereas the HMGCR group had 2.9 mg/dl lower LDL-C and a similar 5.3% lower risk of CHD (OR: 0.947, 95% CI: 0.909 to 0.986). The group with lower LDL-C mediated by both NPC1L1 and HMGCR polymorphisms had 5.8 mg/dl additively lower LDL-C and a 10.8% log-linearly additive lower risk of CHD (OR: 0.892, 95% CI: 0.854 to 0.932).
CONCLUSIONS
The effect of lower LDL-C on the risk of CHD mediated by polymorphisms in NPC1L1, HMGCR, or both is approximately the same per unit lower LDL-C and log-linearly proportional to the absolute exposure to lower LDL-C.
Keywords: ezetimibe, genetic association, PCSK9, statins
Meta-analyses of prospective epidemiologic cohort studies (1,2) and meta-analyses of mendelian randomization genetic studies (3) have shown a consistent, causal, and log-linear association between low-density lipoprotein cholesterol (LDL-C) and the risk of coronary heart disease (CHD). Additionally, meta-analyses of numerous randomized controlled trials have shown that lowering LDL-C by inhibiting 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMGCR) with a statin reduces the risk of CHD and other major vascular events (4). Despite the established causal association between LDL-C and the risk of CHD, several randomized trials have failed to consistently show an incremental clinical benefit from further lowering LDL-C by adding niacin or a fibrate to treatment with a statin (5–7), creating uncertainty as to whether lowering LDL-C by a mechanism other than inhibiting HMGCR with a statin will reduce the risk of CHD.
Ezetimibe is a commonly prescribed medication that effectively lowers LDL-C when used alone or when added to treatment with statin. Importantly, both ezetimibe and statins lower LDL-C through a common final pathway. Ezetimibe inhibits intestinal absorption of cholesterol by binding to the Niemann-Pick C1-Like 1 (NPC1L1) protein, which leads to up-regulation of hepatic LDL-C receptors and increased clearance of circulating LDL-C (8). Similarly, statins reduce hepatic cholesterol synthesis by inhibiting HMGCR, which leads to up-regulation of hepatic LDL-C receptors and increased clearance of circulating LDL-C. Because both ezetimibe and statins reduce LDL-C through the same final common pathway, it is intuitive to hypothesize that lowering LDL-C by inhibiting NPC1L1 with ezetimibe may also reduce the risk of CHD and other major vascular events. IMPROVE-IT (IMProved Reduction of Outcomes: Vytorin Efficacy International Trial) is testing this hypothesis (9,10).
To compare the biological effect of lower LDL-C mediated by inhibition of NCP1L1, HMGCR, or both on the risk of CHD, and to provide a context for interpreting the results of IMPROVE-IT, we sought to compare the effect of naturally random allocation to lower LDL-C on the risk of CHD mediated by genetic polymorphisms in the NPC1L1 gene (as a proxy for ezetimibe treatment), the HMGCR gene (as a proxy for statin treatment), or both (as a proxy for combination treatment) using a novel 2 × 2 factorial mendelian randomization study design.
METHODS
Our study included 108,376 total persons from 14 prospective cohort or case-control studies who provided written informed consent for genetic studies and with individual-level data available as part of the National Center for Biotechnology Information database of Genotypes and Phenotypes program (11). Online Table S1 describes the included studies. As part of a larger project, we first harmonized the definition of all cardiovascular-related exposure and outcome variables across the 14 studies, and then recoded individual-level data for each study subject as necessary to satisfy the harmonized variable definitions. The primary outcome for this study was CHD, defined as the first occurrence of CHD death or a nonfatal myocardial infarction (MI). Among subjects with available genetic information enrolled in the 14 studies, 10,464 first CHD outcome events occurred, all of which were included in the primary analysis.
We first constructed NPC1L1 and HMGCR genetic LDL-C scores to create an instrument that could overcome the weak effect of most NPC1L1 and HMGCR polymorphisms on plasma LDL-C levels (12–14), and that would also allow us to randomly allocate study subjects into approximately equal-sized groups to facilitate the 2 × 2 factorial analysis. We constructed the genetic LDL-C scores by identifying all polymorphisms in or within 100 kb on either side of the NPC1L1 and HMGCR genes, respectively, that were associated with LDL-C at a threshold of p < 5.0 × 10−6, as measured in up to 183,465 persons of European descent in the Global Lipids Genetic Consortium (GLGC) (12). We then ranked the polymorphisms in each gene by the reported p value and iteratively selected all approximately independently inherited polymorphisms, defined by a low degree of linkage disequilibrium (r2 < 0.30 for all comparisons), to include in each gene’s LDL-C score. Depending on the genotyping platform used in each study, we used the selected polymorphism or its nearest proxy. If a close proxy (r2 ≥ 0.95) was not available, we imputed genotypes for the selected polymorphism for all members of that study population using the 1000 Genomes reference panel (15). A total of 5 approximately independent polymorphisms were included in the NPC1L1 genetic LDL-C score, and 3 were included in the HMGCR genetic LDL-C score (Online Tables S2 to S7). For each selected polymorphism, we coded the exposure allele as the allele associated with lower LDL-C.
For each study subject, we calculated a weighted NPC1L1 and HMGCR genetic LDL-C score by summing the number of LDL-C–lowering alleles inherited at each of the polymorphisms included in either score, weighted by its effect on LDL-C measured in mg/dl (as estimated in the GLGC) (12). Because each polymorphism included in either genetic score is inherited approximately randomly at conception and approximately independently of the other polymorphisms included in each score, the number of LDL-C–lowering alleles that a person inherits in either score should also be random.
To conduct the 2 × 2 factorial analyses, we first dichotomized each genetic LDL-C score as above or below the median value for that score (as measured in the study population). Next, study subjects were naturally randomly allocated into 2 groups, depending on whether their HMGCR genetic LDL-C score was above or below the median. Subjects in each of these 2 groups were then randomly allocated into 2 further groups, depending on whether their NPC1L1 genetic LDL-C score was above or below the median. This process naturally randomized all subjects into 1 of 4 groups (Figure 1): the reference group (analogous to a placebo group), a group with lower LDL-C mediated by NPC1L1 polymorphisms (analogous to ezetimibe treatment), a group with lower LDL-C mediated by HMGCR polymorphisms (analogous to statin treatment), and a group with lower LDL-C mediated by the combined effect of polymorphisms in the both the NPC1L1 and HMGCR genes (analogous to treatment with a combination of ezetimibe and a statin).
FIGURE 1. Design of 2 × 2 Factorial Mendelian Randomization Study.
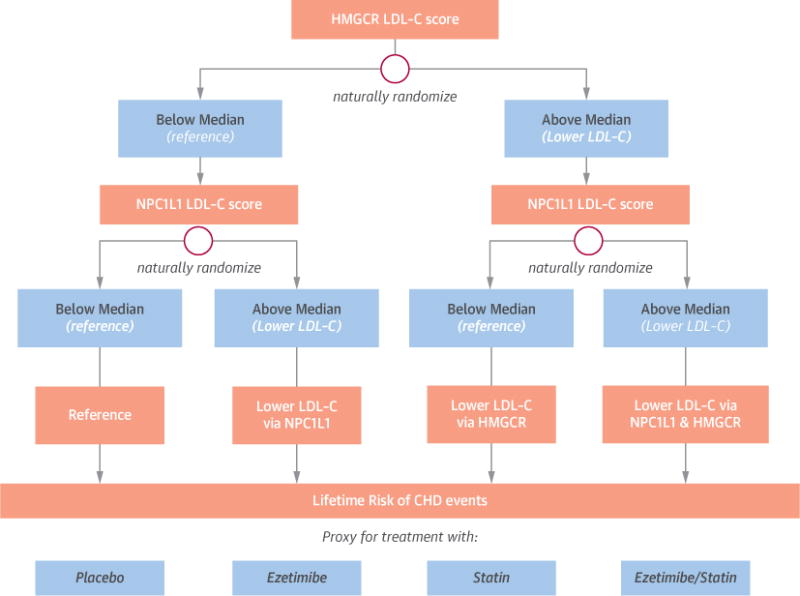
Each subject was first randomly allocated into 1 of 2 groups based on whether their HMGCR LDL-C score was above or below the median, and then randomly allocated into 1 of 2 further groups based on whether their NPC1L1 LDL-C score was above or below the median using a factorial design. This process naturally randomized all subjects into 1 of 4 groups: the reference group (analogous to a placebo group), a group with lower LDL-C mediated polymorphisms in the NPC1L1 gene (analogous to treatment with ezetimibe), a group with lower LDL-C mediated polymorphisms in the HMGCR gene (analogous to treatment with a statin), and a group with lower LDL-C mediated by the combined effect of polymorphisms in both the NPC1L1 and HMGCR genes (analogous to treatment with combination ezetimibe and a statin).
We assessed the success of the naturally random allocation scheme by comparing baseline characteristics among subjects in each of the 4 groups. We measured the difference in LDL-C between each group and the reference group using linear regression and the risk of CHD in each group compared with the reference group using logistic regression (for combined prevalent and incident events) or Cox proportional hazard models (for incident events), adjusted for age and sex. All analyses were performed separately in each of the 14 included study samples, and then combined across studies in a fixed-effects inverse variance-weighted meta-analysis to produce summary estimates of effect. To minimize potential population stratification bias, separate analyses were performed for each included racial group.
To provide external validation, we compared the effect of lower LDL-C on the risk of CHD mediated by the NPC1L1 and HMGCR genetic LDL-C scores in up to 62,240 cases and 127,299 control subjects enrolled in the CARDIoGRAM or CARDIoGRAMplusC4D consortia studies (16). To calculate these scores using available summary data, we looked up the association between each polymorphism included in the NPC1L1 and HMGCR genetic LDL-C scores and the CHD risk, as reported by the CARDIoGRAMplusC4D consortium (17). We adjusted the reported effect size (and corresponding standard error) by the effect of that polymorphism on LDL-C (in mg/dl), as reported by the GLGC (12), using the usual ratio of effect estimates method. We then combined the adjusted effect estimates in a fixed-effects inverse variance-weighted meta-analysis to produce NPC1L1 and HMGCR genetic LDL scores that represent a summary estimate of the effect of each unit lower LDL-C on the risk of CHD mediated by the combined effect of the polymorphisms included in either genetic LDL-C score.
All statistical analyses used a 2-tailed p < 0.05 threshold for nominal statistical significance and all analyses were performed using STATA 12 (StataCorp, LP, College Station, Texas), SNP & Variation Suite (Version 8.1.4; Golden Helix, Bozeman, Montana), or IMPUTE2 (18,19). The Online Appendix provides a detailed description of the methods.
RESULTS
ASSOCIATION BETWEEN GENETIC LDL-C SCORES, PLASMA LDL-C LEVELS, AND RISK OF CHD
There were no significant differences in any baseline characteristics between the 4 groups (Table 1), showing that allocation was indeed random. The apparently random allocation of study subjects into approximately equal-sized groups also internally validates the use of the NPC1L1 and HMGCR LDL-C scores as instrumental variables.
TABLE 1.
Baseline Characteristics
| Characteristic | Reference Group | NPC1L1 LDL-C Score Above Median |
HMGCR LDL-C Score Above Median |
Both LDL-C Scores Above Median |
p Value |
|---|---|---|---|---|---|
| Sample size | 27,744 | 28,611 | 25,577 | 26,444 | |
| LDL-C, mg/dl | 132.5 ± 31.8 | 130.1 ± 33.1 | 129.6 ± 32.7 | 126.7 ± 32.3 | 2.3 × 10−47 |
| Age, yrs | 59.4 ± 6.3 | 59.1 ± 6.7 | 58.9 ± 6.1 | 59.6 ± 5.9 | NS |
| Males | 43.6 | 44.1 | 43.3 | 43.9 | NS |
| Weight, lbs | 166.5 ± 35.8 | 165.2 ± 36.5 | 165.9 ± 36.2 | 167.4 ± 35.4 | NS |
| BMI, kg/m2 | 27.2 ± 5.4 | 27.9 ± 5.7 | 27.1 ± 5.1 | 27.7 ± 4.9 | NS |
| HDL-C, mg/dl | 51.8 ± 14.7 | 51.0 ± 14.8 | 51.7 ± 14.2 | 51.2 ± 15.1 | NS |
| TG, mg/dl | 135.3 (78–158) | 134.4 (77–161) | 134.9 (81–164) | 135.3 (79–156) | NS |
| Lipid treatment | 4.9 | 4.7 | 5.1 | 4.6 | NS |
| SBP, mm Hg | 125.8 ± 16.4 | 125.1 ± 16.1 | 126.0 ± 17.5 | 125.7 ± 16.8 | NS |
| DBP, mm Hg | 73.9 ± 11.2 | 74.2 ± 10.8 | 74.3 ± 11.6 | 73.7 ± 11.0 | NS |
| BP treatment | 36.1 | 37.8 | 36.5 | 36.9 | NS |
| Current smoker | 12.8 | 12.5 | 13.3 | 12.7 | NS |
| Former smoker | 32.1 | 30.9 | 31.4 | 32.6 | NS |
| Diabetes | 6.3 | 6.6 | 6.0 | 5.9 | NS |
Values are n, mean ± SD, %, or median (interquartile range). Sample size includes all subjects in both the prospective cohort and case-control studies. Clinical characteristics are values measured at baseline study visit among subjects enrolled in the prospective cohort studies. Categorical variables were compared with a chi-square test, and continuous variables were compared with either one-way analysis of variance for normally distributed variables or the Kruskal-Wallis test for non-normally distributed variables.
BMI = body mass index; BP = blood pressure; DBP = diastolic blood pressure; HDL-C = high-density lipoprotein cholesterol; HMGCR = hydroxymethyl glutaryl coenzyme A reductase; LDL-C = low-density lipoprotein cholesterol; NPC1L1 = Niemann-Pick C1-Like 1; NS = not significant (p > 0.05); SBP = systolic blood pressure; TG = triglycerides.
The mean age of study subjects was 59 years, only 4.8% were taking a lipid-lowering therapy at baseline, and the mean baseline LDL-C level was 132.5 mg/dl among persons in the reference group (Table 1). Compared with the reference group (both NPC1L1 and HMGCR scores below the median), persons in the group with an NPC1L1 genetic LDL-C score above the median (and an HMGCR score below the median) had 2.4 mg/dl lower LDL-C and a significant 4.8% lower risk of CHD (odds ratio [OR]: 0.952, 95% confidence interval [CI]: 0.920 to 0.985; p = 4.4 × 10−3). Compared with the reference group, persons in the group with an HMGCR genetic LDL-C score above the median (and an NPC1L1 score below the median) had 2.9 mg/dl lower LDL-C and a similar 5.3% lower risk of CHD (OR: 0.947, 95% CI: 0.909 to 0.986; p = 9.1 × 10−3). There was no significant difference in either LDL-C level or risk of CHD among persons with NPC1L1 scores above the median, as compared with persons with HMGCR LDL-C scores above the median (p = 0.84). Compared with the reference group, persons in the group with both NPC1L1 and HMGCR genetic LDL-C scores above the median had 5.8 mg/dl lower LDL-C and a 10.8% lower risk of CHD (OR: 0.892, 95% CI: 0.854 to 0.932; p = 2.5 × 10−7). The combination of both NPC1L1 and HMGCR polymorphisms was associated with a linearly additive effect on plasma LDL-C levels and a log-linearly additive effect on CHD risk (Figure 2). The CHD risk was significantly lower in the group with NPC1L1 and HMGCR scores above the median as compared with both the group with NPC1L1 scores above the median (p = 0.045) and the group with HMGCR scores above the median (p = 0.021).
FIGURE 2. Effect of Lower LDL-C Mediated by Polymorphisms in NPC1L1, HMGCR, or Both.
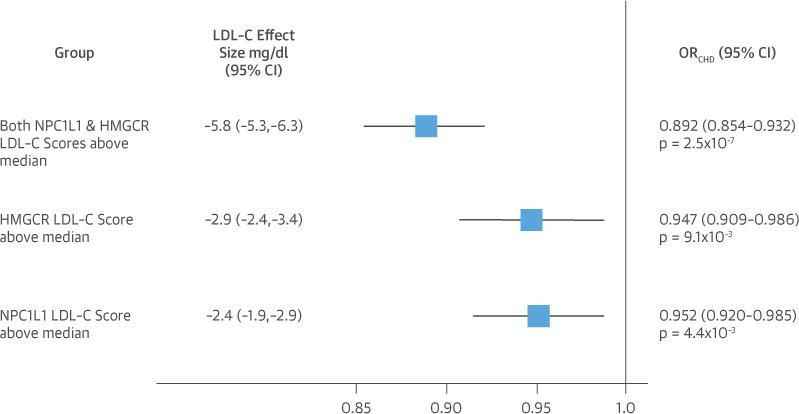
Boxes represent point estimates and lines represent 95% CIs. Reference group is the group with both NPC1L1 and HMGCR LDL-C scores below median. CI = confidence interval; LDL-C = low-density lipoprotein cholesterol; HMGCR = hydroxymethyl glutaryl coenzyme A reductase; NPC1L1 = Niemann-Pick C1-Like 1.
To further compare the effect of lower LDL-C mediated by NPC1L1 and HMGCR polymorphisms, we also evaluated the effect of each genetic LDL-C score in the entire study population (without further partitioning into smaller groups by the other genetic LDL-C score, as occurs in 2 × 2 factorial analysis). Compared with persons with NPC1L1 genetic LDL-C scores below the median, persons with scores above the median had 2.3 mg/dl lower LDL-C (p = 8.2 × 10−12) and a 4.4% lower risk of CHD (OR: 0.956, 95% CI: 0.930 to 0.983; p = 1.3 × 10−3). Similarly, compared with persons with HMGCR genetic LDL-C scores below the median, persons with scores above the median had 3.0 mg/dl lower LDL-C (p = 3.7 × 10−17) and a 5.2% lower risk of CHD (OR: 0.948, 95% CI: 0.920 to 0.977; p = 4.8 × 10−4). These results were essentially unchanged when modeling the genetic LDL-C scores as continuous variables. In these analyses, the effect of lower LDL-C on the risk of CHD mediated by NPC1L1 and HMGCR polymorphisms was very similar per unit lower LDL-C. When both genetic LDL-C scores were included in the same model, each score’s effect remained essentially unchanged, with no evidence for any effect modification on either plasma LDL-C levels or CHD risk in models that included an interaction term.
EXTERNAL VALIDATION ANALYSES
In external validation analyses involving up to 62,240 cases of CHD and 127,299 control subjects, each 10 mg/dl lower LDL-C mediated by polymorphisms in the NPC1L1 genetic LDL-C score was associated with a highly significant 17.7% lower risk of CHD (OR: 0.823, 95% CI: 0.741 to 0.915; p = 3.1 × 10−4) (Online Figures S1A and S1B). Similarly, each 10 mg/dl lower LDL-C mediated by polymorphisms in the HMGCR genetic LDL-C score was associated with a similar and highly significant 17.2% lower risk of CHD (OR: 0.828, 95% CI: 0.775 to 0.885; p = 5.5 × 10−8) (Online Figures S2A and S2B). There was no significant difference in the effect of lower LDL-C on the risk of CHD mediated by NPC1L1 and HMGCR polymorphisms when measured per unit lower LDL-C (p = 0.93) (Figure 3). These data appear to externally validate the finding in our primary analysis that the effect of lower LDL-C on the risk of CHD mediated by NPC1L1 and HMGCR genetic polymorphisms appears to be approximately the same per unit lower LDL-C.
FIGURE 3. Comparison of Effect of 10 mg/dl Lower LDL-C on Risk of CHD Mediated by Polymorphisms in the NPC1L1 and HMGCR Genetic LDL-C Scores in Up to 62,240 Cases of CHD and 127,299 Control Subjects.
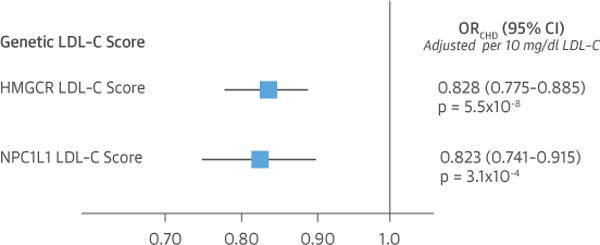
Boxes represent point estimates and lines represent 95% CIs. Abbreviations as in Figure 2.
In additional external validation analyses involving up to 63,746 cases of CHD and 130,681 control subjects, we compared the effect of lower LDL-C mediated by NPC1L1 and HMGCR gene polymorphisms and the NPC1L1 and HMGCR genetic scores with the effect of lower LDL-C mediated by polymorphisms in other genes that lower circulating LDL-C through the common final pathway involving the hepatic LDL-C receptor. This analysis included both common and rare polymorphisms in the pro-protein convertase subtilisin/kexin type 9 gene (PCSK9). The effect of lower LDL-C on the risk of CHD mediated by each of these polymorphisms was very similar per unit lower LDL-C, with no evidence for any significant heterogeneity, as shown in Figure 4 (Online Tables S8 to S10, Online Figures S3 to S5). Furthermore, when each polymorphism’s (or genetic LDL-C score’s) effect on LDL-C was plotted against its effect on CHD risk, there appeared to be a log-linear relationship between the absolute magnitude of exposure to lower LDL-C and the proportional reduction in the risk of CHD, independent of the mechanism by which LDL-C was lowered (Central Illustration).
FIGURE 4. Comparison of Effect of 10 mg/dl Lower LDL-C on Risk of CHD Mediated by Polymorphisms in the LDL-C Receptor Pathway in Up to 63,746 Cases of CHD and 130,681 Control Subjects.
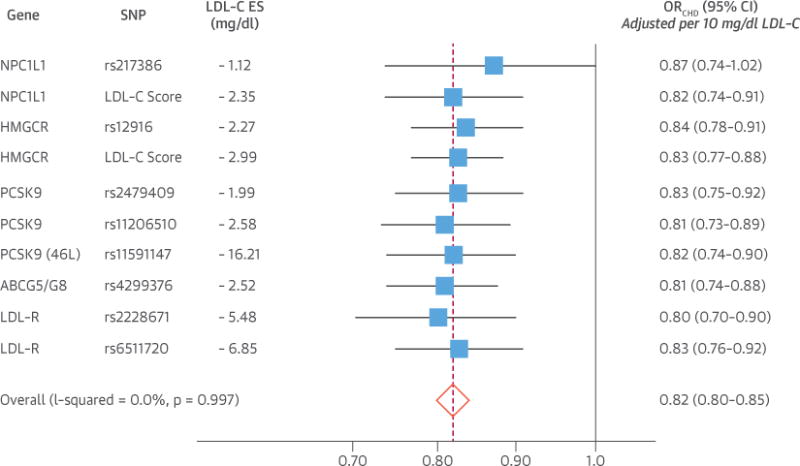
Boxes represent point estimates and lines represent 95% CIs. Point estimates (and CI) adjusted per unit lower LDL-C using the usual ratio of effect estimates method.
CENTRAL ILLUSTRATION. 2 × 2 Factorial Mendelian Randomization Study: Log-Linear Association Between Genetically and Pharmacologically Mediated Lower Low-Density Lipoprotein Cholesterol and Risk of Coronary Heart Disease.
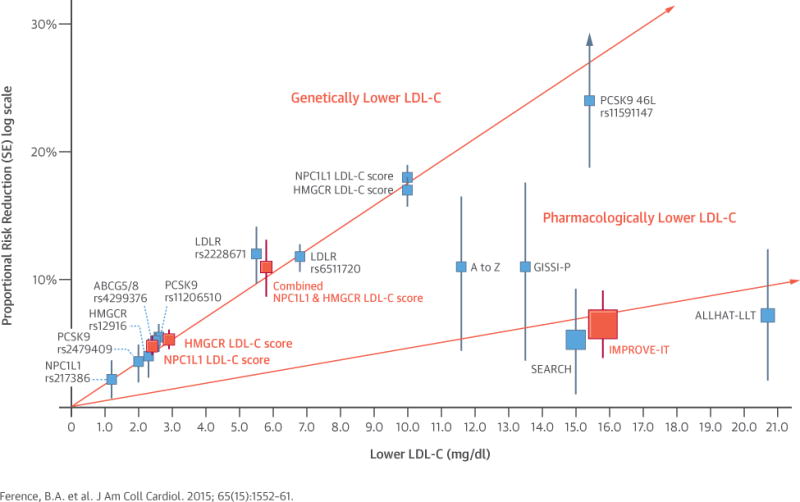
Boxes represent proportional risk reduction (1–OR) of CHD for each exposure allele, genetic score, or randomized trial plotted against the absolute magnitude of lower LDL-C associated with that allele or genetic score; or the absolute difference in LDL-C between treatment groups for each trial. Vertical lines represent 1 SE above and below point estimate of proportional risk reduction. SNPs, genetic scores, and trials are plotted in order of increasing absolute magnitude of effect on lower LDL-C. The lines (which are forced to pass through the origin) represent the increase in proportional risk reduction of CHD per unit lower LDL-C. In the top line, the salmon boxes represent results of the 2 × 2 factorial mendelian randomization study and the blue boxes represent results derived from CARDIoGRAMplusC4D consortia data. In the lower line, the salmon box represents the results of the IMPROVE-IT trial and the blue boxes represent the results of prior statin trials. CHD = coronary heart disease; IMPROVE-IT = IMProved Reduction of Outcomes: Vytorin Efficacy International Trial; LDL-C = low-density lipoprotein cholesterol; OR = odds ratio; SE = standard error; SNP = single-nucleotide polymorphism.
DISCUSSION
We appealed to the principle of mendelian randomization (20) to evaluate the effect of naturally random allocation to lower LDL-C on the risk of CHD mediated by polymorphisms in the NPC1L1 gene (the target of ezetimibe), the HMGCR gene (the target of statins), or both (the target of combination therapy with ezetimibe and a statin) using a 2 × 2 factorial study design. We confirmed that allocation to lower LDL-C was random by showing that there were no significant differences in any baseline characteristics between the groups. Therefore, our results should provide an unconfounded estimate of the causal effect of lower LDL-C mediated by inhibition of NPC1L1, HMGCR, or both on the risk of CHD in a manner analogous to a 2 × 2 factorial randomized trial comparing the effect of treatment with ezetimibe, a statin, or both.
We found that lower LDL-C mediated by common polymorphisms in the NPC1L1 gene is causally associated with a lower CHD risk. This finding was independently externally validated by a recent report that rare NPC1L1 loss-of-function mutations are also associated with both lower LDL-C and a lower risk of CHD (21). Importantly, because we used a 2 × 2 factorial study design, we could directly compare the effect of lower LDL-C on CHD risk mediated by the separate and combined effects of NPC1L1 and HMGCR polymorphisms. We found that NPC1L1 and HMGCR polymorphisms have approximately the same effect on CHD risk when measured per unit lower LDL-C. Furthermore, when present together, NPC1L1 and HMGCR genetic polymorphisms appear to have independent, linearly additive effects on plasma LDL-C levels and log-linearly additive effects on CHD risk. These findings strongly suggest that there is no difference in the biological effect of lower LDL-C on the risk of CHD mediated by inhibition of NPC1L1 or HMGCR. Therefore, our results imply that lowering LDL-C by inhibiting NPC1L1 with ezetimibe, inhibiting HMGCR with a statin, or inhibiting both with the combination of ezetimibe and a statin should each lower the risk of CHD by approximately the same amount per unit lower LDL-C, and that the magnitude of the clinical benefit will be proportional to the absolute magnitude of the achieved reduction in LDL-C, regardless of which treatment is used.
Indeed, our results agree closely with the recently reported results of IMPROVE-IT (22), in which addition of ezetimibe to treatment with simvastatin resulted in a further mean LDL-C reduction of 15.8 mg/dl and a corresponding 6.4% reduction in the primary composite endpoint of cardiovascular death, MI, stroke, coronary revascularization, or hospitalization for unstable angina (p = 0.016); and a 10.0% reduction in the composite endpoint of cardiovascular death, nonfatal MI, or nonfatal stroke (p = 0.003). The magnitude of this risk reduction is consistent with the effect size expected for a similar reduction in LDL-C during statin treatment, as estimated by the Cholesterol Treatment Trialists’ Collaboration meta-analysis of statin trials (4). Therefore, the results of IMPROVE-IT suggest that lowering LDL-C by inhibiting NPC1L1 with ezetimibe has approximately the same effect on CHD risk as inhibiting HMGCR with a statin, when measured per unit lower LDL-C, and that combined NPC1L1 and HMGCR inhibition has independent and additive effects on both plasma LDL-C levels and the corresponding risk of CHD, as anticipated by the naturally randomized genetic evidence in our study.
The close agreement between our study’s results and those of IMPROVE-IT substantially increases confidence in the validity of our findings. It also suggests that it may be reasonable to appeal to the naturally randomized genetic evidence to address questions not specifically tested in IMPROVE-IT; for example, whether the combination of a moderate-dose statin plus ezetimibe will be as effective at reducing the risk of CHD as treatment with a high-dose statin. We found that the effect of lower LDL-C mediated solely by HMGCR gene polymorphisms had approximately the same effect on CHD risk as did the effect of lower LDL-C mediated by the combined effect of polymorphisms in both the HMGCR and NPC1L1 genes, when measured per unit lower LDL-C. Therefore, the naturally randomized genetic evidence suggests that the use of a moderate-dose statin plus ezetimibe should be a reasonable alternative to high-dose statin therapy, particularly among persons unable or unwilling to take a high-dose statin, because the effect of lower LDL-C on the risk of CHD appears to be independent of the mechanism by which LDL-C is lowered.
To further challenge our finding that the effect of lower LDL-C on the risk of CHD appears independent of the mechanism by which LDL-C is lowered, we compared the effect of polymorphisms in multiple different genes, each of which acts to lower LDL-C through the common final pathway of the hepatic LDL-C receptor. This analysis not only included NPC1L1 and HMGCR polymorphisms, but also both common and rare polymorphisms in the PCSK9 gene (which encodes the target of a new class of LDL-C-lowering therapy under active investigation) (23). We found that each of these polymorphisms had a remarkably similar effect on CHD risk when measured per unit lower LDL-C (Figure 4). Together, these data strongly suggest that the effect of lower LDL-C on the risk of CHD appears to be independent of the mechanism by which LDL-C is lowered, at least among pathways involving the LDL-C receptor as the final common LDL-C–lowering mechanism. Instead, the clinical benefit of exposure to both genetically and pharmacologically mediated lower LDL-C appears to be largely determined by the absolute magnitude of exposure to lower LDL-C (Central Illustration). These findings are consistent with the results of prior mendelian randomization studies (3,24), and may explain why treatment with niacin or a fibrate has failed to consistently reduce the risk of CHD when added to a statin in randomized trials. In these trials, the absolute magnitude of the difference in LDL-C between the 2 treatment arms was very small, and likely too small to translate into a numerically stable reduction in the risk of CHD (5–7).
STUDY LIMITATIONS
Our study has several limitations. We measured the effect of lower LDL-C mediated by polymorphisms in the NPC1L1 and HMGCR genes, not the effect of lower LDL-C mediated by treatment with ezetimibe and a statin. The effect of treatments designed to inhibit NPC1L1 and HMGCR may not have the same effect as polymorphisms in the genes encoding the targets of these treatments. However, numerous prior studies have shown that adding ezetimibe to treatment with a statin further reduces LDL-C by approximately 15% to 20%, independent of the dose or type of statin used (25). This is consistent with our finding that combined genetic polymorphisms in the NPC1L1 and HMGCR genes are associated with a linearly additive exposure to lower LDL-C. More importantly, the close agreement between the results of our study and those of IMPROVE-IT suggests that genetically lower LDL-C mediated by polymorphisms in the NPC1L1 and HMCGR genes are reasonable proxies for treatment with ezetimibe and a statin. Additionally, our study found that lifetime exposure to small differences in LDL-C mediated by NPC1L1 and HMGCR polymorphisms was associated with much larger than expected reductions in the risk of CHD than would be predicted by quantitatively similar reductions in LDL-C observed in the statin trials or the IMPROVE-IT trial. This finding is, however, consistent with prior mendelian randomization studies that showed that long-term exposure to lower LDL-C appears to have a cumulative effect on the risk of CHD and is associated with up to a 3-fold greater reduction in the risk of CHD per unit lower LDL-C, as compared to short-term treatment with a statin started later in life (3).
CONCLUSIONS
We found that the effect of lower LDL-C on the risk of CHD mediated by polymorphisms in the NPC1L1 gene, the HMGCR gene, or both is approximately the same per unit lower LDL-C and log-linearly proportional to the absolute magnitude of the exposure to lower LDL-C. We conclude that there appears to be no difference in the biological effect of lower LDL-C on the risk of CHD mediated by inhibition of NPC1L1 or HMGCR. Therefore, lowering LDL-C with ezetimibe, a statin, or combination therapy with both ezetimibe and a statin should each reduce the risk of CHD by approximately the same amount per unit lower LDL-C and the magnitude of the observed clinical benefit should be proportional to the absolute magnitude of the reduction in LDL-C, regardless of which treatment is used. More generally, our results suggest that the effect of lower LDL-C on the risk of CHD appears to be determined by the absolute magnitude of exposure to lower LDL-C, independent of the mechanism by which LDL-C is lowered.
Supplementary Material
PERSPECTIVES.
COMPETENCY IN MEDICAL KNOWLEDGE
Observations based upon mendelian randomization suggests that the clinical benefit of lower LDL-C levels may be more closely related to the magnitude of reduction than to the mechanism of LDL-C lowering.
TRANSLATIONAL OUTLOOK
Further studies using the natural genetic randomization of factors that govern blood lipid metabolism may provide information adjunctive to that from randomized clinical trials.
Acknowledgments
This research was funded in part by an investigator-initiated grant from Merck & Co. The funding body had no role in the design, conduct, or analysis of the study; did not have access to any of the data; did not participate in drafting of the manuscript; and did not participate in the decision to submit for publication. Dr. Ference has reported significant research grants, modest service as a consultant and advisory board member, and has received modest honoraria from Merck. All other authors have reported that they have no relationships relevant to the contents of this paper to disclose.
ABBREVIATIONS AND ACRONYMS
- CHD
coronary heart disease
- HMGCR
3-hydroxy-3-methylglutaryl-coenzyme A reductase
- LDL-C
low-density lipoprotein cholesterol
- MI
myocardial infarction
- NPC1L1
Niemann-Pick C1-Like 1
APPENDIX
For supplemental tables and figures, please see the online version of this article.
Footnotes
Listen to this manuscript’s audio summary by JACC Editor-in-Chief Dr. Valentin Fuster.
You can also listen to this issue’s audio summary by JACC Editor-in-Chief Dr. Valentin Fuster.
References
- 1.Prospective Studies Collaboration. Lewington S, Whitlock G, et al. Blood cholesterol and vascular mortality by age, sex, and blood pressure: a meta-analysis of individual data from 61 prospective studies with 55,000 vascular deaths. Lancet. 2007;370:1829–39. doi: 10.1016/S0140-6736(07)61778-4. [DOI] [PubMed] [Google Scholar]
- 2.Emerging Risk Factors Collaboration. Di Angelantonio E, Sarwar N, et al. Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302:1993–2000. doi: 10.1001/jama.2009.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ference BA, Yoo W, Alesh I, et al. Effect of long-term exposure to lower low-density lipoprotein cholesterol beginning early in life on the risk of coronary heart disease: a Mendelian randomization analysis. J Am Coll Cardiol. 2012;60:2631–9. doi: 10.1016/j.jacc.2012.09.017. [DOI] [PubMed] [Google Scholar]
- 4.Cholesterol Treatment Trialists’ (CTT) Collaboration. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376:1670–81. doi: 10.1016/S0140-6736(10)61350-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.AIM-HIGH Investigators. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–67. doi: 10.1056/NEJMoa1107579. [DOI] [PubMed] [Google Scholar]
- 6.HPS2-THRIVE Collaborative Group. Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med. 2014;371:203–12. doi: 10.1056/NEJMoa1300955. [DOI] [PubMed] [Google Scholar]
- 7.ACCORD Study Group. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010;362:1563–74. doi: 10.1056/NEJMoa1001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garcia-Calvo M, Lisnock J, Bull HG, et al. The target of ezetimibe is Niemann-Pick C1-Like 1 (NPC1L1) Proc Natl Acad Sci U S A. 2005;102:8132–7. doi: 10.1073/pnas.0500269102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cannon CP, Giugliano RP, Blazing MA, et al. for the IMPROVE-IT Investigators Rationale and design of IMPROVE-IT (IMProved Reduction of Outcomes: Vytorin Efficacy International Trial): comparison of ezetimibe/simvastatin versus simvastatin monotherapy on cardiovascular outcomes in patients with acute coronary syndromes. Am Heart J. 2008;156:826–32. doi: 10.1016/j.ahj.2008.07.023. [DOI] [PubMed] [Google Scholar]
- 10.Blazing MA, Giugliano RP, Cannon CP, et al. Evaluating cardiovascular event reduction with ezetimibe as an adjunct to simvastatin in 18,144 patients after acute coronary syndromes: final baseline characteristics of the IMPROVE-IT study population. Am Heart J. 2014;168:205–12.e1. doi: 10.1016/j.ahj.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 11.Mailman MD, Feolo M, Jin Y, et al. The NCBI dbGaP database of genotypes and phenotypes. Nat Genet. 2007;39:1181–6. doi: 10.1038/ng1007-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Global Lipids Genetics Consortium. Willer CJ, Schmidt EM, et al. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45:1274–83. doi: 10.1038/ng.2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Teslovich TM, Musunuru K, Smith AV, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–13. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kathiresan S, Melander O, Guiducci C, et al. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat Genet. 2008;40:189–97. doi: 10.1038/ng.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.1000 Genomes Project Consortium. Abecasis GR, Auton A, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.CARDIoGRAMplusC4D Consortium. Deloukas P, Kanoni S, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013;45:25–33. doi: 10.1038/ng.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.CARDIOGRAMPLYSC4D Consortium. Wellcome Trust Sanger Institute. Available at: http://www.CARDIOGRAMPLUSC4D.org. Accessed February 10, 2015.
- 18.SNP & Variation Suite (Version 8.1.4). Golden Helix, Inc. Available at: http://www.goldenhelix.com/SNP_Variation/index.html. Accessed February 10, 2015.
- 19.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genetics. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lawlor DA, Harbord RM, Sterne JA, et al. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27:1133–63. doi: 10.1002/sim.3034. [DOI] [PubMed] [Google Scholar]
- 21.Myocardial Infarction Genetics Consortium Investigators. Inactivating mutations in NPC1L1 and protection from coronary heart disease. N Engl J Med. 2014;371:2072–82. doi: 10.1056/NEJMoa1405386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cannon CP, for the IMPROVE-IT Investigators IMPROVE-IT Trial: A Comparison of Ezetimibe/Simvastatin versus Simvastatin Monotherapy on Cardiovascular Outcomes After Acute Coronary Syndromes. Paper presented at: American Heart Association Scientific Sessions; November 17, 2014; Chicago, IL. [Google Scholar]
- 23.Dadu RT, Ballantyne CM. Lipid lowering with PCSK9 inhibitors. Nat Rev Cardiol. 2014;11:563–75. doi: 10.1038/nrcardio.2014.84. [DOI] [PubMed] [Google Scholar]
- 24.Voight BF, Peloso GM, Orho-Melander M, et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomization study. Lancet. 2012;380:5572–80. doi: 10.1016/S0140-6736(12)60312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morrone D, Weintraub WS, Toth PP, et al. Lipid-altering efficacy of ezetimibe plus statin and statin monotherapy and identification of factors associated with treatment response: a pooled analysis of over 21,000 subjects from 27 clinical trials. Atherosclerosis. 2012;223:251–61. doi: 10.1016/j.atherosclerosis.2012.02.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.