

Abstract
Modified amino acids are useful synthetic components in both chemistry and biology. Here we describe a simple, scalable two-step procedure to generate α-thio aromatic acids from aromatic amino acids with yields of up to 96%. Diazotization and α-lactone mediated bromination efficiently form the α-bromo acid with retention of configuration. Thiol substitution with mild reagents such as sodium hydrosulfide or sodium trithiocarbonate provides the inverted, free α-thio acid. The mildly acidic soft nucleophile can then be utilized in many synthetic applications.
Keywords: thiol synthesis, α-thio acid, diazonium compound, α-bromo acid, aromatic amino acids
Graphical Abstract
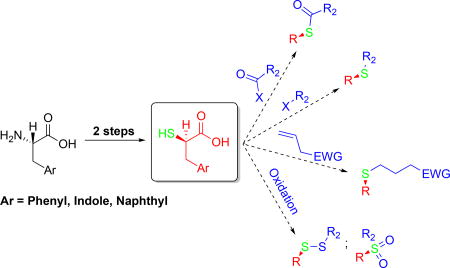
Introduction
Modified amino acids are valuable synthetic tools in numerous fields from total synthesis, chemical and synthetic biology, and medicinal chemistry. One such method of modification involves transformation of the α-amine to bromine, which can be utilized as an electrophile in a range of synthetic applications1,2. Notwithstanding, little has been reported on the direct synthesis of free α-thio acids, particularly from α-bromo acids.
In general, free thiols are useful moieties involved in an assortment of reactions from conjugate addition to polymer cross-linking. The soft, reactive nucleophile is frequently involved in click chemistry, bioconjugation, and materials chemistry3,4. Moreover, α-thio acids can be utilized in a variety of proteomic techniques beyond cysteine conjugation and even peptidomimetic chemistry. For our purposes, α-thio aromatic acids are useful backbone thioether intermediates in the synthesis of ritonavir like CYP3A4 inhibitors5.
Previous synthetic methods to generate free α-thio acids from α-bromo acids typically involve protected thiol nucleophiles, such as 1,1-diphenylmethanethiol, which must be subsequently deprotected6. Here we describe a simple and effective two-step synthesis of free α-thio aromatic acids through diazotization, bromination, and nucleophilic substitution that has been optimized accordingly.
α-Bromination of amino acids was initially reported by Izumiya and Nagamatsu, who diazotized the α-amine with sodium nitrite and brominated with KBr in the presence of H2SO47. The resulting α-bromo acids maintained retention of configuration. Since then several adjustments have been reported to generate a variety of α-bromo acids1,2,8,9. We discovered that HBr alone was a sufficient bromine source for phenylalanine, whereas KBr was required for bromination of tryptophan and 1-naphthylalanine. α-Bromo phenylalanine was easily converted into α-thio phenylalanine in the presence of aqueous sodium hydrosulfide10. However, the more nucleophilic sodium trithiocarbonate was necessary to thiolate tryptophan and 1-naphthylalanine (Scheme 1). Conveniently, both reagents were able to form the free thio acid in a one-pot manner. Due to the retentive nature of α-bromination, thiolation resulted in an inversion of configuration. Therefore, an opposite aromatic amino acid enantiomer should be utilized to produce the desired α-thio aromatic amino acid.
Scheme 1.

Synthesis of α-thio acids from aromatic amino acids
The optimized syntheses of the aforementioned α-bromo and α-thio acids were found to be not only straightforward, but also highly robust. Aside from α-bromo tryptophan and naphthylalanine, liquid-liquid extraction was sufficient to obtain the pure α-bromo or α-thio acid in excellent yields. Another advantage is that all reagents - with the exception of sodium trithiocarbonate, which needs to be freshly prepared - are commonly found in most organic chemistry laboratories.
Results and Discussion
Diazotization of aromatic amines commonly occurs through a radical SRN1 or standard SN1 fashion, with a free radical or aryl cation intermediate respectively11,12,13. However, few mechanical examples of diazotization of aliphatic amines exist. Previous studies with amino acids have shown that halide substitution of a formed alpha diazonium compound results in retention of configuration. Despite this, little has been postulated regarding the mechanism. We presume that the reaction occurs via a double inversion, with an alpha lactone intermediate (Scheme 2). The mechanism is comparable to that of γ-butyrolactone-γ-carboxylic acid synthesis from glutamic acid14,15. Following diazotization, the carboxylic acid displaces the nitrogen to form an alpha lactone with an inversion of configuration. The second inversion occurs when aqueous hydrogen halide (with or without a metal halide) protonates the lactone while haloginating the alpha carbon in a SN2 fashion. Lactone formation is presumably more stable than tertiary carbocation or alpha radical formation - as with other diazonium compounds - because the product retains stereochemistry, rather than forming a racemic mixture.
Scheme 2.

Proposed mechanism of α-halogenation of amino acids via α-lactone intermediate
D- or L-phenylalanine were the only amino acids in the series that a) effectively brominated with HBr alone, and b) substituted more efficiently with NaSH than sodium trithiocarbonate. For bromination, addition of KBr did produce the desired product, however, higher yields were obtained with salt-free, aqueous HBr9. The resulting α-bromo-acid was then treated with aqueous NaSH, heated, and acidified10 to produce the thiol with typical yields of around 75%. Interestingly, large-scale synthesis (40 mmol) of D-α-thio-phenylalanine produced the highest yield, 92% overall, denoting that the synthesis is highly efficient.
Reactions with unprotected L-tryptophan were unsuccessful, presumably due to the semi-reactive indole ring. Boc protection of the indole however, allowed for synthesis of the bromide and subsequent thiol. As reported previously, a mixture of KBr with aqueous HBr was more effective than HBr alone or with NaBr1. This was also the case for DL-1-naphthylalanine, despite the similarity to phenylalanine. Likewise, NaSH failed to produce the final thiol of tryptophan and naphthylalanine, so the more sensitive sodium trithiocarbante was prepared as described previously, and used as the thiol nucleophile16,17. The synthesis was highly robust, with yields ≥90% achieved in both small and large scale.
For tryptophan, the thiol product was expediently a mixture of Boc protected and free amine. In an effort to deprotect the Boc product in one pot, longer exposure to H2SO4 during wash was attempted. However, protected product still remained. Lingering Boc product was simply deprotected with HCl in dioxane to form the unprotected D-α-thio-tryptophan.
In summary, a simple and effective method for the production of α-thio acids from several aromatic amino acids has been developed. For other aromatic amino acids with different R-functionalities, i.e. tyrosine and histidine, the bromination and thiolation process developed for tryptophan could be applicable, but Boc protection of the histidine imidazole would be necessary. The final thiol can be used in numerous applications: as a starting material in thioether/thioester synthesis, to conjugate addition in click chemistry. Moreover, an alternate soft nucleophile at the α-carbon can be very useful in the synthesis of new peptide-like pharmaceuticals, or humbly as a modified amino acid in chemical/synthetic biology.
Supplementary Material
Highlights.
Two-step procedure to produce α-thio aromatic acids from aromatic amino acids.
Stereospecific synthesis that results in free thiols, with yields above 90%.
The free thiol can be used in numerous applications.
Acknowledgments
This work was supported by National Institute of Health grant ES025767. The authors thank A. R. Chamberlin, T. L. Poulos, and D. J. Hogenkamp for materials, discussion and advice.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Experimental details, optical rotation data, and 1H NMR, 13C NMR, and HRMS spectra are provided in the supporting information.
References
- 1.Souers AJ, Schurer S, Kwack H, Virgilio AA, Ellman JA. Preparation of enantioenriched alpha-bromo acids incorporating diverse functionality. Synthesis-Stuttgart. 1999;(4):583–585. [Google Scholar]
- 2.Moumne R, Lavielle S, Karoyan P. Efficient synthesis of beta(2)-amino acid by homologation of alpha-amino acids involving the Reformatsky reaction and Mannich-type imminium electrophile. J Org Chem. 2006;71(8):3332–3334. doi: 10.1021/jo060316a. [DOI] [PubMed] [Google Scholar]
- 3.Nair DP, Podgorski M, Chatani S, Gong T, Xi WX, Fenoli CR, Bowman CN. The Thiol-Michael Addition Click Reaction: A Powerful and Widely Used Tool in Materials Chemistry. Chem Mater. 2014;26(1):724–744. [Google Scholar]
- 4.Stenzel MH. Bioconjugation Using Thiols: Old Chemistry Rediscovered to Connect Polymers with Nature's Building Blocks. Acs Macro Lett. 2013;2(1):14–18. doi: 10.1021/mz3005814. [DOI] [PubMed] [Google Scholar]
- 5.Kaur P, Chamberlin AR, Poulos TL, Sevrioukova IF. Structure-Based Inhibitor Design for Evaluation of a CYP3A4 Pharmacophore Model. J Med Chem. 2016;59(9):4210–4220. doi: 10.1021/acs.jmedchem.5b01146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lanthier CM, MacKinnon GR, Dmitrienko GI. Inhibition of carboxypeptidase a by (S)-2-mercapto-3-phenplpropanoic acid. Chem Commun. 1997;(23):2309–2310. [Google Scholar]
- 7.Izumiya NNa. Walden Inversion of Amino Acids. VI. The Synthesis of D-Surinamine (N-Methyl-D-tyrosine) Bulletin of the Chemical Society of Japan. 1952;25(4):265–267. [Google Scholar]
- 8.Tka N, Kraiem J, Ben Hassine B. Synthesis of Enantiomerically Enriched Alpha-Bromonitriles from Amino Acids. Synthetic Commun. 2013;43(5):735–743. [Google Scholar]
- 9.Badiola E, Fiser B, Gomez-Bengoa E, Mielgo A, Olaizola I, Urruzuno I, Garcia JM, Odriozola JM, Razkin J, Oiarbide M, Palomo C. Enantioselective Construction of Tetrasubstituted Stereogenic Carbons through Bronsted Base Catalyzed Michael Reactions: alpha '-Hydroxy Enones as Key Enoate Equivalent. J Am Chem Soc. 2014;136(51):17869–17881. doi: 10.1021/ja510603w. [DOI] [PubMed] [Google Scholar]
- 10.Kudelko A. Reactions of alpha-mercaptocarboxylic acid hydrazides with triethyl orthoesters: synthesis of 1,3,4-thiadiazin-5(6H)-ones and 1,3,4-oxadiazoles. Tetrahedron. 2012;68(18):3616–3625. [Google Scholar]
- 11.Hodgson HH. The Sandmeyer Reaction. Chem Rev. 1947;40(2):251–277. doi: 10.1021/cr60126a003. [DOI] [PubMed] [Google Scholar]
- 12.Kochi JK. The Mechanism of the Sandmeyer and Meerwein Reactions. J Am Chem Soc. 1957;79(11):2942–2948. [Google Scholar]
- 13.Martinez AG, Cerero SD, Barcina JO, Jimenez FM, Maroto BL. The Mechanism of Hydrolysis of Aryldiazonium Ions Revisited: Marcus Theory vs. Canonical Variational Transition State Theory. Eur J Org Chem. 2013;2013(27):6098–6107. [Google Scholar]
- 14.Smith LR, Williams HJ. Glutamic-Acid in Pheromone Synthesis - Useful Chiral Synthon. J Chem Educ. 1979;56(10):696–698. [Google Scholar]
- 15.Gringore ORF. (S)-(+)-γ-BUTYROLACTONE-γ-CARBOXYLIC ACID. Organic Syntheses. 1990;7:99. [Google Scholar]
- 16.Martin DJ, Greco CC. A Simple Thiol Synthesis. J Org Chem. 1968;33(3):1275-&. [Google Scholar]
- 17.Kurth MJ, Tahir SH, Olmstead MM. A Thioxanone-Based Chiral Template - Asymmetric Induction in the [2,3]-Sigmatropic Rearrangement of Sulfur Ylids - Enantioselective Preparation of C-Beta-Chiral Pent-4-Enoic Acids. J Org Chem. 1990;55(8):2286–2288. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.