

Abstract
Malignant transformation of melanocytes, the pigment cells of human skin, causes formation of melanoma, a highly aggressive cancer with increased metastatic potential. Recently, mono-chemotherapies continue to improve by melanoma specific combination therapies with targeted kinase inhibitors. Still, metastatic melanoma remains a life-threatening disease because tumors exhibit primary resistance or develop resistance to novel therapies, thereby regaining tumorigenic capacity. In order to improve the therapeutic success of malignant melanoma, the determination of molecular mechanisms conferring resistance against conventional treatment approaches is necessary; however, it requires innovative cellular in vitro models. Here, we introduce an in vitro three-dimensional (3D) organotypic melanoma spheroid model that can portray the in vivo architecture of malignant melanoma and may warrant new insights into intra-tumoral as well as tumor-host interactions. The model incorporates defined numbers of mature and differentiated melanoma spheroids in a 3D human full skin reconstruction model consisting of primary skin cells. The cellular composition and differentiation status of the embedded melanoma spheroids is similar to the one of cutaneous melanoma metastasis in vivo. Using this organotypic melanoma spheroid model as a drug screening platform may support the identification of responders to selected combination therapies, while sparing the unnecessary treatment burden for non-responders, thereby increasing the benefit of therapeutic interventions.
Keywords: Cancer Research, Issue 135, Melanoma, spheroids, organotypic, full skin model, 3D melanoma screening, screening system
Introduction
The human skin is composed of two distinct compartments that serve different functions in protecting the body from adverse environmental effects1. The lower dermal compartment consists of a fibro-elastic connective tissue. It is composed of loosely connected collagen and elastin fibers synthesized by fibroblasts, serving a mechanical barrier function. The dermis is separated from the upper epidermis by the basal lamina which is produced as an extracellular matrix due to a constant communication between both skin compartments. In contrast to the dermis, the epidermis is a squamous epithelium which mainly consists of keratinocytes and can be differentiated into four layers. The stratum basale consists of undifferentiated basal keratinocytes which constantly derive from skin progenitor cells stratifying through the stages of the stratum spinosum and stratum granulosum into the stratum corneum to protect the body from dehydration and infections2. Melanocytes are aligned at the basal membrane, and communicate through dendritic extensions with multiple keratinocytes. They produce the pigment melanin to protect the skin tissue from the adverse effects of UV radiation, like skin ageing, immunosuppression, inflammation, and induction of non-melanoma skin cancer. UV radiations contribution to the transformation of melanocytes to malignant melanoma, however, is still under debate3.
Melanoma development is differentiated into different tumor progression stages, and characterized by certain genetic, morphologic, and histologic changes4. They originate either de novo or from an innate or acquired nevus due to a local increase of melanocyte proliferation causing benign neoplasia. This precursor lesion may convert into structurally modified dysplastic tissue containing atypic cells, which may continue to the first malignant stage, the radial growth phase (RGP). This early tumor progression phase is characterized by cells radially proliferating within the epidermis, showing few locally invasive cells within the papillary dermis. During the subsequent vertical growth phase (VGP) melanoma cells already show a metastatic and invasive phenotype by breaking through the basal lamina to infiltrate the deeper parts of the dermis as well as the subcutaneous tissue5. Finally, the metastatic melanoma (MM) represents the most aggressive progression stage with metastatic cells systemically spreading throughout the blood- and lymph system to invade distally organs like liver, lung, and brain6.
To date, early diagnosis followed by surgery still remains the most effective therapy of malignant melanoma. The prognosis for patients with distant metastasis, however, remains particularly poor7, because classical chemotherapy regimens confer only little survival benefit8,9. However, after decades of stagnation, recent advances in targeted therapies have considerably improved the prognosis of malignant melanoma.
Dysregulation of two major mitogen activated pathways, the RAS-RAF-MEK-ERK and the PI3K-AKT-PTEN signaling pathways, present key drivers of melanoma progression, especially when constitutively activating point mutations of the proto-oncogenes BRAFV600 and NRAS are present10. Accordingly, the invention of targeted kinase inhibitors promised therapeutic benefit for patients suffering from metastatic melanoma. A multitude of clinical trials conducted to this point has not achieved significant benefit for patients with metastatic melanoma. Nearly all responses are partial, with a subpopulation of patients showing primary resistance. Moreover, the acquisition of secondary resistance leading to relapse was observed in the majority of patients11,12.
It becomes clear that analysis of the mutation status alone is not sufficient to develop the most potent therapeutic strategy. New fast and reliable diagnostic tools are necessary to systematically record and analyze the responsiveness of individual cancer cells. The vast majority of currently available data on human melanoma have been obtained from two-dimensional (2D) melanoma cell cultures. Tumor cells, however, grown in 3D allow intercellular crosstalk between differentiated cancer cell subpopulations as well as between cancer cells and the non-transformed surrounding host tissue. Therefore, it would be best to reconstruct the 3D environment in which the melanoma developed, to be used as a preclinical screening model13,14.
Protocol
All human tissue work was performed using approved institutional protocols.
1. Separation of Dermis and Epidermis from Human Skin (Juvenile Foreskin from Circumcision)
Place the foreskin (usually 1 - 2 cm2) into a non-adhesive sterile cell culture dish (Ø 10 cm) and cover it with 10 - 12 mL of phosphate-buffered saline+ (PBS, 0.5 mM MgCl2, 0.9 mM CaCl2, pH 7.2 = PBS+).Remove all excess (adipose) tissue using sterile scalpel and forceps.
Cut the skin into 3 mm x 5 mm pieces, wash them with PBS, and transfer them to a non-adhesive sterile cell culture dish (Ø 6 cm). Cover the tissue pieces with 5 - 7 mL of dispase solution (2 U/mL in PBS). Seal the cell culture dish with paraffin film and incubate for 16 h at 4 °C.
Retract the epidermal from the dermal part with two sterile forceps. Transfer the dissected tissue pieces to individual non-adhesive cell culture dishes (Ø 6 cm) and cover each of them with PBS+.
2. Isolation of Primary Keratinocytes from the Epidermis
Carefully remove the PBS+ from the cell culture dish using a Pasteur pipette and wash the epidermal tissue pieces (section 1.3) with PBS. Cut the epidermis into smaller pieces (1 mm2) and collect them in a 50-mL conical tube. Add 10 mL 1x Trypsin-EDTA (preheated to 37 °C) and incubate for 5 min in a water bath at 37 °C. Vortex the tissue suspension every 2 min.
Stop the enzymatic reaction by adding 1 mL fetal calf serum (FCS). Resuspend the cells by pipetting up and down with a 10-mL plastic pipette for 4 min. Try to avoid bubbles and foam formation.
Pipet the cell suspension into a cell strainer (pore size 100 µm), placed on top of a fresh 50 mL conical tube, and rinse 3x with 5 mL PBS. Centrifuge the cells at 200 x g for 5 min. Resuspend the cell pellet in 2 - 6 mL culture medium containing 1% (v/v) gentamycin. Determine the cell number manually by counting cells in a hemocytometer and seed 6 x 105 cells into a T75 cell culture flask.
3. Cultivation of Primary Keratinocytes
Incubate primary keratinocytes isolated in step 2.3 in a cell incubator at 37 °C and 5% CO2 in keratinocyte medium without FCS, to 50 - 70% confluency.
To passage the keratinocytes carefully remove the medium, add 5 mL PBS-EDTA, and incubate the cells for 10 min at 37 °C. Check if cells have started to detach from the culture flask under a light microscope (4X magnification: cells will appear rounded but are still attached!). If not, replace old PBS-EDTA with fresh PBS-EDTA and re-incubate for another 10 min at 37 °C.
Add 5 mL 1x Trypsin-EDTA to the rounded cells still covered with 10 mL PBS-EDTA and re-incubate them for 1 - 3 min at 37 °C. Stop the enzymatic reaction by adding 1 mL FCS and collect the detached cells in a 50-mL conical tube. Centrifuge the cells at 200 x g for 5 min, resuspend the pellet in keratinocyte medium, and seed 6 x 105 cells into a fresh T75 cell culture flask. NOTE: To generate organotypic skin reconstructs, the primary keratinocytes should be used no later than passage 3 - 4.
4. Isolation of Primary Fibroblasts from the Dermis
Cut the dermal tissue (section 1.3) into smaller pieces (1 mm2) and transfer them into a 50-mL conical tube. Add 10 mL collagenase-solution (5 U/mL in PBS+) and incubate for 45 min in a water bath at 37 °C. Centrifuge the mixture of tissue pieces and cells at 200 x g for 5 min.
Wash the cell pellet twice with 10 mL DMEM containing 4.5 g/L glucose and L-Glutamine, but without L-Pyruvate. Finally resuspend the tissue pieces in 2 mL of DMEM containing 1% (v/v) gentamycin and 10% FCS. Transfer them into a T25 cell culture flask and incubate them in a cell incubator at 37 °C and 5% CO2 overnight. NOTE: The small volume allows the fibroblasts to migrate out of the tissue-debris and forces them to attach to the culture flask.
The next morning, add another 6 mL DMEM/Gentamycin/FCS and incubate for 2 - 3 days at 37 °C until the cells have reached 80 - 90% confluency.
5. Cultivation of Primary Fibroblasts
To passage the fibroblasts, remove the medium and wash adherent cells with PBS. Add 5 mL 1x Trypsin-EDTA and incubate for 3 min at 37 °C.
Stop the enzymatic reaction by adding 5 mL DMEM/FCS and collect the detached cells in a 50-mL conical tube. Centrifuge the cells at 200 x g for 5 min, resuspend the pellet in DMEM medium, and seed 6 x 105 cells into a fresh T75 cell culture flask. NOTE: To generate organotypic skin reconstructs, the primary fibroblasts should be used no later than passage 4 - 6.
6. Generation of 3D Melanoma Spheroids Via the Hanging Drop Method
- Culture melanoma cells (e.g., 451-LU, or any melanoma cell line of interest) according to general protocols, using RPMI containing 10% FCS15.
- To generate melanoma spheroids of similar size and quality, wash the melanoma cells in PBS, add 5 mL of 1x Trypsin-EDTA in PBS to the cells in a T175 cell culture flask, and incubate for 3 - 5 min at room temperature (RT). Neutralize the Trypsin by adding 5 mL RPMI/10% FCS. Harvest the cells by centrifugation at 200 x g for 5 min. Re-suspend the cell pellet in RPMI/FCS at a final concentration of 10,000 cells/mL as determined by counting in a hemocytometer.
- Spot 40 x 25 µL (= 250 cells) of the cell suspension onto the inner surface of the lid of a sterile non-adhesive cell culture dish (Ø 10 cm) using an electronic multi-pipette. With a fast but smooth movement, turn the lid around and place it on the respective cell culture dish containing 5 mL PBS.
- Culture "hanging drop dishes" in a cell incubator at 37 °C and 5% CO2 for 10 - 14 days, depending on the cell type used. NOTE: Cells from the MM growth phase typically grow faster and form more solid spheroids in the hanging drop compared to melanoma cells derived from the RGP or VGP. For individual evaluation, observe spheroid growth under a pair of binoculars or under a light microscope (4X magnification). Usually, spheroids become detectable after 48 h under a pair of binoculars or under a light microscope (4X magnification). After 10 - 14 days, they are visible without any magnification device.
5 days after the initial drop spotting, add 10 µL of fresh RPMI/10% FCS medium to each drop. Subsequently, exchange 10 µL of medium every other day. The use of an electronic dispenser is very helpful in this step.
Depending on the cell type, harvest the spheroids (see section 10 for details) after 10 - 15 days by gently rinsing them off the cell culture dish lid with PBS. Collect them in a fresh non-adhesive cell culture dish. NOTE: The cultivation period depends on the tumor growth phase of the melanoma cells when they were initially derived, e.g., spheroids derived from 451-LU cells grow to approximately 500 µm in diameter within 12 days of culturing in the hanging drop15.
7. Generation of the Dermal Compartment of Organotypic Full Skin Reconstructs
Generate skin models using 24-well cell microporous membrane inserts (pore size 8 µm) placed in 24-well plates. NOTE: Make sure not to use hanging but standing inserts, because they will be placed into a 6-well plate for air-liquid cultivation at a later stage.
Prepare the gel neutralization solution (GNL)15, as well as the culture media referred to as MM and endothelial growth media (EGM)16,17. To prevent coagulation, store the collagen type I (usually from rat tail, 3.5 - 4 mg/mL in 0.02 N acetic acid) on ice until use, because it starts to gel at RT.
Re-suspend 1 x 105 fibroblasts per insert in GNL and quickly mix the cell suspension with collagen at a ratio of 1:3 in a final volume of 500 µL/insert. Mix by gently pipetting up and down to avoid bubble formation as bubbles may impair the quality of the skin reconstruct.
Allow the individual dermal gels to settle by keeping them without medium at RT for 30 min in a sterile hood. Subsequently cover each gel with DMEM containing 4.5 g/L glucose, 1% L-glutamine, 10% FCS, and without L-pyruvate, and incubate overnight at 37 °C.
8. Generation of the Epidermal Compartment of Organotypic Full Skin Reconstructs
The next day remove the medium from the dermal gels and equilibrate them with EGM media (10% FCS, 1% PenStrep, 10 mg/mL gentamicin) for 2 h at 37 °C. Withdraw the medium and carefully seed 1 x 105 keratinocytes resuspended in 100 µL EGM on top of the dermal gel.
Incubate the reconstructs for 1.5 h at 37 °C to allow the keratinocytes to adhere to the dermal compartment. NOTE: During this incubation time, the gels will start to shrink due to fibroblast-induced contraction, and thus, at least partially detach from the insert walls.
Cover the skin equivalents with approximately 800 µL EGM and carefully remove the residual gel from the insert wall with a small (white) pipette tip.Culture the skin equivalents submerged in EGM for 7 days in a cell incubator at 37 °C, 5% CO2, and change the medium every other day. NOTE: During this time the skin equivalents will shrink significantly.
9. Air-liquid Cultivation of Organotypic Full Skin Reconstructs
At day 8, transfer each insert into an individual well of a 6-well plate. Only add 1.2 - 1.4 mL of MM medium to each well, so that the skin reconstruct is supplied with medium from the bottom of the well but is not covered with medium. NOTE: Cultivation at the air-liquid interface allows stratification of the epidermal part and establishment of a full cornified layer (stratum corneum).
During the next 10 - 17 days, change the medium as stated in section 9.1 every other day. During this period, add drugs or other stimuli as needed to the medium. Carefully remove the full skin reconstruct from the microporous membrane insert using curved tweezers for further analysis, e.g., immunohistochemical analysis (Figure 1).
10. Generation of the Organotypic Melanoma Spheroid Skin Models
To integrate the melanoma spheroids into the dermal compartment of the organotypic full skin reconstructs, carefully rinse the spheroids (after step 6.4) off the lid of the hanging drop cell culture dish with PBS. Collect 10 - 20 spheroids/insert in a sterile non-adhesive cell culture dish. Carefully remove excessive PBS with a Pasteur pipette.
Aspirate the spheroids in the smallest volume possible of EGM. At this point observe and count the spheroids with the naked eye, without any further magnification device. Take 10 - 20 spheroids per skin model/gel, as stated in step 10.1.
Transfer them to the desired volume of GNL containing fibroblasts (during step 7.3), and mix with the collagen type I. From this step on proceed as described in sections 7.3 - 9.1. NOTE: Tumor spheroids become visible in the dermal gel as white spots (Figure 2)

Representative Results
Successful treatment of melanoma metastasis can be influenced by the cross-talk between tumor cells as well as between tumor and non-transformed host cells. The purpose of developing organotypic models of cancer in vitro is to provide suitable preclinical test systems that recapitulate the 3D organization and complexity of human melanoma in vivo. This allows the study of the therapeutic impact on the tumor within an organotypic environment and the adverse effects on the surrounding primary tissue in parallel.
To develop the best organotypic skin models, the quality of the primary cells is crucial. It is advantageous to use juvenile primary fibroblasts and keratinocytes, because they are typically less differentiated compared to adult primary skin cells. Juvenile skin cells can either be isolated from juvenile foreskin as described in the protocol sections 1 - 5, but can also be purchased from companies as pre-natal primary fibroblasts and keratinocytes. If purchasing, it is necessary to order cells from different donors to avoid donor-specific results, e.g., for drug sensitivity. The whole protocol is displayed as a scheme in Figure 3.
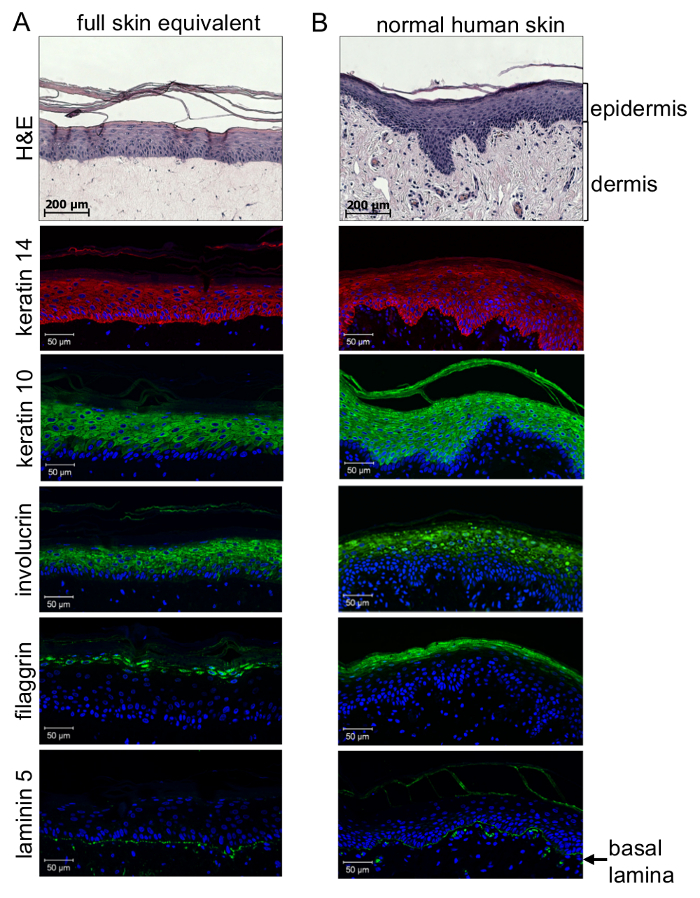
Quality control of 3D full-skin equivalents requires immunohistochemical analysis. A first impression can be obtained by Hematoxylin-Eosin (H&E) staining of paraffin embedded sections (3 µm). Detailed analysis of the quality of epidermal differentiation and formation of the basal lamina between the dermis and epidermis requires immunohistochemical analysis using specific antibodies against an epidermal stratification marker. This allows distinguishing between undifferentiated, highly proliferative cells located close to the basal membrane and highly differentiated and keratinized cells at the stratum corneum through the formation of distinct epidermal layers in between. As shown by immune-histological staining(Figure 4), differentiation of keratinocytes throughout the epidermis could be achieved similar to normal skin: mainly the undifferentiated cells from the lower epidermal layers (stratum basale and stratum spinosum) stain positive for keratin 14, while the more differentiated cells from the supra-basal layers (stratum granulosum and stratum corneum) stain positive for keratin 10 and involucrin. Accordingly, filaggrin staining could only be observed in highly differentiated cells of the stratum corneum. Most importantly, laminin 5 staining reveals that a basal lamina was generated to physiologically connect the epidermal to the dermal compartment of the artificial skin reconstruct. This proves that a communicating organotypic microenvironment has been generated to host melanoma cells or spheroids for physiologic and pathophysiologic analysis.
For the purpose of melanoma drug screening, single melanoma cells can also be integrated into the dermis of full skin equivalents to allow de novo melanoma nest formation18,19. Therefore, melanoma cells are combined with primary fibroblasts at a ratio of 5:1, centrifuged together at 200 x g for 5 min and resuspended in GNL prior to mixing with collagen. As a result, the melanoma cell nests will spontaneously form in the dermal compartment. According to our experience, only cells of the metastatic growth phase form proper nests, compared to melanoma cells of the RGP or VGP15. One major drawback of these types of models is the fact that the number and size of melanoma nests formed cannot be predicted, and may vary between individual skin reconstructs, independently of any treatment. For example, 1,000 cells seeded into the dermal compartment may gain 10 nests consisting of 100 cells or 100 nests consisting of 10 cells each (Figure 5). These biologic variables present with three deficiencies: first, the number and size of melanoma nests formed are unpredictable; second, the metastases in vivo are usually larger than melanoma nests and exhibit a more complex intra-tumoral diversity; and third, due to the limited life-span of tumor-nest models, treatment is initiated early, and consequently rather inhibits tumor outgrowth instead of causing regression of existing tumor nests.
To overcome these limitations organotypic melanoma spheroid skin models can be generated. By culturing 250 metastatic melanoma cells in a hanging drop for 14 days20, spheroids are reproducibly generated consisting of viable melanoma cells presenting a compact structure with a final diameter of approximately 500 µm mimicking non vascularized tumor nodes, micro-metastasis, or inter-capillary micro regions of solid tumors21,22. In general, any melanoma cell line is suitable for the generation of spheroids via the hanging drop method; however, cells derived from more advanced metastatic tumor stages form more solid spheroids compared to cell lines derived from early progression stages, e.g., the RGP.
For some cells, it is advantageous for proper spheroid formation to enhance the viscosity of the hanging drop culture medium. This can be achieved by the addition of 10 - 50% methyl-cellulose to the culture medium. For the methyl-cellulose stock solution, autoclave 1.2 g methyl-cellulose together with a magnetic stir bar in a 100 mL glass bottle. Add 100 mL preheated (60 °C) medium and stir for 20 min at room temperature, and another 1 - 2 h at 4 °C. Centrifuge the stock solution for 2 h at 5,000 x g and store the viscous supernatant at 4 °C until use.
Proper validation of full skin melanoma spheroid models is provided by the fact that a defined number of melanoma spheroids can - at least statistically - be integrated into the dermal fibroblast/collagen I scaffold at day 1 of the skin model construction, allowing them to co-develop during epidermal differentiation for 25 - 27 more days. The yield of spheroids can be analyzed directly after seeding, because spheroids appear as white spots within the transparent dermal gel and can be seen without any magnification device (Figure 2). As a result, a 3D skin model is generated that harbors mature melanoma spheroids, which had been cultured in vitro for a total of approximately 42 days, showing the highest level of intra-tumoral cell differentiation15.
H&E staining of the skin melanoma spheroid model reveals the histological appearance and cellular distribution of melanoma spheroids to be very similar to the one of non-vascularized human melanoma skin metastases in vivo15 (Figure 6). Two subpopulations of melanoma cells are clearly distinguishable under these conditions: a peripheral proliferating subpopulation and a central subpopulation mainly consisting of shrunken, apoptotic or necrotic cells, forming the so-called "necrotic" center. Immunohistochemically living and proliferating subpopulations can be detected using antibodies against the proliferation marker KI-67, whereas cells of the necrotic center can be visualized by TUNEL-staining15. This particular distribution of tumor cell subpopulations is warranted by the spheroid size (≥500 µm), resulting from a lack of nutrients and oxygen in the central part where catabolic waste accumulates. Following the protocol provided here will allow the generation of a reliable and reproducible organotypic human full-thickness skin model with embedded human melanoma spheroids that mimic human melanoma skin metastasis. Applications of this model include drug testing, screening of toxins, influence of cosmetic compounds or laser therapy on melanoma outgrowth, and treatment.
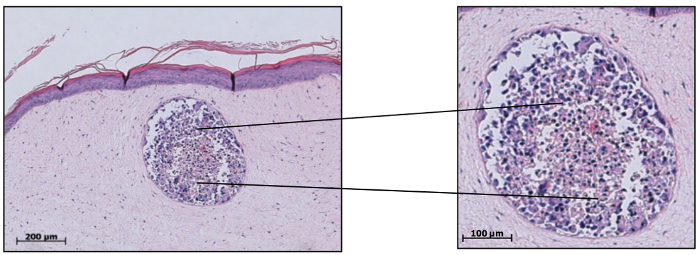
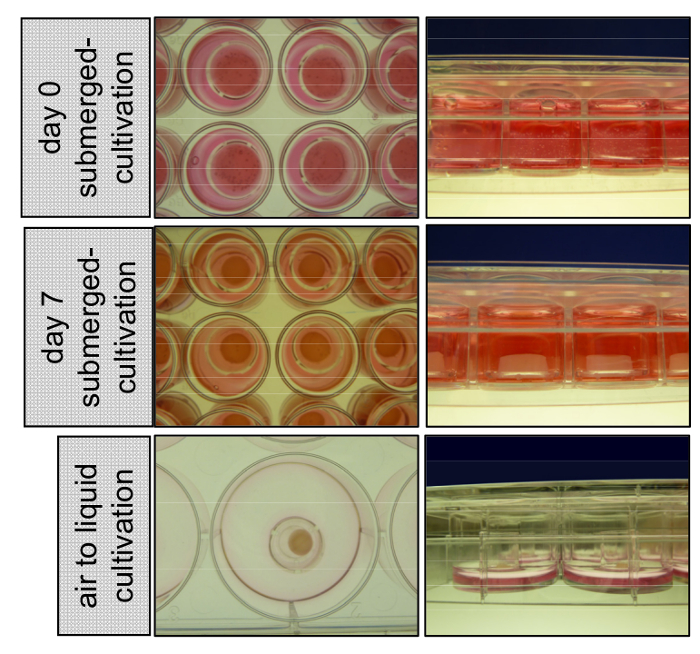
Figure 1: Cultivation of 3D organotypic skin reconstructs submerged with medium and at the air-liquid interface. At day 0 primary keratinocytes are seeded on top of the dermal compartment consisting of primary fibroblasts embedded into a collagen type I matrix. 3D skin reconstructs stay cultivated submerged with EGM for 7 days, detach from the insert wall, and start shrinking. At day 8 the inserts are transferred to 6-wells and cultivated at the air-liquid interface to allow epidermal stratification. Please click here to view a larger version of this figure.
Figure 2: Melanoma spheroids embedded into the dermal compartment appear as white spots. While preparing the dermal compartment of the 3D skin reconstruct, a defined number of melanoma spheroids can be added to the fibroblast collagen type I mix. Once the dermal gel has settled, melanoma spheroids become visible as white spots.
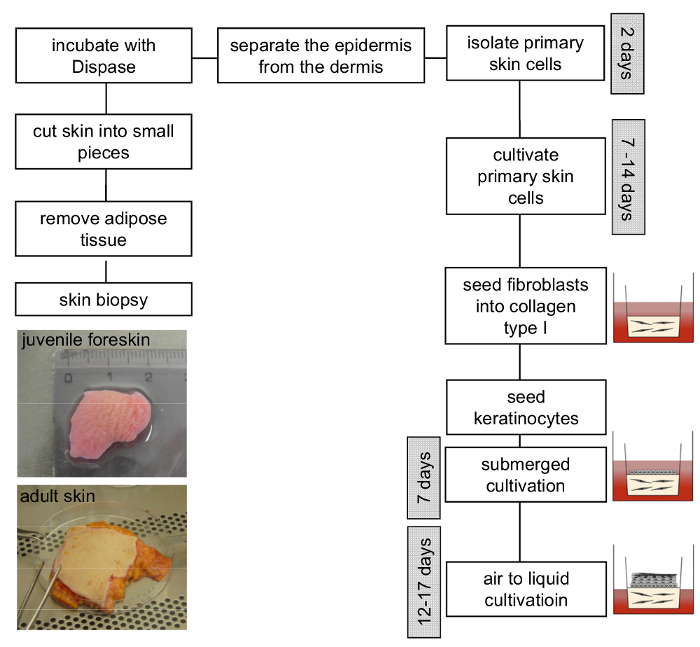
Figure 3: Scheme of 3D organotypic skin model construction. Remove adipose tissue from the skin sample and cut it into smaller pieces. Incubation with dispase solution overnight at 4 °C facilitates the separation of the epidermis from the dermis. Isolated primary fibroblasts and keratinocytes should be cultivated separately and used between passage 4 - 6 and 3 - 4, respectively. Subsequently, the generation of the 3D skin model can proceed as described in the protocol. Please click here to view a larger version of this figure.
Figure 4: 3D organotypic skin reconstructs show a differentiation level similar to normal human skin. Paraffin sections of skin equivalents (A) compared to normal human skin (B) were stained for the expression of keratins 14 (red: λex 554 nm; λem 568 nm) and 10, involucrin (green: λex 490 nm; λem 525 nm), filaggrin (green: λex 490 nm; λem 525 nm), and laminin 5 (green: λex 490 nm; λem 525 nm), and analyzed with a confocal fluorescence microscope. Cell nuclei were visualized by DAPI staining (blue: λex 340 nm; λem 488 nm). Immunohistochemical examination of the 3D full-thickness of skin equivalents revealed proper epidermal stratification forming distinct layers of the epidermis as seen in normal human skin. While cells from the lower epidermal layers stained positive for keratin 14, the more differentiated cells from the supra-basal layer showed keratin 10 and involucrin staining. Highly differentiated cells close to the stratum corneum expressed filaggrin. Laminin 5 staining shows that a basal lamina is generated to physiologically connect the epidermal to the dermal compartment (lowest panel). This figure has been taken from Voersmann et al.15 with permission. Please click here to view a larger version of this figure.
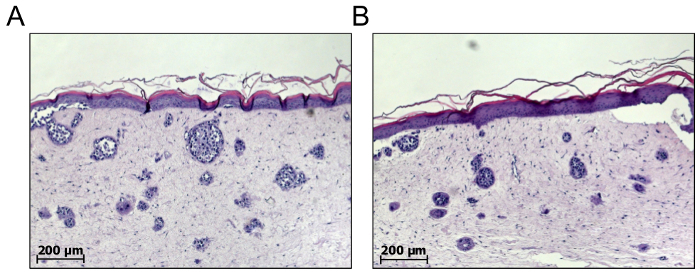
Figure 5: The number and size of spontaneously formed melanoma nests cannot be predicted. De novo melanoma nest formation in the dermal compartment of full skin equivalents can be achieved by mixing a defined number of melanoma cells with primary fibroblasts to embed both cell types in the collagen type I matrix. The number and size of spontaneously formed melanoma nests can only be analyzed from a mature 3D skin reconstruct after about 21 days. As depicted from two samples (A) and (B), the numbers and sizes of the melanoma nests may vary between individual skin reconstructs. As a consequence, it is difficult to validate these models and to predict the therapeutic impact. Please click here to view a larger version of this figure.
Figure 6: Melanoma spheroids integrated into skin equivalents recapitulate key features of human cutaneous melanoma metastasis. H&E stained paraffin sections of tumor spheroids embedded into skin equivalents revealed spheroids to share key features with non-vascularized human cutaneous melanoma metastases in vivo. Two subpopulations of cells are clearly discernible: a peripheral living subpopulation and central subpopulation mainly consisting of shrunken, apoptotic or necrotic cells, forming the "necrotic" center. This distribution of tumor cell subpopulations is guaranteed by the spheroid size. This figure has been modified from Voersmann et al.15 with permission. Please click here to view a larger version of this figure.
Discussion
The organotypic melanoma spheroid skin model introduced here warrants new insights for a deeper understanding of intra-tumoral and tumor-host interaction, and may provide an advanced screening platform to study molecular mechanisms of tumor development and therapy resistance in the future.
To guarantee the best physiologic and in vivo mimicking conditions of skin reconstructs, the quality of the primary cells is of utmost importance. As stated above, juvenile or pre-natal skin cells show the lowest differentiation grade and are therefore best suitable to generate full-thickness skin equivalents. The first possible quality control is the extent of dermal contraction at day 2 after seeding the keratinocytes and equilibrating the gel to EGM (Protocol sections 8.2 and 8.3). Dermal gels will shrink the most with the best quality of fibroblasts. Also, the integration of too few or too many fibroblasts may impair dermal contraction and consequently attachment of the epidermal to the dermal compartment. This in turn will compromise epidermal differentiation, because this process requires an extensive cross-talk between dermal and epidermal cells.
The quality of primary cells also critically depends on the cell confluency as well as the passage of the cells. If keratinocytes are grown to ≥80% confluency they immediately stop proliferating and start to differentiate. It is therefore important to culture them to 40 - 70% confluency during passaging and use them no later than passage 3 - 4. Fibroblasts are less sensitive but should not be used later than passage 4 - 6. Please also note that all primary cells and cell lines used to generate organotypic melanoma spheroid skin models are cultured free of antibiotics, and therefore the contamination risk is high. However, the addition of antibiotics changes the physiology of the individual cells and therefore reduces the quality of the skin equivalents.
In general, tumor spheroid formation is possible from almost all kinds of tumor cell lines and also from tumor cells freshly isolated from patient material. The initial cell number and/or cultivation time for gaining optimal spheroid sizes may vary depending on the cell type used. Up to a size of 150 - 200 µm, all cells included in a spheroid can still be sufficiently supplied with nutrients via simple diffusion. Only spheroids with sizes ≥500 µm show a high degree of cellular differentiation and represent typical features of non-vascularized tumor tissue22.
The organotypic melanoma-spheroid-skin-model developed here is particularly suitable to study tumor-host interactions and for melanoma drug testing under in vivo-like conditions15. Still, the environment of melanoma in vivo is even more complex, harboring a variety of tumor associated cell types, including immune cells and endothelial cells. Since the addition of proper primary immune cells faces problems with histocompatibility, these models are not yet able to adequately monitor immune-therapeutic approaches.
Nevertheless, melanoma spheroids can be generated from freshly isolated patient material and once included into the full skin reconstruct, may serve as individual drug screening platforms. Even the integration of pieces of melanoma metastasis into skin reconstructs is conceivable to test drug combinations in a tailored therapeutic approach. Including the organotypic 3D skin-melanoma model in preclinical testing is likely to help ensure that only the most promising novel therapeutic concepts are taken forward into clinical testing, thus reducing the attrition rate of potential new treatments for this disease and increasing the rate of therapeutic success in clinical trials.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
The authors thank Hanna Voersmann for establishing the 3D organotypic melanoma-spheroid-skin-model and providing excellent protocols. The authors also thank Silke Busch for valuable technical support. The work was supported by BMBF e:Med program "Melanoma Sensitivity" 031A423A.
References
- Boulais N, Misery L. The epidermis: a sensory tissue. Eur. J. Dermatol. 2008;18(2):119–127. doi: 10.1684/ejd.2008.0348. [DOI] [PubMed] [Google Scholar]
- Blanpain C, Fuchs E. Epidermal homeostasis: a balancing act of stem cells in the skin. Nat.Rev. Mol. Cell Biol. 2009;10(3):207–217. doi: 10.1038/nrm2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shain AH, Bastian BC. From melanocytes to melanomas. Nat. Rev. Cancer. 2016;16(6):345–358. doi: 10.1038/nrc.2016.37. [DOI] [PubMed] [Google Scholar]
- Clark WH., Jr Human cutaneous malignant melanoma as a model for cancer. Cancer Metastasis Rev. 1991;10(2):83–88. doi: 10.1007/BF00049406. [DOI] [PubMed] [Google Scholar]
- Hsu MY, Meier F, Herlyn M. Melanoma development and progression: a conspiracy between tumor and host. Differentiation. 2002;70(9-10):522–536. doi: 10.1046/j.1432-0436.2002.700906.x. [DOI] [PubMed] [Google Scholar]
- Miller AJ, Mihm MC., Jr Melanoma. N. Engl. J. Med. 2006;355(1):51–65. doi: 10.1056/NEJMra052166. [DOI] [PubMed] [Google Scholar]
- Balch CM, et al. Final version of 2009 AJCC melanoma staging and classification. J. Clin. Oncol. 2009;27(36):6199–6206. doi: 10.1200/JCO.2009.23.4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eigentler TK, Caroli UM, Radny P, Garbe C. Palliative therapy of disseminated malignant melanoma: a systematic review of 41 randomised clinical trials. Lancet Oncol. 2003;4(12):748–759. doi: 10.1016/s1470-2045(03)01280-4. [DOI] [PubMed] [Google Scholar]
- Kim T, Amaria RN, Spencer C, Reuben A, Cooper ZA, Wargo JA. Combining targeted therapy and immune checkpoint inhibitors in the treatment of metastatic melanoma. Cancer Biol. Med. 2014;11(4):237–246. doi: 10.7497/j.issn.2095-3941.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499(7457):214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty KT, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010;363(9):809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauschenberg R, Garzarolli M, Dietrich U, Beissert S, Meier F. Systemic therapy of metastatic melanoma. J. Dtsch. Dermatol. Ges. 2015;13(12):1223–1235. doi: 10.1111/ddg.12891. [DOI] [PubMed] [Google Scholar]
- Kunz-Schughart LA, Freyer JP, Hofstaedter F, Ebner R. The use of 3-D cultures for high-throughput screening: the multicellular spheroid model. J. Biomol. Screen. 2004;9(4):273–285. doi: 10.1177/1087057104265040. [DOI] [PubMed] [Google Scholar]
- Pampaloni F, Reynaud EG, Stelzer EH. The third dimension bridges the gap between cell culture and live tissue. Nat. Rev. Mol. Cell Biol. 2007;8(10):839–845. doi: 10.1038/nrm2236. [DOI] [PubMed] [Google Scholar]
- Vorsmann H, Groeber F, Walles H, Busch S, Beissert S, Walczak H, Kulms D. Development of a human three-dimensional organotypic skin-melanoma spheroid model for in vitro drug testing. Cell Death. Dis. 2013;4:e719. doi: 10.1038/cddis.2013.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CS, Lavker RM, Rodeck U, Risse B, Jensen PJ. Use of a serum-free epidermal culture model to show deleterious effects of epidermal growth factor on morphogenesis and differentiation. J. Invest. Dermatol. 1995;104(1):107–112. doi: 10.1111/1523-1747.ep12613595. [DOI] [PubMed] [Google Scholar]
- Meier F, et al. Human melanoma progression in skin reconstructs : biological significance of bFGF. Am. J. Pathol. 2000;156(1):193–200. doi: 10.1016/S0002-9440(10)64719-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnberg T, et al. Inhibition of PI3K-AKT-mTOR signaling sensitizes melanoma cells to cisplatin and temozolomide. J. Invest. Dermatol. 2009;29(6):1500–1515. doi: 10.1038/jid.2008.379. [DOI] [PubMed] [Google Scholar]
- Niessner H, et al. The farnesyl transferase inhibitor lonafarnib inhibits mTOR signaling and enforces sorafenib-induced apoptosis in melanoma cells. J. Invest. Dermatol. 2011;131(2):468–479. doi: 10.1038/jid.2010.297. [DOI] [PubMed] [Google Scholar]
- Foty RA. A simple hanging drop cell culture protocol for generation of 3D spheroids. J. Vis. Exp. 2011. p. e2720. [DOI] [PMC free article] [PubMed]
- Hirschhaeuser F, Menne H, Dittfeld C, West J, Mueller-Klieser W, Kunz-Schughart LA. Multicellular tumor spheroids: an underestimated tool is catching up again. J. Biotechnol. 2010;148(1):3–15. doi: 10.1016/j.jbiotec.2010.01.012. [DOI] [PubMed] [Google Scholar]
- Lin RZ, Chang HY. Recent advances in three-dimensional multicellular spheroid culture for biomedical research. Biotechnol. J. 2008;3(9-10):1172–1184. doi: 10.1002/biot.200700228. [DOI] [PubMed] [Google Scholar]