

Abstract
Immunofluorescent staining is currently the method of choice for determination of protein expression levels in cell-culture systems when morphological information is also necessary. The protocol of immunocytochemical staining on paraffin-embedded cell blocks, presented herein, is an excellent alternative to immunofluorescent staining on non-paraffin-embedded fixed cells. In this protocol, a paraffin cell block from HeLa cells was prepared using the thromboplastin-plasma method, and immunocytochemistry was performed for the evaluation of two proliferation markers, CKAP2 and Ki-67. The nuclei and cytoplasmic morphology of the HeLa cells were well preserved in the cell-block slides. At the same time, the CKAP2 and Ki-67 staining patterns in the immunocytochemistry were quite similar to those in immunohistochemical staining in paraffin cancer tissues. With modified cell-culture conditions, including pre-incubation of HeLa cells under serum-free conditions, the effect could be evaluated while preserving architectural information. In conclusion, immunocytochemistry on paraffin-embedded cell blocks is an excellent alternative to immunofluorescent staining.
Keywords: Cancer Research, Issue 135, Immunocytochemistry, immunofluorescent staining, cell block, paraffin embedding, paraffin block, thromboplastin-plasma, Cytoskeleton-Associated Protein 2, KI-67
Introduction
In most laboratories, paraffin-embedded cell blocks are not commonly used. Rather, fixed cultured cells, not paraffin-embedded cells, are employed in subcellular localization studies. For those fixed cultured cells, fluorescence instead of chromogen has been used. Therefore, immunofluorescent staining is currently the method of choice for determination of protein expression levels in research employing cell cultures1. Slides prepared for immunofluorescent staining, however, can be observed only under immunofluorescent microscopy, which might show images quite different from those indicated under plane microscopy2. Additionally, preservation of slides for immunofluorescent staining requires protection from bright light, and fluorescent signals become weaker with repeated exposure for imaging due to the loss of the fluorescent signal3. Results from immunocytochemistry on paraffin-embedded cell blocks are quite similar to those from immunohistochemistry on paraffin-embedded tissues4, and they can easily be translated into clinical information. Therefore, immunocytochemistry can be an excellent alternative. However, cell-block preparation has not been popular in basic research laboratories. In this protocol, then, cell-block preparation and immunocytochemical staining are provided to promote the use of this method in the field of cell-culture studies.
Cell-block preparation and immunocytochemistry are not unique methods, and they have already been applied from clinical diagnosis to basic research4,5. Although various cell-block preparation methods have been reported4,6 the thromboplastin-plasma method is simple, cost-effective, and readily adaptable. Therefore, in the protocol presented in this paper, the thromboplastin-plasma method4,5,6,7,8 is used for preparation of paraffin-embedded cell blocks.
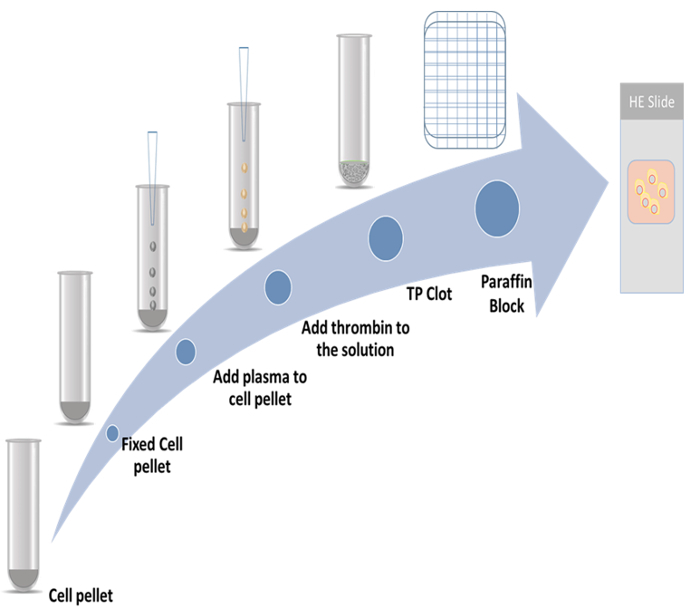
In the present study, the detailed procedures for the preparation of thromboplastin-plasma cell blocks and the immunocytochemistry method employing two proliferation markers were demonstrated. One marker is cytoskeleton-associated protein 2 (CKAP2), which has recently been reported as a mitotic marker9,10,11; the other one is Ki-67, which is the best-known proliferation marker12. The representative scheme is shown in Figure 1.
Protocol
The study protocol was approved by the Institutional Review Board of National Cancer Center, Korea (NCCNCS-12-630).
1. Sample Preparation (30 min)
For the culture of HeLa cells (CCL-2, ATCC), use 10 mL complete DMEM medium (10% fetal bovine serum, 1% pen-strep) in a 100 mm culture dish. To prepare serum-starved HeLa cells, incubate confluent HeLa cells in DMEM without fetal bovine serum for 48 hours, as described in a previous study11.
To detach the cells, wash them with 2 mL phosphate-buffered saline (PBS), and add 2 mL 0.25% EDTA trypsin per 100 mm culture dish. Then, incubate for 2 - 3 min in a CO2 incubator at 37 °C.
Add 5 mL of complete DMEM medium to the culture dish to deactivate trypsin, and transfer the detached cells to a 15 mL tube. Then, centrifuge the cells at 300 x g for 10 min to form a pellet.
Remove the supernatant, and wash the pellet twice with 2 mL cold PBS by centrifugation at 300 x g for 10 min.
After removal of the supernatant, add 1 mL of 95% ethanol to the cell pellet, and mix the cell pellet with ethanol by vortexing. Keep the fixed cells on ice until the cell-block preparation.
2. Cell-block Preparation (1 h 30 min)
For the preparation of frozen plasma aliquots, collect EDTA plasma from healthy donor blood and centrifuge at 13,000 x g for 10 min. Collect the supernatant plasma, and aliquot 200 - 400 µL each into microfuge tubes. Store the aliquots at -80 °C until use. When the frozen plasma aliquots are ready, retrieve them and thaw the plasma by incubation at 37 °C for 5 min.
Centrifuge the fixed cells at 700 x g for 10 min, then discard the supernatant.
Mix the cell pellet with 2 drops (about 200 µL) of the plasma, 2 drops of thromboplastin, and 2 drops of 0.025 M calcium chloride. Then, mix them by pipetting.
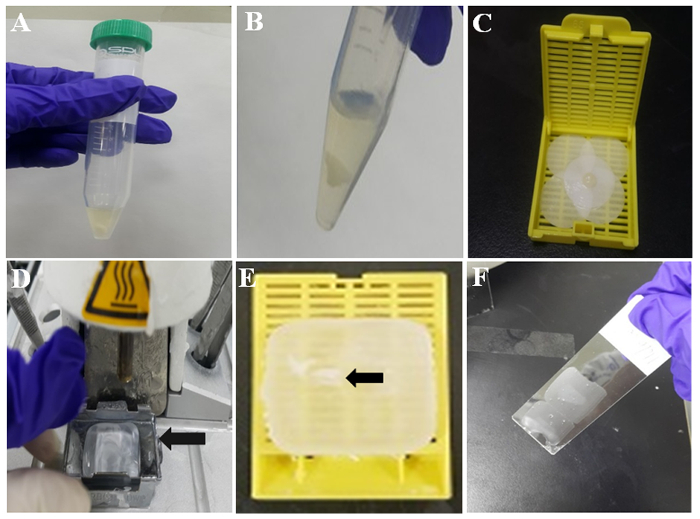
Allow the mixture to form a cell clot at room temperature for 10 min, as shown in Figure 2A. Then, wash the cell clot with PBS two times by pouring and removing PBS by pipette. The representative white cell clot is shown in Figure 2B.
Moisten a piece of filter paper with formalin, and wrap the cell clot with the pre-moistened filter paper. Then, place the clotted mixture with a pincette in a tissue cassette as shown in Figure 2C.
Place the tissue cassette in a glass jar containing 50 mL of buffered formalin (10% formalin in PBS) overnight at 4 °C for formalin fixation.
3. Tissue Processing and Paraffin Embedding (Overnight)
Load the tissue cassette containing the formalin-fixed cell clot in a tissue processor overnight as described previously11. The tissue processing is for water removal and conditioning of fixed cells prior to paraffin embedding. For research without tissue processing, perform the tissue processing procedure as shown in Table 1.
Turn on the heated embedding station, and 60 minutes later, check that there is molten paraffin in the metal mold therein (Figure 2D).
Take the formed cell clot from the tissue cassette and place it in the molten paraffin within the metal mold.
Place the new tissue cassette without the lid into the metal mold containing the cell clot in the middle of the molten paraffin, and pour more molten paraffin into the tissue cassette and onto the metal mold. Then, let the paraffin solidify in the cold plate for 30 - 60 seconds.
Separate the tissue cassette from the metal mold. Then, the paraffin cell block is ready for immunocytochemistry. The embedded cell clot in the paraffin-embedded cell block is indicated by the arrow in Figure 2E.
4. Preparation of Slides for Immunocytochemistry (1 h)
Check the area for the cell clot in the paraffin cell block, and cut the cell block into slices with thicknesses of 3 - 4 µm using a microtome. Put the paraffin sections on silane-coated glass slides, as shown in Figure 2F.
Place the section slides in a 37 °C oven for 30 min to make the sections adhere to the slides.
5. Immunocytochemistry of Cell Blocks (6 h)
NOTE: For immunocytochemical staining on cell-block sections, various kits can be used (see Table of Materials). All such kits have various sensitivities and specificities depending on their modifications.
Incubate the sections in 50 mL of xylene for 4 min to de-paraffinize.
Incubate the slides in 100% ethanol for 2 min for dehydration, and two times in 95% ethanol, and in 80% ethanol for 2 min. Then, remove the ethanol in running water for 10 min.
For antigen retrieval, place the slides in a jar containing 40 mL Tris-EDTA retrieval buffer, pH 9.0, and boil in a cooker for 30 min.
Wash the slides under running water, and incubate them in 95% ethanol for 10 min at 4 °C. Then, mark the cell-staining area on the slides with a Pap pen for easy identification.
Wash the slides in Tris-buffered saline containing 0.2% Tween 20 (TBS-T), and incubate in the hydrogen peroxide block (see Table of Materials) at room temperature for 15 min to remove any remnant peroxidase activity. Then, wash the slides in TBS-T three times for 2 min each.
Prepare diluted antibody solution by diluting primary antibody in the protein block (see Table of Materials). For example, the CKAP2 primary antibody can be diluted 1:100, and the Ki-67 antibody 1:500. Then, incubate the slides in 100 µL of diluted antibody solution for 1 h.
After washing in TBS-T five times for 2 min each, incubate the slides in the primary antibody enhancer in the kit for 15 min at room temperature in the dark.
After the washing in TBS-T four times for 2 min each, add 2 drops of HRP polymer (a secondary antibody labeled with horseradish peroxidase), and incubate the slides at room temperature for 30 min.
After washing in TBS-T five times for 2 min each, add 100 µL of diaminobenzidine (DAB) solution (see Table of Materials), and incubate the slides for 3 min.
After washing in TBS-T two times, add 100 µL of Hematoxylin solution (the mixture of 100 µL of Hematoxylin and 600 µL of distilled water), and incubate the slides for 1 min.
After washing the slides in TBS-T once, dehydrate by incubating in 95% ethanol for 2 min, dipping in 95% ethanol once, and dipping in 100% ethanol two times. Then, incubate the slides in 40 mL xylene in a glass jar for 5 min.
When the slides are dry from xylene, mount the respective coverslips.
Observe the staining patterns using light microscopy.
Representative Results
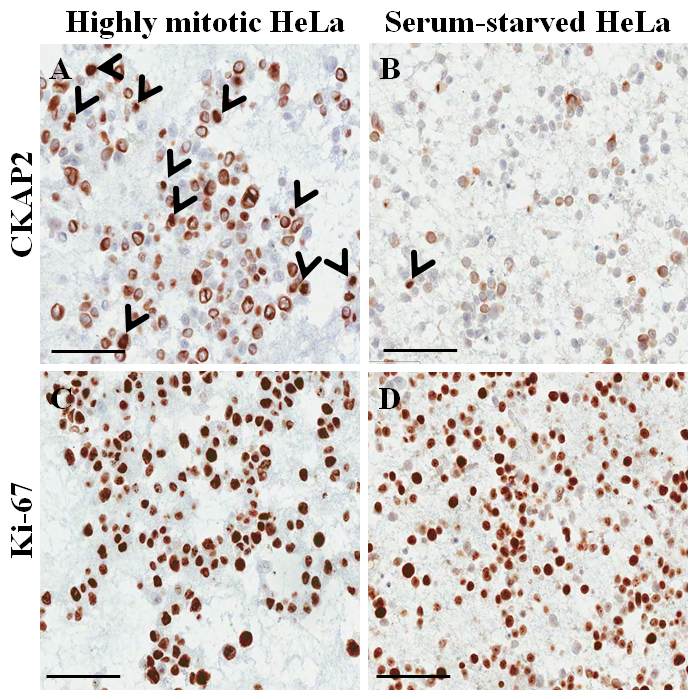
In a hematoxylin-and-eosin-stained slide from the paraffin-embedded cell block (Figure 3A,B), most of the nuclei and cytoplasm of the cells are intact, suggesting that the morphological preservation is excellent with the current protocol (Figure 3A). In the immunocytochemical staining, positive CKAP2 staining was observed in condensed chromatin, mitotic spindle, and cytoplasm (Figure 4), as previously reported10. Ki-67 staining was observed in the cell nuclei, as expected (Figure 4). Only the cells with CKAP2 staining in condensed chromatin (see the arrows in Figure 4A,B) were mitotic cells. Many CKAP2-positive cells were shown in the highly mitotic HeLa cells that had been prepared after an incubation in complete medium (Figure 4A). In comparison, there were few CKAP2-positive cells in the serum-starved HeLa cells (Figure 4B). Most of the highly mitotic HeLa cells were Ki-67 positive (Figure 4C). Contrastingly, the Ki-67-positive rate in the serum-starved HeLa cells remained as high as ~50% (Figure 4D). These results are quite comparable to those of a previous report11 which suggests that CKAP2 is a more reliable proliferation marker in cancer cells than is Ki-67.
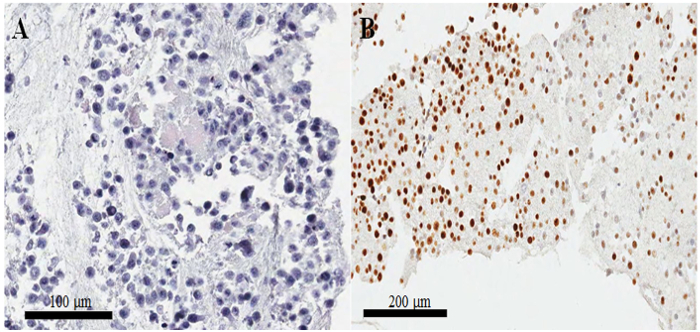
In poorly prepared cell blocks, the nuclei are separated from the cytoplasm, and there is also, resultantly, poor morphologic preservation. Longer-than-overnight incubation of fixed cells in a refrigerator might cause such poor results. Another important problem is that cells are not stained well by immunocytochemistry, notwithstanding the excellent morphology. This problem arises more often when the cell clot is small. Typically, the staining intensity is irregular as shown in Figure 3B; but when the cell clot is larger, there is much less chance of irregular staining. Therefore, in this protocol, we increased the volumes of plasma, thromboplastin, and calcium chloride in order to form a large cell clot.
Figure 1: Cell-block preparation scheme. Please click here to view a larger version of this figure.
Figure 2: Illustration of paraffin cell-block preparation. (A) Thromboplastin-plasma cell clot in tube. (B) TP Cell clot after PBS washing. (C) Cell clot on moistened filter paper. (D) Tissue-embedding station with molten wax. Metal mold (arrow) holds molten wax for solidification. (E) Paraffin-embedded cell clot (arrow) embedded in paraffin or paraffin-embedded cell block. (F) Thin paraffin section on the middle part of a coated slide. Please click here to view a larger version of this figure.
Figure 3: Cell-block preparation and confirmation of quality by hematoxylin and eosin and immunostaining. (A) Hematoxylin-and-eosin-stained image of HeLa cells in a paraffin-embedded cell-block section. (B) Irregular staining of Ki-67 in immunocytochemistry on a poorly prepared paraffin-embedded cell-block section. Scale bars are (A) 100 and (B) 200 μm. Please click here to view a larger version of this figure.
Figure 4: Immunocytochemical staining on paraffin-embedded cell block for HeLa cells. (A) CKAP2 staining under highly mitotic conditions. (B) CKAP2 staining under serum-starved conditions. (C) Ki-67 staining under highly mitotic conditions. (D) Ki-67 staining under serum-starved conditions. 100 μm scale bars are shown. The arrowheads indicate CKAP2-positive cells. Please click here to view a larger version of this figure.
| Procedure | Steps | Solution | Time/Temperature |
| Dehydration | 1 | 70% alcohol | 15 min/RT |
| 2 | 80% alcohol | 15 min/RT | |
| 3 | 95% alcohol | 15 min/RT | |
| 4 | 100% alcohol | 15 min/RT | |
| Clearing | 1 | Xylene | 60 min/4 °C |
| 2 | Xylene | 10 min/RT | |
| 3 | Xylene | 10 min/RT | |
| 4 | Xylene | 10 min/RT | |
| *RT, room temperature |
Table 1. Tissue processing procedure.
Discussion
Immunofluorescent staining on fixed cultured cells is currently the method of choice for determination of protein expression level in cells while preserving morphological information1. However, immunocytochemistry on paraffin-embedded cell blocks can be an excellent alternative. The detailed procedures for the preparation of paraffin-embedded cell blocks and immunocytochemistry have been described in this protocol, and we hope it can facilitate the application of immunocytochemistry in cell studies.
Immunocytochemistry has several advantages over immunofluorescent staining. Immunofluorescent staining for cells usually requires freshly cultured cells, but paraffin cell blocks can be kept at room temperature for several years13. Additionally, immunocytochemistry on cell blocks can explore intracellular expression patterns by employing the same antibody used in routine immunohistochemistry on human tissues4. Furthermore, it can explore the changes in protein levels or posttranslational modifications either by pre-incubating cells with various drugs or under various culture conditions11.
In contrast to the advantages of immunocytochemical staining, the preparation of paraffin-embedded cell blocks takes time and is costly14. Also, most research laboratories lack experience in this technique, and technical errors under such circumstances are common. The most common errors are poor preservation of cell morphology and poor or irregular immunocytochemical staining on the paraffin cell block sections. These and most others can be avoided by making cell blocks under the best cell conditions and using enough solution to form cell clots.
As a demonstration of the present protocol, cell blocks were prepared for HeLa cells, and immunocytochemical staining was performed for two proliferation markers, CKAP2 and Ki-67, as previously reported11. For immunocytochemistry, the cells were manipulated by incubation in media with and without fetal bovine serum, and the effect of serum starvation could be observed. These prepared paraffin-embedded cell blocks can be employed for a large number of antibodies, because many slides can be prepared from a cell block using only a 4 - 5 µm-thick cell-block section per slide. Therefore, expression patterns corresponding to two different conditions can be evaluated with several different antibodies. The immunostaining patterns for CKAP2 and Ki-67 in cancer tissues have already been reported9,10,11,12, and the immunocytochemical staining results could be easily evaluated, because the staining patterns were quite similar to those from immunohistochemistry.
In conclusion, immunocytochemical staining on paraffin cell blocks can be an excellent alternative to immunofluorescent staining; moreover, it can be easily and reliably employed in basic research for expression profiling in cell lines while preserving morphological information.
Disclosures
All authors have declared no conflicts of interest.
Acknowledgments
This work was supported by research grants to K.-M.H. from the National Cancer Center, Korea (1510121) and National Research Foundation, Korea (no. NRF-2015R1A2A2A04007432).
References
- Bhattacharyya D, Hammond AT, Glick BS. High-quality immunofluorescence of cultured cells. Methods Mol Biol. 2010;619:403–410. doi: 10.1007/978-1-60327-412-8_24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CM. Fluorescence microscopy - Avoiding the pit falls. J Cell Sci. 2007;120:1703–1705. doi: 10.1242/jcs.03433. [DOI] [PubMed] [Google Scholar]
- Odell ID, Cook D. Immunofluorescence Techniques. J Invest Dermatol. 2013;133:e4. doi: 10.1038/jid.2012.455. [DOI] [PubMed] [Google Scholar]
- Kulkarni MB, Desai SB, Ajit D, Chinoy RF. The utility of the thromboplastin-plasma cell-block technique for fine-needle aspiration and serous effusions. Diagn Cytopathol. 2009;37(2):86–90. doi: 10.1002/dc.20963. [DOI] [PubMed] [Google Scholar]
- Shivakumarswamy U, Arakeri SU, Karigowdar MH, Yelikar B. Diagnostic utility of the cell block method versus the conventional smear study in pleural fluid cytology. J Cytol. 2012;29(1):11–15. doi: 10.4103/0970-9371.93210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan NA, Narayan E, Smith MM, Horn MJ. Cell block cytology: Improved preparation and its efficacy in diagnostic cytology. Am J Clin Pathol. 2000;114(4):599–606. doi: 10.1309/G035-P2MM-D1TM-T5QE. [DOI] [PubMed] [Google Scholar]
- Keyhani-Rofagha S, Vesey-Shecket M. Diagnostic value, feasibility, and validity of preparing cell blocks from fluid-based gynecologic cytology specimens. Cancer. 2002;96:204–209. doi: 10.1002/cncr.10716. [DOI] [PubMed] [Google Scholar]
- Young NA, Naryshkin S, Katz SM. Diagnostic value of electron microscopy on paraffin-embedded cytologic material. Diagn Cytopathol. 1993;9:282–290. doi: 10.1002/dc.2840090308. [DOI] [PubMed] [Google Scholar]
- Jin Y, Murakumo Y, Ueno K, Hashimoto M, Watanabe T, Shimoyama Y, et al. Identification of a mouse cytoskeleton-associated protein, CKAP2, with microtubule-stabilizing properties. Cancer Sci. 2004;95(10):815–821. doi: 10.1111/j.1349-7006.2004.tb02187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong KU, Choi Y-B, Lee J-H, Kim H-J, Kwon H-R, Seong Y-S, et al. Transient phosphorylation of tumor associated microtubule associated protein (TMAP)/cytoskeleton associated protein 2 (CKAP2) at Thr-596 during early phases of mitosis. Exp Mole Med. 2008;40:377–386. doi: 10.3858/emm.2008.40.4.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim SH, Bae CD, Kwon Y, Hwang H-L, Poojan S, Hong H-I, et al. CKAP2 (cytoskeleton-associated protein2) is a new prognostic marker in HER2-negative luminal type breast cancer. PLoS ONE. 2017;12(8):e0182107. doi: 10.1371/journal.pone.0182107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inwald EC, Klinkhammer-Schalke M, Hofstadter F, Zeman F, Koller M, Gerstenhauer M, et al. Ki-67 is a prognostic parameter in breast cancer patients: results of a large population-based cohort of a cancer registry. Breast Cancer Res Treat. 2013;139(2):539–552. doi: 10.1007/s10549-013-2560-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katikireddy KR, O'Sullivan F. Immunohistochemical and immunofluorescence procedures for protein analysis. Methods Mol Biol. 2011;784:155–167. doi: 10.1007/978-1-61779-289-2_11. [DOI] [PubMed] [Google Scholar]
- Beutner EH. Immunofluorescent staining: The fluorescent antibody method. Microbiol Mol Biol Rev. 1961;25(1):49–76. doi: 10.1128/br.25.1.49-76.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]