

Abstract
The efficient and robust isolation and culture of primary oligodendrocytes (OLs) is a valuable tool for the in vitro study of the development of oligodendroglia as well as the biology of demyelinating diseases such as multiple sclerosis and Pelizaeus-Merzbacher-like disease (PMLD). Here, we present a simple and efficient selection method for the immunomagnetic isolation of stage three O4+ preoligodendrocytes cells from neonatal mice pups. Since immature OL constitute more than 80% of the rodent-brain white matter at postnatal day 7 (P7) this isolation method not only ensures high cellular yield, but also the specific isolation of OLs already committed to the oligodendroglial lineage, decreasing the possibility of isolating contaminating cells such as astrocytes and other cells from the mouse brain. This method is a modification of the techniques reported previously, and provides oligodendrocyte preparation purity above 80% in about 4 h.
Keywords: Neuroscience, Issue 135, oligodendrocytes, in vitro, primary cells, mouse, neuroscience, oligodendrocyte purification, immunomagnetic isolation
Introduction
Oligodendrocytes (OLs) are the myelinating cells of the central nervous system (CNS)1. The isolation and culture of primary oligodendrocytes in a tightly regulated environment is a valuable tool for the in vitro study of the development of oligodendroglia as well as the biology of demyelinating diseases such as multiple sclerosis2. This requires an efficient and robust oligodendrocyte isolation and culture method3. In this study, we took advantage of the expression of a distinctive oligodendrocyte cell surface marker to implement a modified isolation technique that is rapid and specific.
Four distinct stages of oligodendrocyte maturation have been identified, each characterized by the expression of distinctive cell surface markers for each developmental stage (Figure 1). These cell surface markers can be recognized by specific antibodies4,5, and can be used to isolate OLs at specific stages. In the first stage, oligodendrocyte precursor cells (OPCs) have the capacity to proliferate, migrate, and specifically express platelet-derived growth factor receptor (PDGF-Rα)6, ganglioside A2B5, proteoglycan NG27,8, polysialic acid-neural cell adhesion molecule9 and fatty-acid-binding protein 7 (FABP7)10. OPCs have bipolar morphology with few short processes emanating from the opposing poles of the cell body, which is characteristic of neural precursor cells11.
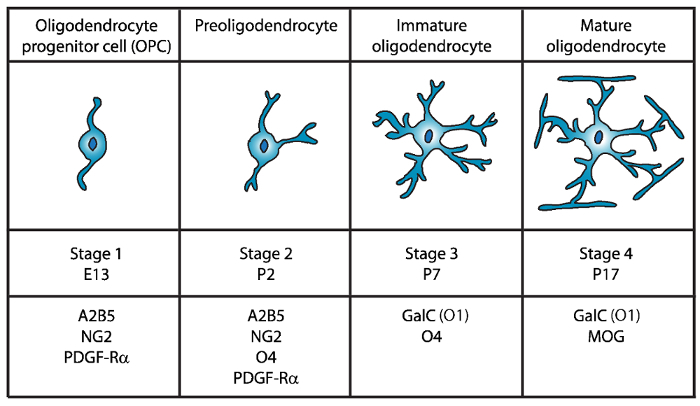
Figure 1: Expression of cell surface markers during the mouse oligodendrocyte development. OLs cell surface markers such as A2B5, GalC (O1), NG2, O4, and PDGF-Rα can be used to specifically isolate oligodendrocytes at specific developmental stage by using specific antibodies. Please click here to view a larger version of this figure.
In the second stage, OPCs give rise to preoligodendrocytes and express at the cell membrane not only OPC markers, but also the sulfatide (a sulfated galactolipid) recognized by the O4 antibody12,13, and the GPR17 protein14, which persists until the immature oligodendrocyte (OL) stage. At this stage, preoligodendrocytes extend multipolar short processes. Preoligodendrocytes are the major OL stage at postnatal day 2 (P2) in the cerebral white matter of both rat and mouse with a minor population of immature OLs15.
During the third stage, immature OLs continue to express O4, lose expression of A2B5 and NG2 markers and begin to express galactocerebroside C16. At this stage, OLs are committed to the oligodendroglial lineage and become post-mitotic cells with long ramified branches17,18. Immature OL constitute more than 80% of the rodent white matter at P7 and at this time the first MBP+ cells are observed15,19,20,21. Therefore, isolation of OLs at P7 could ensure high cellular yield.
In the final and fourth stage of OL development, mature OLs express myelinating proteins (myelin basic protein (MBP), proteolipid protein (PLP), myelin associated glycoprotein (MAG) and myelin oligodendrocyte glycoprotein (MOG)22,23,24,25,26. At this stage, mature OLs extend membranes that form compact enwrapping sheaths around the axons and are able to myelinate. This coincides with the observation that in rat and mouse brain, MBP+ cells become increasingly abundant at P1419,20,21.
Since the first isolation of oligodendrocyte by Fewster and colleagues in 196727, several methods for isolation of OLs from the rodent CNS have been implemented including immunopanning28,29,30, fluorescence-activated cell sorting (FACS) exploiting cell surface-specific antigens28,31, differential gradient centrifugation32,33,34,35 and a shaking method based on differential adherence of different CNS glia36,37. However, existing culture methods have limitations, particularly in terms of purity, yield and time required to perform the procedures38. Therefore, more efficient isolation methods for oligodendrocytes are required.
In this paper, we present a simple and efficient selection method for the immunomagnetic isolation of stage three O4+ preoligodendrocytes cells from neonatal mice pups. This method is a modification of the techniques reported by Emery et al.39 and Dincman et al.40 and provides an oligodendrocyte preparation purity above 80% in about 4 h.
Protocol
The mice used in this study were cared for according to the guidelines of the SUNY Downstate Medical Center Division of Laboratory Animal Resources (DLAR) protocol number 15-10492.
NOTE: Primary oligodendrocytes were isolated from neonatal (P5-P7 wild-type C57Bl/6N) mice. At this stage, immature OLs constitute more than 80% of the rodent white matter ensuring high cellular yield. All buffer and reagent compositions are available at the end of the Table of Materials.
1. Coverslips Preparation
NOTE: Poly-D-lysine (PDL)/laminin coated coverslips should be prepared prior to OL isolation.
Place #1 German glass coverslips into a 50-mL conical tube and add 35 mL of 70% EtOH to clean them.
Close the tube, place it in a nutating mixer and incubate at room temperature for at least 30 min while gently rocking. Coverslips can be incubated overnight.
Discard the 70% EtOH.
Wash the coverslips 3 times with deionized water, remove the water every time with vacuum aspiration.
Add 30-35 mL of PDL 50 µg/mL to the coverslips in the 50-mL tube, enough to cover the coverslips.
Place the 50-mL tube containing the coverslips on a nutating mixer and incubate at room temperature for at least 30 min while rocking gently.
Wash the coverslips 3 times with deionized water, removing the water every time with vacuum aspiration.
Transfer the coverslips into 60-mm Petri dishes containing deionized water.
Use a P200 pipette tip to arrange the coverslips as a layer covering the entire surface of the Petri dish.
Carefully, remove the water from the dishes with vacuum aspiration.
Remove the excess of water around the coverslips and let them dry overnight with lid open and under UV light in the hood. NOTE: PDL coated glass coverslips can be stored at 4 °C in sterile Petri dishes for up to three months.
Place the coverslips into 24-well plates, one per well, using fine forceps.
Move the coverslips to the center of the well making sure the edges do not touch the wall of the well.
Add 100 µL of 10 µg/mL laminin diluted in B27NBMA (see the Table of Materials) starting in the center of each coverslip and moving in a circular fashion towards the edges to cover all the area. NOTE: Since laminin is involved in OL maintenance and promotes differentiation into mature OLs, it is an important OL survival factor for in vitro culture41,42,43,44.
Incubate the plate in a 37 °C incubator at 5% CO2 for at least 1 h or until the OLs are ready for plating.
2. Mouse Brain Cortex Dissociation
Sacrifice postnatal 5-7-day old C57Bl/6N mouse pups by quick decapitation with scissors previously cleaned in 70% ethanol. NOTE: Cerebral cortices from 5-6 neonatal mice pups were used for each preparation. On average, one mouse brain yields 1-1.5 x 106 OLs.
Cut the scalp skin along the midline with small dissecting scissors and retract to expose the skull.
Cut the skull carefully along the midline starting from the opening in the back of the skull towards the frontal area, lifting up with the scissors to avoid damaging the brain. Then, using the opening in the back of the skull cut toward each eye socket along the base of the skull.
Use fine forceps to gently tease the cortices away from the midbrain and transfer them to a 60-mm tissue culture dish containing 7 mL of B27NBMA. Repeat steps 2.2 through 2.4 for each brain, pooling the dissected cortices.
Transfer the cortices to a new 60-mm tissue culture dish containing 5 mL of dissociation buffer (B27NBMA, 20-30 U/mL Papain, and 2,500 U DNase I).
Dice the cortices into small pieces of about 1 mm3 using a #15 scalpel blade and incubate for 20 min in a 37 °C, 5% CO2 incubator.
Add 1 mL of bovine growth serum (BGS) to stop the enzymatic reaction.
Transfer the cortices along with the dissociation media to a 15-mL conical tube using a 10 mL serological pipet.
Gently begin to dissociate the brain tissue by slowly pipetting up and down 6-8 times using a 10-mL serological pipet, using care to minimize bubbles.
Allow the tissue chunks to settle for 2-3 min and transfer the supernatant to a fresh tube.
Add 3 mL of B27NBMA containing 10% BGS/2500 U DNase I to the tissue pellet.
Use a 5 mL serological pipette to gently dissociate the brain tissue while pipetting up and down 6-8 times. Be careful to minimize bubbles.
Allow the tissue chunks to settle for 2-3 min.
Transfer the supernatant to a fresh tube. Add 3 mL of B27NBMA containing 10% BGS/DNase I to the tissue pellet.
Gently dissociate the brain tissue using a P1000 pipet tip, while pipetting the mix of media and brain tissue up and down. Repeat Steps 2.13 and 2.14 until no large chunks of tissue remain or until B27NBMA containing 10% BGS/DNase I is exhausted, whichever comes first. Be careful to minimize bubbles.
Pool cell suspension with previous supernatants.
Place 70-µm cell strainer in a 50-mL tube. Using a 10-mL serological pipet, pass the pooled cell suspension gently over the 70-µm cell strainer.
Wash the cell strainer by adding 1 mL of B27NBMA containing 10% BGS/DNase I.
Discard the cell strainer. Bring the volume to 30-mL with B27NBMA containing 10% BGS/DNase I.
Transfer the cell suspension to two 15-mL conical tubes. Centrifuge the cell suspension for 10 min at 200 x g.
Remove the supernatant without leaving the cells exposed to the air. The supernatant will be cloudy due to cell debris. Add 3 mL of B27NBMA containing 10% BGS to the pellet.
Carefully dissociate the cell pellet using a P1000 pipet. Bring the volume to 15 mL with B27NBMA containing 10% BGS.
Pass the cell suspension over a fresh 40 µm cell strainer.
Wash the cell strainer by adding 1mL of B27NBMA containing 10% BGS. Discard the cell strainer.
Bring the volume to 30 mL with B27NBMA containing 10% BGS.
Transfer the cell suspension to 2 x 15-mL conical tubes. Centrifuge the cell suspension for 10 min at 200 x g.
Remove most of the supernatant without leaving the cells exposed to the air. Resuspend the cell pellets in 5 mL of ice cold magnetic cell sorting (MCS) buffer.
3. Determination of Cell Count and Viability
Dilute 100 µL of the cell suspension with 400 µL of Trypan Blue Solution in a 1.5-mL microcentrifuge tube to achieve a 1:5 dilution in 0.4% (w/v).
Center a cover glass over a hemocytometer chamber and fill the two chambers with 10 µL of the cell dilution using a P10 pipette and avoiding overfill. The solution will pass under the cover glass by capillary action.
Place the hemocytometer on the microscope stage and adjust focus using 40X magnification. NOTE: Cell counts are recorded using a hand-held counter in each of five squares (four corners and one center). Only non-viable cells absorb the dye and appear blue, while live and healthy cells appear round and refractive and do not absorb the blue-colored dye, allowing for determination of the number of viable and total cells per milliliter.
4. Isolation of O4+ Oligodendrocytes
Centrifuge the cell suspension at 200 x g for 10 min.
Carefully discard the supernatant by using vacuum aspiration, avoiding exposure of the cells to the air.
Resuspend the pellet in 90 µL of MCS buffer per 1 x 107 total cells followed by the addition of 10 µL of anti-O4 beads per 1 x 107 cells.
Mix the cell suspension and beads by gently flicking the 15-mL conical tube with the finger 4-5 times.
Incubate the mix for 15 min at 4 °C, flicking the 15-mL conical tube with the finger 4-5 timesevery 5 min.
Wash the mix by gently adding 2 mL of MCS buffer per 1 x 107 cells to the tube. Centrifuge the mix at 200 x g for 10 min.
Discard the supernatant by carefully using vacuum aspiration, avoiding exposure of the cells to the air. Resuspend the cell pellet in 500 µL of MCS buffer for every 1 x 107 cells.
Attach a magnetic separator to a magnetic separator stand. Attach a selection column to the magnetic separator and place a 40 µm cell strainer on top of the column. Place two 15-mL or one 50-mL conical tube below the separation column to collect the flow through.
Pre-rinse the 40-µm cell strainer and separation column with 3 mL of MCS buffer and let the buffer run through the column without letting it dry.
Add the mix of cell suspension and beads to the cell strainer and into the selection column.
Wash the 40 µm cell strainer with 1 mL of MCS buffer.
Discard the cell strainer and let the mix of cells, beads and buffer run through the column without letting it dry.
Wash the separation column 3 times with 3 mL of MCS buffer and 1 time with OL proliferation media.
Remove the separation column from the magnetic separator, quickly place it into a 15-mL conical tube, and immediately add 5 mL of OL proliferation media.
Place a plunger on top of the column and firmly push to flush out the labeled cells into the 15-mL conical tube.
Remove 100 µL of cell suspension to determine cell count and viability using Trypan Blue and hemocytometer as indicated in section 3. NOTE: Cell viability above 80% is considered acceptable to proceed with the culture of OLs, but viability greater than 90% is optimal.
5. Plating of Isolated O4+ Oligodendrocytes
Dilute the cell suspension to the desire plating density (5 x 105 cells per coverslip) using OL proliferation media. NOTE: OL seeding density is very important, so appropriate cell density should be confirmed using a tissue culture microscope prior to proceeding. Seeding density below 10,000 cells/coverslips may lead to poor cell survival. At plating density at 10,000 to 25,000, OL morphological differentiation is slow45. We plate OLs at a seeding density of at least 50,000 cells/coverslips to ensure cell survival.
Move the 24-well plates containing coverslips coated with laminin from incubator to the tissue culture hood.
Remove laminin from each coverslip and replace it with 100 µL of OL suspension.
Incubate OLs in the 37 °C/5% CO2 incubator for a maximum of 45 min to promote OL adhesion to the coverslips.
After incubation, remove the 24-well plates from the incubator. Flood each well of the 24-well plate with 500 µL of OL proliferation media.
Place the OLs back in the 37 °C/5% CO2 incubator for additional 24 h.
24 h after plating of OLs, remove proliferation media and substitute with OL differentiation media to induce differentiation.
Place the OLs back in the incubator and do not remove until the cells are ready for fixation. NOTE: Changes in pH are detrimental for OLs and decrease cell viability, therefore unnecessary removal of OLs cultures from the incubator should be avoided.
6. Immunofluorescence Staining
For the detection of cell surface markers (NG2, O1 and O4) remove the media from the wells.
Add 250 µL of mouse hybridoma supernatants containing primary antibodies against O1 (undiluted)13 and O4 (undiluted)13 and rabbit polyclonal against NG2 (1:150 diluted in B27NBMA). NOTE: If commercially available anti-O1 and anti-O4 antibodies are to be used, it is advisable to utilize dilutions suggested by the manufacturer.
Incubate the cells with primary antibody for 45 min maximum in a 37 °C/5% CO2 incubator.
Wash twice with 0.05% Tween-20/1X PBS. Fix the cells with 4% paraformaldehyde (4% PFA) for 10 min at room temperature.
Wash three times for 5 min each with 0.05% Tween-20/1X PBS.
Dilute Alexa 488-conjugated goat anti-mouse IgM or anti-rabbit IgG secondary antibody (1:400) in B27NBMA.
Remove the 0.05% Tween-20/1X PBS from the coverslips. Incubate the cells with secondary antibody for 1 h at room temperature in the dark.
Wash three times for 5 min each with 0.05% Tween-20/1X PBS.
For double staining for the detection of internal markers (GFAP), remove the 0.05% Tween-20/1X PBS and block/permeabilize cells with block/permeabilization buffer for 15 min at room temperature. Otherwise proceed to step 6.17.
Remove block/permeabilization buffer and add 250 µL chicken anti-GFAP (1:100) diluted in block/permeabilization buffer.
Incubate the cells with anti-GFAP for 1 h at room temperature. Wash three times for 5 min each with 0.05% Tween-20/1X PBS.
Dilute Alexa 594-conjugated goat anti-chicken IgG secondary antibody (1:400) in B27NBMA.
Remove the 0.05% Tween-20/1X PBS from the coverslips. Incubate the cells with secondary antibody for 1 h at room temperature in the dark.
Wash three times for 5 min each with 0.05% Tween-20/1X PBS.
Counterstain with 4',6-diamidino-2-phenylindole (DAPI) and mount the cells with antifade mounting media containing DAPI.
Let the coverslips cure overnight in the dark at room temperature.
Image the stained cells with an epifluorescence microscope. NOTE: In this protocol, the cells were imaged at 40X magnification using a uniform exposure time.
Representative Results
The purpose of this study was to establish an improved isolation method for O4+ primary mouse oligodendrocytes requiring the least possible manipulation of the target cells. The entire procedure from euthanasia of the pups to plating of the cells in coverslips takes about 4 h and data shown here represent three independent experiments. After tissue dissociation, an average of 4.3 ± 0.46 x 107 cells were isolated for each independent experiment, with a viability of 91% ± 5.6%. After immunomagnetic isolation using anti-O4 tagged magnetic microbeads an average of 6.9 ± 0.38 x 106 OLs were obtained (1-1.5 x 106 per mouse) which is 16.2% ± 1.6% of the total cells initially dissociated; viability was 96.3% ± 3.5%.
Figure 2: Immunomagnetically isolated OLs express immature markers at 1DIV. (A) OLs under phase contrast microscope show bi- and tri-polar morphology. OLs express NG2 (B), O4 (C), and O1 (D). Very few astrocytes were present at 1DIV (C, red), scale bar = 50 µm. Data were collected from 3 independent experiments. Please click here to view a larger version of this figure.
To examine the antigenic phenotype of the immunomagnetically isolated cells, immunofluorescence staining was performed 24 h (1DIV) and 72 h (3DIV) after plating the cells. At 1DIV, the cell appeared to be bi- or tripolar under the phase contrast microscope (Figure 2A-B), a characteristic of feature of early stage proliferating OLs (OPCs and or preoligodendrocytes). 66.1 ± 8.4% of these cells were NG2-labeled (Figure 2B), and 58% ± 9.4% of the cells were O4-labeled (Figure 2C). O1 (GalC)-labeling, a marker of more mature cells in the oligodendrocyte lineage, was much less common (7.4% ± 6.0%, Figure 2D). This marker profile suggests that the majority of cells at this point are pre-oligodendrocytes with smaller percentages immature or mature oligos and possibly some oligodendrocyte progenitor cells (OPCs). Astrocytes, as evidenced by GFAP staining, comprised of the 0.57 ± 1.0% (Figure 2C). These data suggest that the oligodendrocytes isolated with the modified technique are highly purified and the majority show expression pattern for a marker typical of immature oligodendrocytes.
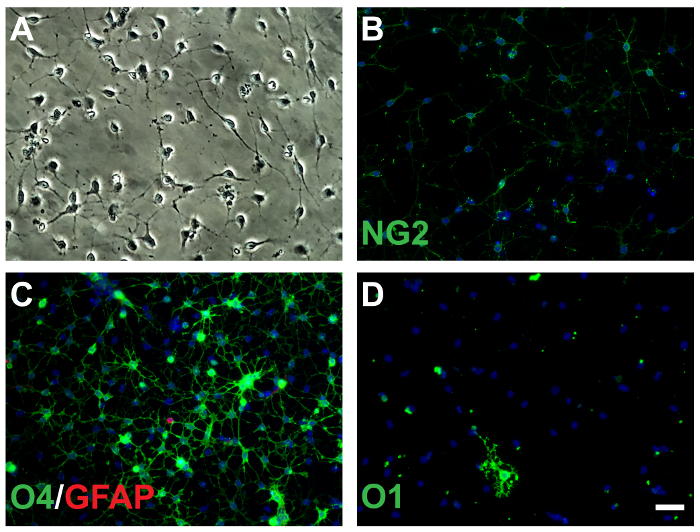
Figure 3: Expression of mature OLs markers at 3DIV. (A) OLs under phase contrast microscope show more complex morphology. At 3DIV, expression of NG2 (B) decreases dramatically, while O4 (C) and O1 (D) increase. Few astrocytes are visible at 3DIV (C, red), scale bar = 50 µm. Data were collected from 3 independent experiments. Please click here to view a larger version of this figure.
At 3DIV, oligodendrocyte morphology appeared more complex than the cells at 1DIV (Figure 3A). At this time point, the frequency of cells positive for the early oligodendrocyte marker NG2 was much lower compared to 1DIV. NG2-labeled cells comprised 31.2 ± 17.1% (versus 66.1 ± 8.4% at 1DIV, t-test p <0.0001, Figure 3B,Figure 4A). Although not analyzed here, the average intensity of staining in cells deemed to be positive for NG2 was reduced at 3DIV compared to 1DIV. Most cells (81.9% ± 4.884%, Figure 3C) showed O4-labeling, which was higher than at 1DIV (58% ± 9.4%, t-test p <0.0001, Figure 4B). Also, almost half the cells (47.2% ± 16.4%, Figure 3D) showed staining for O1 (GalC), a marker of more mature oligodendrocyte lineage, suggesting that the cells are differentiating to a more mature phenotype. The increase of O1+ cells at 3DIV was also significant compared to 1DIV (7.4% ± 6.0%, t-test p <0.0001, Figure 4C). The presence of astrocytes at this time point remained extremely low (0.8% ± 1.5%, Figure 3C) and it was not significantly different compared to 1DIV (0.57% ± 1.0%, not significant (ns), Figure 4D). These results indicate that over time, immunomagnetically isolated oligodendrocytes are able to differentiate in vitro into stage three of maturation with very little contamination of other cell types such as astrocytes.
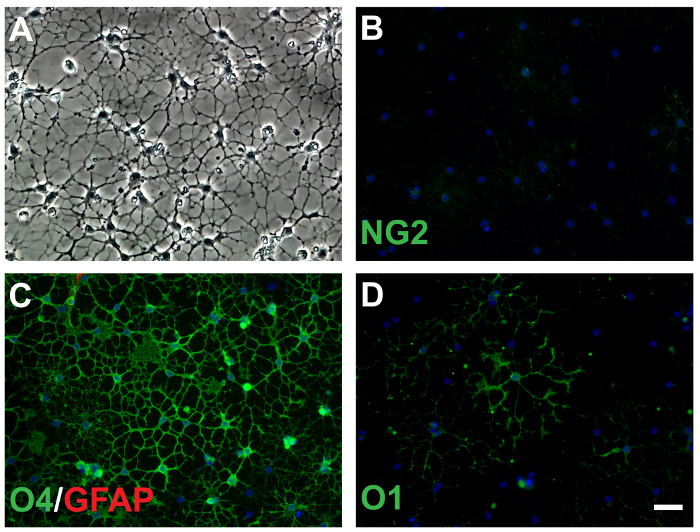
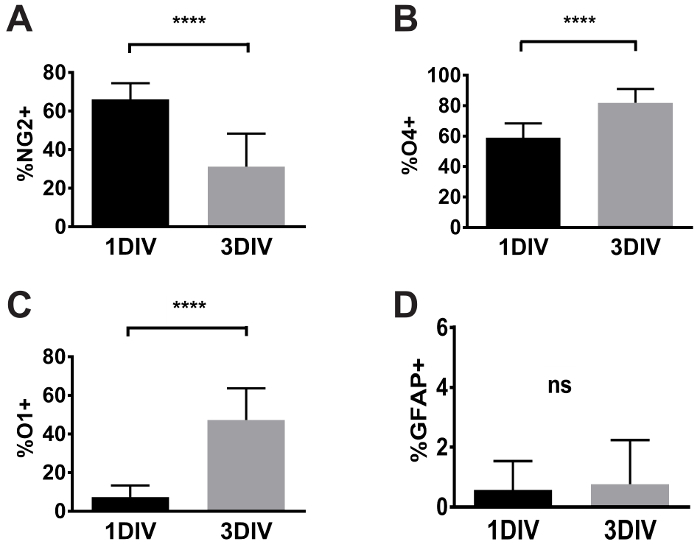
Figure 4: Quantification of the expression of OL markers at 1DIV and 3DIV (A-D). At 1DIV, 66% of cells were NG2+, 58% of the cells were O4+, 7% of the cells were O1+ and 0.5% of the cells were GFAP+. At 3DIV, 31% of cells were NG2+, 81% of the cells were O4+, 47% of the cells were O1+ and 0.7% of the cells were GFAP+. Mean values ± SD are presented here, **** indicates p <0.0001, ns= not significant. Data were collected from 3 independent experiments. Please click here to view a larger version of this figure.
Discussion
In this communication, we present a method for the efficient isolation of highly purified immature mouse oligodendrocyte cultures. Compared to previously published protocols39,40, this method yielded a higher purity with a much lower level of GFAP-positive astrocytes and a very low percentage of other non-characterized cells. It is important to point out that these are immature OLs already committed to the oligodendroglial lineage. Thus, these cells would not be useful for study of early phases of differentiation.
One of the modifications from the Dincman protocol40 was the removal of basic fibroblast growth factor (bFGF) from our proliferation media. It has been reported that in vitro, bFGF acting as a strong mitogenic factor not only inhibits OPCs from differentiation, but also decreases the number of processes46,47. Moreover, bFGF increases oligodendrocyte dedifferentiation and proliferation in passaged cells of the oligodendroglial lineage 48. Therefore, it is plausible that the reduced numbers of O4+ cells and increased numbers of A2B5+ and NG2+ cells observed in Dincman study40 may be due to the effect of bFGF in culture.
Despite using O4 binding as the method for isolating the cells, only 59% of the cells are O4+ at 1DIV. These results are qualitatively similar to those of Dincman and coworkers40 who found that using O4 binding for isolation, slightly less than 50% of cells were O4+ at 1DIV. It has been shown that PDGF delays post-mitotic development by transiently reverting O4+GalC- progenitors to A2B5+O4- pre-progenitor-like cells that subsequently differentiate even in the continued presence of PDGF 49. Therefore, it is likely that during the process of proliferation in the first day in vitro some immature OLs may experience a reversal from O4+NG2+ to O4-NG2+, while still remaining committed to the oligodendrocyte lineage. This hypothesis is supported by Dincman's finding40 that immediately after isolation about 80% of the cells were O4+.
At 3DIV, this protocol produces a somewhat higher OL purity than that described by others39,40 though we did not make a head-to-head comparison with their methods. Compared to protocol of Dincman et al.40, the lower number of GFAP-positive cells in our cultures could be due to the lack of FGF in the proliferation media. The Emery and Dugas protocol39 relies on the sequential passage of cells through culture dishes; astrocytes can attach easily to culture dishes50 and could be carried over after each sequential passage of cells during the immunopanning protocol. The presented method does not risk introducing contaminating astrocytes in our purification fraction.
Our modified method is faster compared to the other two taking about 4 h to perform. We achieve this by eliminating unnecessary steps and reagents incorporated by the other protocols. For example, compared to the Dincman protocol40, which takes 5-6 h, we omitted both the use of a kit for neural tissue dissociation and the incubation of the OL cell suspension with rat anti-mouse IgM beads to remove dead cells. Instead, we performed the dissociation of mouse brain cortices with papain and dead cells were removed by performing low speed centrifugations (200 x g).
A major modification from the Emery protocol39, which takes about 9 h, was the removal of the immunopanning selection and the removal of the requirement for a direct O2/CO2 line used to pre-equilibrate the Papain Buffer during tissue digestion. Instead, we used immunomagnetic selection and direct incubation of the mix of mouse brain cortices and Papain buffer in the CO2 incubator.
Although not directly tested in this study, the anti-O4 microbeads can also be used to isolate OLs from rat, human and transgenic mouse brain tissue. This highlights the versatility of the technique to use multiple tissue sources for OLs isolation with the potential to benefit research labs interested in working with OLs from sources other than mouse.
In conclusion, the modified OPC isolation method described in this study provides a rapid and specific alternative to obtain primary mouse oligodendrocytes that could be applied in studies of myelination and demyelinating disease.
Disclosures
The authors have no disclosures.
Acknowledgments
This study was supported by grants from the National Multiple Sclerosis Society (RG4591A1/2) and the National Institutes of Health (R03NS06740402). The authors thank Dr. Ivan Hernandez and his lab members for providing laboratory space, equipment and advice.
References
- Emery B. Regulation of oligodendrocyte differentiation and myelination. Science. 2010;330(6005):779–782. doi: 10.1126/science.1190927. [DOI] [PubMed] [Google Scholar]
- Yang Z, Watanabe M, Nishiyama A. Optimization of oligodendrocyte progenitor cell culture method for enhanced survival. J Neurosci Methods. 2005;149(1):50–56. doi: 10.1016/j.jneumeth.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Niu J, et al. An efficient and economical culture approach for the enrichment of purified oligodendrocyte progenitor cells. J Neurosci Methods. 2012;209(1):241–249. doi: 10.1016/j.jneumeth.2012.05.032. [DOI] [PubMed] [Google Scholar]
- Zhang SC. Defining glial cells during CNS development. Nat Rev Neurosci. 2001;2(11):840–843. doi: 10.1038/35097593. [DOI] [PubMed] [Google Scholar]
- Pfeiffer SE, Warrington AE, Bansal R. The oligodendrocyte and its many cellular processes. Trends Cell Biol. 1993;3(6):191–197. doi: 10.1016/0962-8924(93)90213-k. [DOI] [PubMed] [Google Scholar]
- Hart IK, Richardson WD, Heldin CH, Westermark B, Raff MC. PDGF receptors on cells of the oligodendrocyte-type-2 astrocyte (O-2A) cell lineage. Development. 1989;105(3):595–603. doi: 10.1242/dev.105.3.595. [DOI] [PubMed] [Google Scholar]
- Nishiyama A, Lin XH, Giese N, Heldin CH, Stallcup WB. Interaction between NG2 proteoglycan and PDGF alpha-receptor on O2A progenitor cells is required for optimal response to PDGF. J Neurosci Res. 1996;43(3):315–330. doi: 10.1002/(SICI)1097-4547(19960201)43:3<315::AID-JNR6>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Pringle NP, Mudhar HS, Collarini EJ, Richardson WD. PDGF receptors in the rat CNS: during late neurogenesis, PDGF alpha-receptor expression appears to be restricted to glial cells of the oligodendrocyte lineage. Development. 1992;115(2):535–551. doi: 10.1242/dev.115.2.535. [DOI] [PubMed] [Google Scholar]
- Grinspan JB, Franceschini B. Platelet-derived growth factor is a survival factor for PSA-NCAM+ oligodendrocyte pre-progenitor cells. J Neurosci Res. 1995;41(4):540–551. doi: 10.1002/jnr.490410414. [DOI] [PubMed] [Google Scholar]
- Sharifi K, et al. Differential expression and regulatory roles of FABP5 and FABP7 in oligodendrocyte lineage cells. Cell Tissue Res. 2013;354(3):683–695. doi: 10.1007/s00441-013-1730-7. [DOI] [PubMed] [Google Scholar]
- Chittajallu R, Aguirre A, Gallo V. NG2-positive cells in the mouse white and grey matter display distinct physiological properties. J Physiol. 2004;561(Pt 1):109–122. doi: 10.1113/jphysiol.2004.074252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal R, Warrington AE, Gard AL, Ranscht B, Pfeiffer SE. Multiple and novel specificities of monoclonal antibodies O1, O4, and R-mAb used in the analysis of oligodendrocyte development. J Neurosci Res. 1989;24(4):548–557. doi: 10.1002/jnr.490240413. [DOI] [PubMed] [Google Scholar]
- Sommer I, Schachner M. Monoclonal antibodies (O1 to O4) to oligodendrocyte cell surfaces: an immunocytological study in the central nervous system. Dev Biol. 1981;83(2):311–327. doi: 10.1016/0012-1606(81)90477-2. [DOI] [PubMed] [Google Scholar]
- Boda E, et al. The GPR17 receptor in NG2 expressing cells: focus on in vivo cell maturation and participation in acute trauma and chronic damage. Glia. 2011;59(12):1958–1973. doi: 10.1002/glia.21237. [DOI] [PubMed] [Google Scholar]
- Dean JM, et al. Strain-specific differences in perinatal rodent oligodendrocyte lineage progression and its correlation with human. Dev Neurosci. 2011;33(3-4):251–260. doi: 10.1159/000327242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu WP, Collarini EJ, Pringle NP, Richardson WD. Embryonic expression of myelin genes: evidence for a focal source of oligodendrocyte precursors in the ventricular zone of the neural tube. Neuron. 1994;12(6):1353–1362. doi: 10.1016/0896-6273(94)90450-2. [DOI] [PubMed] [Google Scholar]
- Armstrong RC, Dorn HH, Kufta CV, Friedman E, Dubois-Dalcq ME. Pre-oligodendrocytes from adult human CNS. J Neurosci. 1992;12(4):1538–1547. doi: 10.1523/JNEUROSCI.12-04-01538.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gard AL, Pfeiffer SE. Oligodendrocyte progenitors isolated directly from developing telencephalon at a specific phenotypic stage: myelinogenic potential in a defined environment. Development. 1989;106(1):119–132. doi: 10.1242/dev.106.1.119. [DOI] [PubMed] [Google Scholar]
- Bjelke B, Seiger A. Morphological distribution of MBP-like immunoreactivity in the brain during development. Int J Dev Neurosci. 1989;7(2):145–164. doi: 10.1016/0736-5748(89)90065-8. [DOI] [PubMed] [Google Scholar]
- Hardy RJ, Friedrich VL., Jr Progressive remodeling of the oligodendrocyte process arbor during myelinogenesis. Dev Neurosci. 1996;18(4):243–254. doi: 10.1159/000111414. [DOI] [PubMed] [Google Scholar]
- Hartman BK, Agrawal HC, Kalmbach S, Shearer WT. A comparative study of the immunohistochemical localization of basic protein to myelin and oligodendrocytes in rat and chicken brain. J Comp Neurol. 1979;188(2):273–290. doi: 10.1002/cne.901880206. [DOI] [PubMed] [Google Scholar]
- Wei Q, Miskimins WK, Miskimins R. Stage-specific expression of myelin basic protein in oligodendrocytes involves Nkx2.2-mediated repression that is relieved by the Sp1 transcription factor. J Biol Chem. 2005;280(16):16284–16294. doi: 10.1074/jbc.M500491200. [DOI] [PubMed] [Google Scholar]
- Stolt CC, et al. Terminal differentiation of myelin-forming oligodendrocytes depends on the transcription factor Sox10. Genes Dev. 2002;16(2):165–170. doi: 10.1101/gad.215802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery B, et al. Myelin gene regulatory factor is a critical transcriptional regulator required for CNS myelination. Cell. 2009;138(1):172–185. doi: 10.1016/j.cell.2009.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds R, Wilkin GP. Development of macroglial cells in rat cerebellum. II. An in situ immunohistochemical study of oligodendroglial lineage from precursor to mature myelinating cell. Development. 1988;102(2):409–425. doi: 10.1242/dev.102.2.409. [DOI] [PubMed] [Google Scholar]
- Scolding NJ, et al. Myelin-oligodendrocyte glycoprotein (MOG) is a surface marker of oligodendrocyte maturation. J Neuroimmunol. 1989;22(3):169–176. doi: 10.1016/0165-5728(89)90014-3. [DOI] [PubMed] [Google Scholar]
- Fewster ME, Scheibel AB, Mead JF. The preparation of isolated glial cells from rat and bovine white matter. Brain Res. 1967;6(3):401–408. doi: 10.1016/0006-8993(67)90054-6. [DOI] [PubMed] [Google Scholar]
- Gard AL, Williams WC, 2nd, Burrell MR. Oligodendroblasts distinguished from O-2A glial progenitors by surface phenotype (O4+GalC-) and response to cytokines using signal transducer LIFR beta. Dev Biol. 1995;167(2):596–608. doi: 10.1006/dbio.1995.1051. [DOI] [PubMed] [Google Scholar]
- Gard AL, Pfeiffer SE. Glial cell mitogens bFGF and PDGF differentially regulate development of O4+GalC- oligodendrocyte progenitors. Dev Biol. 1993;159(2):618–630. doi: 10.1006/dbio.1993.1269. [DOI] [PubMed] [Google Scholar]
- Barres BA, Raff MC. Proliferation of oligodendrocyte precursor cells depends on electrical activity in axons. Nature. 1993;361(6409):258–260. doi: 10.1038/361258a0. [DOI] [PubMed] [Google Scholar]
- Behar T, McMorris FA, Novotny EA, Barker JL, Dubois-Dalcq M. Growth and differentiation properties of O-2A progenitors purified from rat cerebral hemispheres. J Neurosci Res. 1988;21(2-4):168–180. doi: 10.1002/jnr.490210209. [DOI] [PubMed] [Google Scholar]
- Vitry S, Avellana-Adalid V, Lachapelle F, Baron-Van Evercooren A. Migration and multipotentiality of PSA-NCAM+ neural precursors transplanted in the developing brain. Mol Cell Neurosci. 2001;17(6):983–1000. doi: 10.1006/mcne.2001.0987. [DOI] [PubMed] [Google Scholar]
- Duncan ID, Paino C, Archer DR, Wood PM. Functional capacities of transplanted cell-sorted adult oligodendrocytes. Dev Neurosci. 1992;14(2):114–122. doi: 10.1159/000111655. [DOI] [PubMed] [Google Scholar]
- Goldman JE, Geier SS, Hirano M. Differentiation of astrocytes and oligodendrocytes from germinal matrix cells in primary culture. J Neurosci. 1986;6(1):52–60. doi: 10.1523/JNEUROSCI.06-01-00052.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Althaus HH, Montz H, Neuhoff V, Schwartz P. Isolation and cultivation of mature oligodendroglial cells. Naturwissenschaften. 1984;71(6):309–315. doi: 10.1007/BF00396614. [DOI] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85(3):890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szuchet S, Yim SH. Characterization of a subset of oligodendrocytes separated on the basis of selective adherence properties. J Neurosci Res. 1984;11(2):131–144. doi: 10.1002/jnr.490110203. [DOI] [PubMed] [Google Scholar]
- Chew LJ, DeBoy CA, Senatorov VV., Jr Finding degrees of separation: experimental approaches for astroglial and oligodendroglial cell isolation and genetic targeting. J Neurosci Methods. 2014;236:125–147. doi: 10.1016/j.jneumeth.2014.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery B, Dugas JC. Purification of oligodendrocyte lineage cells from mouse cortices by immunopanning. Cold Spring Harb Protoc. 2013;2013(9):854–868. doi: 10.1101/pdb.prot073973. [DOI] [PubMed] [Google Scholar]
- Dincman TA, Beare JE, Ohri SS, Whittemore SR. Isolation of cortical mouse oligodendrocyte precursor cells. J Neurosci Methods. 2012;209(1):219–226. doi: 10.1016/j.jneumeth.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttery PC, ffrench-Constant C. Laminin-2/integrin interactions enhance myelin membrane formation by oligodendrocytes. Mol Cell Neurosci. 1999;14(3):199–212. doi: 10.1006/mcne.1999.0781. [DOI] [PubMed] [Google Scholar]
- Chun SJ, Rasband MN, Sidman RL, Habib AA, Vartanian T. Integrin-linked kinase is required for laminin-2-induced oligodendrocyte cell spreading and CNS myelination. J Cell Biol. 2003;163(2):397–408. doi: 10.1083/jcb.200304154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colognato H, Ramachandrappa S, Olsen IM, ffrench-Constant C. Integrins direct Src family kinases to regulate distinct phases of oligodendrocyte development. J Cell Biol. 2004;167(2):365–375. doi: 10.1083/jcb.200404076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ffrench-Constant C, Colognato H. Integrins: versatile integrators of extracellular signals. Trends Cell Biol. 2004;14(12):678–686. doi: 10.1016/j.tcb.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Oh LY, Yong VW. Astrocytes promote process outgrowth by adult human oligodendrocytes in vitro through interaction between bFGF and astrocyte extracellular matrix. Glia. 1996;17(3):237–253. doi: 10.1002/(SICI)1098-1136(199607)17:3<237::AID-GLIA6>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Besnard F, Perraud F, Sensenbrenner M, Labourdette G. Effects of acidic and basic fibroblast growth factors on proliferation and maturation of cultured rat oligodendrocytes. Int J Dev Neurosci. 1989;7(4):401–409. doi: 10.1016/0736-5748(89)90061-0. [DOI] [PubMed] [Google Scholar]
- Armstrong R, Friedrich VL, Holmes KV, Dubois-Dalcq M. In vitro analysis of the oligodendrocyte lineage in mice during demyelination and remyelination. J Cell Biol. 1990;111(3):1183–1195. doi: 10.1083/jcb.111.3.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinspan JB, Stern JL, Franceschini B, Pleasure D. Trophic effects of basic fibroblast growth factor (bFGF) on differentiated oligodendroglia: a mechanism for regeneration of the oligodendroglial lineage. J Neurosci Res. 1993;36(6):672–680. doi: 10.1002/jnr.490360608. [DOI] [PubMed] [Google Scholar]
- Mason JL, Goldman JE. A2B5+ and O4+ Cycling progenitors in the adult forebrain white matter respond differentially to PDGF-AA, FGF-2, and IGF-1. Mol Cell Neurosci. 2002;20(1):30–42. doi: 10.1006/mcne.2002.1114. [DOI] [PubMed] [Google Scholar]
- Schildge S, Bohrer C, Beck K, Schachtrup C. Isolation and culture of mouse cortical astrocytes. J Vis Exp. 2013. [DOI] [PMC free article] [PubMed]