

Abstract
Quantification of DNA methylation can be achieved using bisulfite sequencing, which takes advantage of the property of sodium bisulfite to convert unmethylated cytosine into uracil, in a single-stranded DNA context. Bisulfite sequencing can be targeted (using PCR) or performed on the whole genome and provides absolute quantification of cytosine methylation at the single base-resolution. Given the distinct nature of nuclear- and mitochondrial DNA, notably in the secondary structure, adaptions of bisulfite sequencing methods for investigating cytosine methylation in mtDNA should be made. Secondary and tertiary structure of mtDNA can indeed lead to bisulfite sequencing artifacts leading to false-positives due to incomplete denaturation poor access of bisulfite to single-stranded DNA. Here, we describe a protocol using an enzymatic digestion of DNA with BamHI coupled with bioinformatic analysis pipeline to allow accurate quantification of cytosine methylation levels in mtDNA. In addition, we provide guidelines for designing the bisulfite sequencing primers specific to mtDNA, in order to avoid targeting undesirable NUclear MiTochondrial segments (NUMTs) inserted into the nuclear genome.
Keywords: Genetics, Issue 135, Epigenetics, DNA methylation, bisulfite sequencing, mitochondria, mitochondrial DNA, mitochondrial DNA methylation, mitoepigenetics
Introduction
The mitochondrial genome is a circular, double-stranded structure of approximately 16.5-kilo base (kb) long, constituting of a heavy and a light strand. The mitochondrial genome is present in multiple copies within each cell, maternally-inherited, and encodes essential components of the respiratory chain complexes1. Similar to bacterial genomes and unlike the nuclear genome, the mitochondrial genome is organized in numerous secondary and tertiary structures, such as in coiled and supercoiled structures2, which can render access difficult during sequencing experiments3.
In the nucleus, methylation of the DNA is an extensively studied epigenetic mark that plays a role in numerous processes, notably in the regulation of gene expression. In mammalian genomes, DNA methylation occurs primarily on the 5-position of the pyrimidine ring of deoxycytidines, mostly on CG dinucleotides (or CpG). Cytosine methylation is found at 70% of all CpG in the genome of somatic cells and accounts for ~1% of total DNA bases4. DNA methylations has also been described in non-CpG contexts, such as CpA, CpT, and CpC and exist in various amounts in nuclear DNA, with values up to 25% of all methylated cytosines in embryonic stem cells5,6,7.
While cytosine methylation of the nuclear genome is widely accepted, the existence of mitochondrial DNA (mtDNA) methylation is still controversial. The first study investigating mtDNA methylation was performed in cultured cells where mtDNA methylation was readily detected, although at lower levels compared to nuclear DNA8. In both human and murine cells, mtDNA methylation was also detected at low levels (2-5%). Using assays relying on 5 methylcytosine capture such as methylated DNA immunoprecipitation (MeDIP) followed by quantitative PCR, mtDNA methylation was also detected in various mouse and human and cells lines9,10,11,12. Using antibodies against 5-methylcytosine in an ELISA assay or mass spectrometry, substantial levels of DNA methylation were detected from purified mitochondrial fractions13,14,15,16. However, most of the assays in the aforementioned studies used techniques that were not designed to provide absolute quantification of DNA methylation at the single base-resolution.
Quantitative and resolutive DNA methylation analysis can be achieved by a technique named "bisulfite sequencing", which takes advantage of the property of sodium bisulfite to convert unmethylated cytosine into uracil in single-stranded DNA context17. Using bisulfite sequencing, a constellation of studies has detected the presence of cytosine methylation at various levels. Methylation mtDNA in the D-loop region, the 12S or the 16S region was readily detected in human18,19,20,21,22,23 and mouse24 tissues and cells, however, with an intriguing variability, of 1-20% of total cytosines across studies.
In comparison to these numerous studies, only a few studies, including from our group, have disputed the presence of mtDNA methylation3,25,26,27 or questioned the biological relevance of very low levels of mtDNA levels (below 2%)28. Recently, we reported the observation of a potential bisulfite-sequencing artifact in whole mitochondrial bisulfite sequencing3. We provided evidence that the secondary structure of mitochondrial DNA could lead to false positives in bisulfite sequencing, thereby overestimating methylation levels. We provide here a protocol to prevent an artifact of bisulfite-conversion of mtDNA. This protocol uses a simple enzymatic digestion of DNA to disrupt mtDNA secondary structures and allow full access to bisulfite following a bisulfite sequencing protocol. In addition, we provide an accompanying bioinformatic pipeline for the analysis of bisulfite sequencing.
Protocol
1. Restriction Enzyme Treatment
Linearize mtDNA by treating total human DNA with the restriction enzyme BamHI, that cuts at position 14258 in the human mitochondrial DNA. NOTE: For mouse DNA, use the restriction enzyme BglII under identical conditions as below.
For each sample where mtDNA methylation is to be assessed, prepare one 0.2 mL reaction tube and add the following mix:3 μg genomic DNA (quantified by fluorometry), 15 μL Buffer 3, 3 μL BamHI and water up to of 150 μL
Place tubes in a thermal cycler for 4 h at 37 °C.
2. Bisulfite Conversion
Convert BamHI-treated DNA using sodium bisulfite as described elsewhere29.
Use 200-500 ng BamHI-treated DNA for each conversion reaction in a total volume of 20 μL. The DNA recovery is 50-70%.
Elute the Bisulfite-converted DNA in 10 μL elution buffer.
Quantify the single-stranded DNA by fluorometry.
Optional: Store bisulfite-converted DNA at -20 °C before proceeding the remaining of the protocol. For long-term storage, store bisulfite-converted DNA at -80 °C
3. Design of Bisulfite Sequencing Primers
Design primers for bisulfite sequencing using the online tools Methprimer30 and BiSearch31,32. NOTE: Methprimer and BiSearch allow to search for primer binding sequences on genomic sequences where cytosines are converted to thymines, similarly to post-bisulfite treatment.
For optimal and unbiased amplification, avoid CpG dinucleotides in the primers and design the amplicon size between 100-300 bp.
Ensure that designed primers are specific to the mitochondrial DNA using BiSearch's function Primer Search. Insert the already designed bisulfite converted primers, tick the bisulfite box and include a reference genome and PCR parameters. NOTE: A list of possible PCR products will be displayed.
Copy the top 1-5 primer sequences and paste them into an ordering form for oligonucleotide synthesis. NOTE: A list of human and mouse mtDNA primer sequences that has been validated in the lab is displayed in Table 1.
Test primers using bisulfite converted PCR following agarose gel electrophoresis as described in step 4. Test and optimize all primer pairs separately and ensure the correct amplicon size appears on the agarose gel. NOTE: If multiplexing primers are needed, add multiple primers to one single PCR reaction and visualize the PCR products on agarose gel electrophoresis or, a more resolutive method such as polyacrylamide gel electrophoresis, to distinguish products of similar size.
4. Bisulfite Converted PCR and Gel Extraction
To amplify the regions of interest, perform PCR using a hot start Taq polymerase. Briefly, mix 100 ng converted DNA, 500 µM sense and anti-sense primers, 1 µL dNTP mix (10 µM of each dNTP), and polymerase at 7.5 U/reaction in a total volume of 50 µL. Run PCR with the following conditions: 5 min at 95 °C (60 s 94 °C; 60 s 55 °C*; 60 s 72 °C)
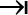 35 cycles; 10 min 72 °C. Use primers either separately or multiplexed.
35 cycles; 10 min 72 °C. Use primers either separately or multiplexed.Optional: When multiplexing primers, use 200 ng converted DNA and the following cycling conditions: 5 min 95 °C (60 s 94 °C; 90 s 55 °C*; 90 s 72 °C)
35 cycles; 10 min 72 °C. NOTE: Temperature may vary depending on the melting temperature of primers.Subject PCR products to gel electrophoresis on 2% agarose at 100 volts.
Under gentle UV light, extract PCR products with a scalpel and add to microtubes. Do not expose PCR products to excessive UV light to avoid DNA damage. Avoid exposure time over 30 s.
Purify the PCR products using gel purification as described previously3.
Quantify the purified DNA from step 4.5 using fluorometry. Proceed to step 5 or store samples at -20°C.
5. Bisulfite Sequencing Library Preparation
Perform library preparation of bisulfite-converted PCR products. Optional: multiplex PCR products at this stage.
Perform end-repair using up to 100 ng of PCR product. Add 3 µL End prep enzyme and 6.5 µL End repair reaction buffer (10x) to 55.5 µL PCR product in a 0.2 mL tube. Place and incubate tube in a thermal cycler for 30 min at 20 °C followed by 30 min at 65 °C.
Ligate sequencing adaptors by mixing the end-repaired DNA with 15 µL Blunt/TA ligase mix, 2.5 µL Adaptor and 1 µL Ligation enhancer. If using <100 ng PCR product as input, dilute adaptor 10 times.
Place the mix (step 5.3) in a thermal cycler and incubate 15 min at 20 °C. Pause the thermal cycler and add 3 µl USER Enzyme to the tube. Mix and place the tube back in the Thermal cycler and incubate 15 min at 37 °C.
Size select the adaptor-ligated DNA by using Solid Phase Reversible Immobilization (SPRI) beads with varying ratios of beads to DNA depending on PCR product size.
Add 13.5 µL H2O to adaptor-ligated DNA from step 5.4 and 55 µL resuspended SPRI beads. Incubate at room temperature for 5 min and place tube on a magnetic stand. When the solution is clear, transfer the supernatant to a new tube and discard tube containing the beads. NOTE: The supernatant contains the adaptor-ligated DNA and large unwanted fragments are bound to the discarded beads.
Add 25 µL resuspended SPRI beads to the supernatant from step 5.6, mix and incubate for 5 min at room temperature. Place the tube on the magnetic stand and discard the supernatant when the solution is clear. NOTE: The discarded supernatant contains unwanted DNA, whereas the adaptor-ligated DNA is bound to beads.
While the tube is on the magnetic stand, add 200 µL 80% ethanol (freshly prepared) to wash the beads. Incubate 30 s and remove and discard the supernatant. Repeat this step for a total of 2 washes.
Air dry beads for 5 min while the tube is on magnetic stand with an open lid.
Remove the tube from the magnet, elute the DNA in 23 µL elution buffer and mix. Incubate at room temperature for 2 min.
Place the tube on a magnetic stand and when the solution is clear, transfer 2 µL to a 96-well PCR plate for the pre-PCR amplification (step 5.14).
Transfer the rest approximately 21 µL to a new 0.2 mL tube for PCR amplification (step 5.17). NOTE: Be sure NOT to transfer any beads as beads can inhibit enzymatic reactions of next steps.
Proceed to step 5.14 or store samples at -20°C.
Perform pre-PCR amplification by RT-qPCR to estimate the number of cycles needed to amplify the adaptor-ligated DNA. Per reaction, add the following in the 96-well PCR plate containing 2 µL DNA (step 5.11): 0.4 µL Index primer, 0.4 µL universal PCR primer, 7.2 µL H2O, 10 µL RT-qPCR master mix.
Place the plate in a real-time PCR machine and subject the plate to the following conditions: 30 s at 98 °C (10 s 98 °C; 75 s 65 °C)
20 cycles; 5 min 65 °C.Estimate the number of amplification cycles needed for each sample by subtracting one Ct-value to the Ct-value of the pre-amplification RT-qPCR corresponding to the end of the linear phase of the PCR reaction.
Amplify the adaptor-ligated DNA from step 5.12 by mixing: 21 µL DNA, 25 µL PCR master mix, 1 µL Index primer, 1 µL Universal PCR primer and 2 µL H2O. In a thermal cycler, subject each sample to the following conditions: 30 s at 98 °C (10 s 98 °C; 75 s 65 °C)
[calculated Ct from step 5.16] cycles; 5 min 65 °C. NOTE: Remember to only use one Index primer per sample to allow for multiplexing. The index is inserted during the PCR.Purify the amplified DNA SPRI beads and elute in 22 µL elution buffer.
Control library quality by gel electrophoresis or alternative method. Look for correct library peak sizes (PCR product(s) size plus adaptor).
Estimate the average base-pair size of the library product(s) by smear analysis: In advanced global settings, double-click on "Table" under smear analysis. Define the region from 100-1000 bp and click OK. Under Region Table, the defined region appears and library average size (bp) is calculated.
Quantify libraries by fluorometry. Proceed to step 6 or store libraries at -20 °C.
6. Next Generation Sequencing
Calculate the nM concentration of libraries using the formula:
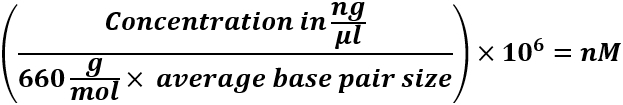
Dilute libraries to the same nM concentration e.g. 2 nM (choose 4 nM, 2 nM, 1 nM, or 0.5 nM depending on library concentrations). Pool all libraries to achieve a pool of 2 nM libraries.
Denature and dilute libraries following the sequencing instrument guide. In short: Denature the 2 nM pool by adding 0.2 N NaOH and incubate at room temperature for 5 min. Dilute the denatured pool to a final dilution of 10 pM. Denature and dilute a commercially available Control library to a final dilution of 12.5 pM. To a new tube, mix the 10 pM pool with 20% 12.5 pM Control library. NOTE: Bisulfite conversion introduces very low complexity. 20% Control library spike-in will result in better sequencing quality.
Run a 150 bp paired-end sequencing using a deep sequencing instrument.
7. Computational Analysis - Estimate Methylation Levels
Generate .fastq files (here called sample1_R1.fastq.gz and sample1_R2.fastq.gz), generate a bismark index of the entire genome of interest (here in a folder called bismarkIndex) and make sure that the genomic sequence of chrM is available in fasta format (here in a folder called chrM).
Pre-process reads to remove adapters and bias introduced by the primers: Use the tool Trim_galore33. Remove two nucleotides from the 5' end of both the forward and reverse reads. trim_galore --clip_R1 2 --clip_R2 2 -o ./ --trim1 --paired sample1_R1.fastq.gz sample1_R2.fastq.gz
Align pre-processed reads to the entire genome using Bismark34 bismark --dovetail --bam -o ./ ./bismarkIndex / -1 ./sample1_R1_val_1.fq.gz -2 ./sample1_R2_val_2.fq.gz NOTE: If another aligner is used, make sure that reads that cannot be uniquely aligned are discarded.
Extract reads mapping to the chromosome M, here using Samtools35 samtools sort -l 0 -O BAM -o sample1_sorted.bam sample1_R1_val_1_bismark_bt2_pe.bam samtools index sample1_sorted.bam samtools view -u sample1_sorted.bam chrM | samtools sort -n -O BAM -o sample1_chrM.bam
Extract the methylation information bismark_methylation_extractor -p --CX --cytosine_report --comprehensive --genome_folder ./chrM ./sample1_chrM.bam
Use the .bedGraph, the .cov or the CX_report.txt files for further analysis. Use the sample1_chrM.CX_report.txt file for visualization and testing.
Perform further clipping in the preprocessing step, if multiple regions are interrogated and a primer bias may be visible in the M-bias plots generated during the mapping. Refer to the Bismark documentation for more information.
8. Computational Analysis - Test of differences
Ensure that the CX_report.txt file contains 7 columns, chromosome (here chrM), position, strand, number of methylated reads, number of unmethylated reads, C context and surrounding sequence, for every C on chromosome M.
As the CX_report covers chrM in its entirety, check the subset to the region(s) which is amplified.
Use non-parametric tests for differences between samples, such as a Fishers's exact test for individual C's or a sign-test for an entire region. NOTE: Quantification will often be close to 0% methylation, which is the reason why assuming normality is not appropriate.
Similarly, for visualization, either visualize quantiles or calculate binomial proportion confidence intervals.
Representative Results
Two steps in this protocol are crucial when investigating mtDNA methylation. 1) The opening of the secondary structure and 2) the design of mitochondrial DNA-specific primers.
By digesting the human genomic DNA with the restriction enzyme BamHI (Figure 1), the mitochondrial DNA structure will be cut at nucleotide position 14,258 and the secondary structure will be opened.
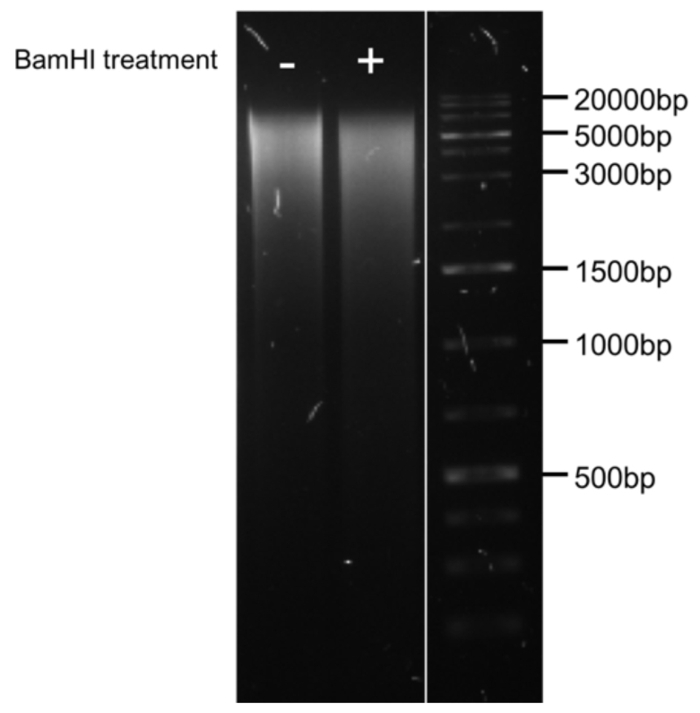
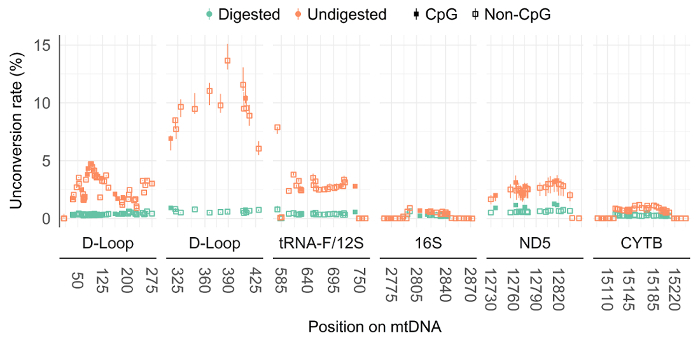
The possible impairment of bisulfite conversion with an intact mtDNA secondary structure is very likely to overestimate the mtDNA unconversion rate. By bisulfite sequencing, the mtDNA unconversion rate was investigated, in undigested and digested total DNA from human skeletal muscle cells, at 5 different regions of the mitochondrial genome: the displacement Loop (D-Loop), tRNA phenylalanine and 12S ribosome (tRNA-F+12S), 16S ribosome (16S), NADH dehydrogenase 5 (ND5) and cytochrome b (CYTB) mRNA-encoding gene (Figure 2). Unconversion rate ranged from 0 - 15.1% across all investigated regions for undigested mtDNA. However, when the DNA was digested prior to bisulfite conversion, the unconversion rate dropped to a maximum of 1% across all regions (Figure 3). Thus, illustrating the importance of BamHI digestion prior to bisulfite conversion to avoid overestimation of mtDNA methylation levels.
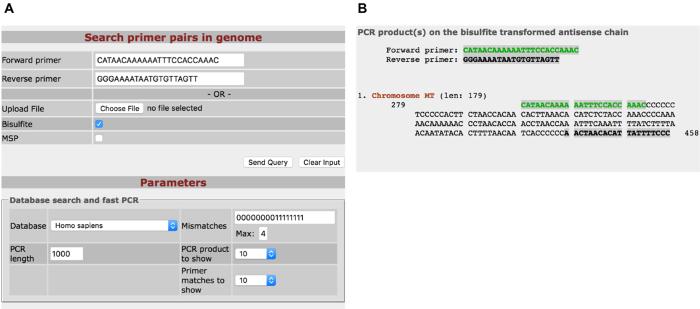
Contamination of nuclear DNA is inevitable when isolating the mitochondrial genome and since almost the entire mitochondrial genome has replicated into the nuclear genome, it has given rise to nuclear insertions of the mitochondrial genome termed NUclear MiTochondrial segments (NUMTs)36. To ensure that the analyzed DNA methylation levels correspond to mitochondrial DNA and not NUMTs, it is of outermost importance to design primers that are specific to the mitochondrial genome. Figure 4 shows how to check the primer specificity and Table 1 displays human and mouse mtDNA primer sequences.
| Name | Sequence | Position | Amplicon size | Annealing temperature | Strand |
| Human | |||||
| D-Loop | F: AAATCTATCACCCTATTAAC | ||||
| R: GTGGAAATTTTTTGTTATGATGT | 6-298 | 292 | 55°C | Antisense | |
| D-Loop | F: CATAACAAAAAATTTCCACCAAAC | ||||
| R: GGGAAAATAATGTGTTAGTT | 279-458 | 179 | 55°C | Antisense | |
| 12S+TF | F: TTTATATAACTTACCTCCTC | ||||
| R: GTGTTTGATGTTTGTTTTTTTTG | 577-765 | 188 | 55°C | Antisense | |
| 16S | F: AATAAATTTATAGGTTTTTAAATTATTAAAT | ||||
| R: TAACTAATAAAATCTTAACATATACTACTC | 2763-2873 | 110 | 53°C | Sense | |
| CYTB | F: GGTATTATTTTTTTGTTTGTAATTATAGTA | ||||
| R: CCTCAAATTCATTAAACTAAATCTATCC | 15091-15243 | 152 | 55°C | Sense | |
| Mouse | |||||
| 12S | F: ACACATACAAACCTCCATAAAC | ||||
| R: GAGGTATAATTTAGTTAAAT | 111-287 | 176 | 53°C | Antisense | |
| 16S | F: AAATTCCAATTCTCCAAACATAC | ||||
| R: TTTGGATTTTTTTTTTAGGT | 1749-1896 | 147 | 53°C | Antisense | |
| CYTB | F: CTACAAAAACACCTAATAACAAAC | ||||
| R: AGTGTATGGTTAAGAAAAGA | 14129-14307 | 178 | 55°C | Antisense | |
| 16S | F: AGAGAAATAGAGTTATTTTATAAATAAG | ||||
| R: CTAAAAACAAAATTTTAAATCTTAC | 2576-2727 | 151 | 55°C | Sense | |
| ND5 | F: TTAAGTTAATTAGGTTTGATAATAGTGA | ||||
| R: TAAAACCCTATTAAAAATAATATTCCTATA | 12659-12910 | 251 | 55°C | Sense | |
| CYTB | F: TGGGTTTTTTTTAGGAGTTTGTTTA | ||||
| R: CAATAAAAATACTCCAATATTTCAAATTTC | 14242-14504 | 262 | 55°C | Sense |
Table 1: Primer sequences for human and mouse mtDNA. Note that annealing temperatures of PCR primers vary due to differences in the melting temperature of primers.
Figure 1: Gel electrophoresis of BamHI-digested, or undigested DNA. Genomic DNA from human skeletal muscle was untreated or treated with BamHI for 4h at 37°C and gel electrophoresis was run on a 1.5% agarose gel. Please click here to view a larger version of this figure.
Figure 2: Map of the human mitochondrial genome. Regions of the mitochondrial genome investigated by bisulfite sequencing and the BamHI restriction enzyme site are displayed. The mitochondrial DNA contains 13 mRNAs, 2 ribosomal RNAs, and 22 transfer RNAs. Abbreviations: PH1: heavy-strand promoter 1; PH2: Heavy-strand promote 2; PL: Light-strand promoter; OH: Origin of replication from heavy-strand; OL: Origin of replication from light-strand. Please click here to view a larger version of this figure.
Figure 3: BamHI digestion prior to bisulfite conversion reduces the mtDNA unconversion rate in human skeletal muscle cells. By bisulfite sequencing, undigested and digested mitochondrial DNA unconversion percentage were interrogated at five different regions of the mitochondrial genome in human skeletal muscle cells (N=3). The full square represents CpG sites, and the open square represents non-CpG sites. Results are presented with a min-max interval and a sign test was used to test for significant unconversion differences. D-Loop (6-298) P= 1.83E-42; D-Loop (279-458): P= 4.55E-13; tRNA-F+12S: P= 8.38E-16; 16S: P= 5.91E-06; ND5: P=1.70E-05; CYTB: P= 2.69E-10. D-Loop (6-298) includes the origin of replication and tRNA-F+12S includes heavy strand promoter 2. These data have been published elsewhere 3. Please click here to view a larger version of this figure.
Figure 4: The use of Bisearch to verify primer specificity towards the mitochondrial genome. A) Bisulfite-converted primer sequences are added to the BiSearch function Primer Search (http://bisearch.enzim.hu/?m=genompsearch). The bisulfite box is ticked, and a reference genome is selected. B) Example of potential PCR products generated on sense and antisense chains. Please click here to view a larger version of this figure.
Discussion
Here, we provide a bisulfite-sequencing protocol which is specifically designed to interrogate mtDNA methylation. The differences with bisulfite-sequencing protocols used for genomic DNA lies in the utilization of a prior restriction enzyme digestion step and a bioinformatic analysis ruling out false positive arising from NUMT sequences.
We provide a protocol to avoid bisulfite-sequencing artifacts when investigating mtDNA methylation. Bisulfite sequencing artifacts leading to false-positives were previously reported37. Insufficient removal of DNA accessory proteins by proteinase K has been described37. Incomplete denaturation or partial reannealing can lead to stretches of incomplete conversion, possibly by lowering access of bisulfite to single-stranded DNA37. The later artifact could be at play in mtDNA where a secondary structure would impair access to cytosines. To the best of our knowledge, the addition of a restriction enzyme digestion step prior to bisulfite conversion in the context of estimating mtDNA methylation has been first reported in 20163,28. In our hands too, prior digestion with a single-cutter restriction enzyme dramatically lowered detection of unconverted cytosines3,28.
Here, we provide a bioinformatic pipeline to analyze bisulfite sequencing results and to detect bisulfite sequencing artifacts in the context of whole mitochondrial genome bisulfite sequencing. This method originates from our observation that mtDNA non-conversion rate was inversely correlated with sequencing depth3,28. This observation suggests that the secondary and even tertiary structure of mtDNA impairs the fragmentation of mtDNA at specific regions during the construction of sequencing libraries and could also impair full access of sodium bisulfite to mtDNA. Since digestion with a one-cutter enzyme releases secondary and tertiary structures, our results indicate that mtDNA structure is a source of bisulfite artifact and validate the methods described herein for the quantification of cytosine methylation in mtDNA.
The inevitable contamination of nuclear DNA in mitochondrial preparation constitutes a challenge. This leads to contamination in NUMT sequences in sequencing reads, which may bias estimation of cytosine methylation in mtDNA, even when performing targeted and not whole mitochondrial genome bisulfite sequencing. To avoid such bias, the design of unique bisulfite sequencing primers allows to exclude contamination with methylation at NUMTs. NUMTs span certain regions of the human and mouse mitochondrial genome with 100% identity, and it is, therefore, impossible to design unique primers at certain mtDNA regions. When performing whole mitochondrial genome bisulfite sequencing, sequencing long reads can ease discrimination between mtDNA and NUMTs. Aligning reads to the whole genome and only use reads uniquely mapped to the mitochondrial genome can further ensure detection of mtDNA methylation.
Finally, this protocol can be used beyond mitochondrial genomes, for example, in other instances of bisulfite sequencing where the secondary structure of DNA represents a potential source of artifact. The association between methylation levels and sequencing depth can be investigated to assess the need for a digestion with a restriction enzyme prior to bisulfite sequencing of complex DNA sequences.
Disclosures
The authors have no competing financial interests to declare.
Acknowledgments
The Novo Nordisk Foundation Centre for Basic Metabolic Research is an independent research center at the University of Copenhagen partially funded by an unrestricted donation from the Novo Nordisk Foundation.
References
- Falkenberg M, Larsson N-G, Gustafsson CM. DNA replication and transcription in mammalian mitochondria. Annu Rev Biochem. 2007;76:679–699. doi: 10.1146/annurev.biochem.76.060305.152028. [DOI] [PubMed] [Google Scholar]
- Kolesar JE, Wang CY, Taguchi YV, Chou S-H, Kaufman BA. Two-dimensional intact mitochondrial DNA agarose electrophoresis reveals the structural complexity of the mammalian mitochondrial genome. Nucleic Acids Res. 2013;41(4):e58. doi: 10.1093/nar/gks1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechta M, Ingerslev LR, Fabre O, Picard M, Barres R. Evidence Suggesting Absence of Mitochondrial DNA Methylation. Front Genet. 2017;8:166. doi: 10.3389/fgene.2017.00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich M, Gama-Sosa MA, et al. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res. 1982;10(8):2709–2721. doi: 10.1093/nar/10.8.2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462(7271):315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Zierath JR, Barres R. Evidence for non-CpG methylation in mammals. Exp Cell Res. 2011;317(18):2555–2561. doi: 10.1016/j.yexcr.2011.08.019. [DOI] [PubMed] [Google Scholar]
- Patil V, Ward RL, Hesson LB. The evidence for functional non-CpG methylation in mammalian cells. Epigenetics. 2014;9(6):823–828. doi: 10.4161/epi.28741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nass MK. Differential methylation of mitochondrial and nuclear DNA in cultured mouse, hamser and virus trasnforemd hamser cells in vivo and in vitro methylation. J Mol Biol. 1973;80(1):155–175. doi: 10.1016/0022-2836(73)90239-8. [DOI] [PubMed] [Google Scholar]
- Shock LS, Thakkar PV, Peterson EJ, Moran RG, Taylor SM. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. PNAS. 2011;108(9):3630–3635. doi: 10.1073/pnas.1012311108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellizzi D, D'Aquila P, et al. The Control Region of Mitochondrial DNA Shows an Unusual CpG and Non-CpG Methylation Pattern. DNA Res. 2013;20(6):537–547. doi: 10.1093/dnares/dst029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Y, Li R, et al. Maternal Low-Protein Diet Affects Epigenetic Regulation of Hepatic Mitochondrial DNA Transcription in a Sex-Specific Manner in Newborn Piglets Associated with GR Binding to Its Promoter. PLoS ONE. 2013;8(5):e63855. doi: 10.1371/journal.pone.0063855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Y, Song H, Gao G, Cai D, Yang X, Zhao R. Maternal Betaine Supplementation during Gestation Enhances Expression of mtDNA-Encoded Genes through D-Loop DNA Hypomethylation in the Skeletal Muscle of Newborn Piglets. J Agric Food Chem. 2015;63(46):10152–10160. doi: 10.1021/acs.jafc.5b04418. [DOI] [PubMed] [Google Scholar]
- Infantino V, Castegna A, Iacobazzi F, Spera I. Impairment of methyl cycle affects mitochondrial methyl availability and glutathione level in Down's syndrome. Mol Genet Metab. 2011;102(3):378–382. doi: 10.1016/j.ymgme.2010.11.166. [DOI] [PubMed] [Google Scholar]
- Chen H, Dzitoyeva S, Manev H. Effect of valproic acid on mitochondrial epigenetics. Eur J Pharmacol. 2012;690(1-3):51–59. doi: 10.1016/j.ejphar.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzitoyeva S, Chen H, Manev H. Effect of aging on 5-hydroxymethylcytosine in brain mitochondria. Neurobiol Aging. 2012;33(12):2881–2891. doi: 10.1016/j.neurobiolaging.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menga A, Palmieri EM, et al. SLC25A26 overexpression impairs cell function via mtDNA hypermethylation and rewiring of methyl metabolism. FEBS J. 2017;284(6):967–984. doi: 10.1111/febs.14028. [DOI] [PubMed] [Google Scholar]
- Frommer M, McDonald LE, et al. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. PNAS. 1992;89(5):1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byun H-M, Panni T, et al. Effects of airborne pollutants on mitochondrial DNA Methylation. Part Fibre Toxicol. 2013;10(1):1. doi: 10.1186/1743-8977-10-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen BG, Byun H-M, Gyselaers W, Lefebvre W, Baccarelli AA, Nawrot TS. Placental mitochondrial methylation and exposure to airborne particulate matter in the early life environment: An ENVIRONAGE birth cohort study. Epigenetics. 2015;10(6):536–544. doi: 10.1080/15592294.2015.1048412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng LD, Linarelli LE, et al. Insulin resistance is associated with epigenetic and genetic regulation of mitochondrial DNA in obese humans. Clin Epigenetics. 2015;7(1):1739. doi: 10.1186/s13148-015-0093-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byun H-M, Colicino E, Trevisi L, Fan T, Christiani DC, Baccarelli AA. Effects of Air Pollution and Blood Mitochondrial DNA Methylation on Markers of Heart Rate Variability. J Am Heart Assoc. 2016;5(4) doi: 10.1161/JAHA.116.003218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchessi V, Vinci MC, et al. Mitochondrion. MITOCH. 2016;27(C):40–47. doi: 10.1016/j.mito.2016.02.004. [DOI] [PubMed] [Google Scholar]
- Wijst MGP, van Tilburg AY, Ruiters MHJ, Rots MG. Experimental mitochondria-targeted DNA methylation identifies GpC methylation, not CpG methylation, as potential regulator of mitochondrial gene expression. Sci Rep. 2017;7(1):177. doi: 10.1038/s41598-017-00263-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong M, Gertz B, Chestnut BA, Martin LJ. Mitochondrial DNMT3A and DNA methylation in skeletal muscle and CNS of transgenic mouse models of ALS. Front cellr Neurosci. 2013;7:279. doi: 10.3389/fncel.2013.00279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawid IB. 5-methylcytidylic acid: absence from mitochondrial DNA of frogs and HeLa cells. Science. 1974;184(4132):80–81. doi: 10.1126/science.184.4132.80. [DOI] [PubMed] [Google Scholar]
- Gama-Sosa MA. Levels and distribution of 5-methylcytosine in Chordate DNA. 1985. pp. 1–201.
- Hong EE, Okitsu CY, Smith AD, Hsieh C-L. Regionally specific and genome-wide analyses conclusively demonstrate the absence of CpG methylation in human mitochondrial DNA. Mol Cell Biol. 2013;33(14):2683–2690. doi: 10.1128/MCB.00220-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Du Q, et al. CpG methylation patterns ofhuman mitochondrial DNA. Nature Publishing Group. 2016. pp. 1–10.
- Donkin I, Versteyhe S, et al. Obesity and Bariatric Surgery Drive Epigenetic Variation of Spermatozoa in Humans. Cell Metab. 2016;23(2):369–378. doi: 10.1016/j.cmet.2015.11.004. [DOI] [PubMed] [Google Scholar]
- Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18(11):1427–1431. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
- Tusnády GE, Simon I, Váradi A, Arányi T. BiSearch: primer-design and search tool for PCR on bisulfite-treated genomes. Nucleic Acids Res. 2005;33(1):e9. doi: 10.1093/nar/gni012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arányi T, Váradi A, Simon I, Tusnády GE. The BiSearch web server. BMC bioinformatics. 2006;7:431. doi: 10.1186/1471-2105-7-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F. Trim Galore! 2018. Available from: http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/
- Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27(11):1571–1572. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos A, Barbena E, et al. Nuclear insertions of mitochondrial origin: Database updating and usefulness in cancer studies. Mitochondrion. 2011;11(6):946–953. doi: 10.1016/j.mito.2011.08.009. [DOI] [PubMed] [Google Scholar]
- Warnecke PM, Stirzaker C, Song J, Grunau C, Melki JR, Clark SJ. Identification and resolution of artifacts in bisulfite sequencing. Methods (San Diego, Calif) 2002;27(2):101–107. doi: 10.1016/s1046-2023(02)00060-9. [DOI] [PubMed] [Google Scholar]