

Abstract
Organ-to-organ communication by endocrine signaling, for example, from the periphery to the brain, is essential for maintaining homeostasis. As a model animal for endocrine research, Drosophila melanogaster, which has sophisticated genetic tools and genome information, is being increasingly used. This article describes a method for the calcium imaging of Drosophila brain explants. This method enables the detection of the direct signaling of a hormone to the brain. It is well known that many peptide hormones act through G-protein-coupled receptors (GPCRs), whose activation causes an increase in the intracellular Ca2+concentration. Neural activation also elevates intracellular Ca2+ levels, from both Ca2+ influx and the release of Ca2+ stored in the endoplasmic reticulum (ER). A calcium sensor, GCaMP, can monitor these Ca2+ changes. In this method, GCaMP is expressed in the neurons of interest, and the GCaMP-expressing larval brain is dissected and cultured ex vivo. The test peptide is then applied to the brain explant, and the fluorescent changes in GCaMP are detected using a spinning disc confocal microscope equipped with a CCD camera. Using this method, any water-soluble molecule can be tested, and various cellular events associated with neural activation can be imaged using the appropriate fluorescent indicators. Moreover, by modifying the imaging chamber, this method can be used to image other Drosophila organs or the organs of other animals.
Keywords: Neuroscience, Issue 136, Calcium imaging, GCaMP, brain explant, peptide hormone, G-protein-coupled receptor, Drosophila
Introduction
Organ-to-organ communication is an evolutionarily conserved strategy for maintaining homeostasis to cope with environmental changes. In humans, a variety of hormones arereleased from the endocrine glands into the circulation. Many of these hormones target the hypothalamus of the brain, which regulates metabolic processes and fundamental behaviors such as feeding1,2. Many hormones have been discovered using mammalian models. However, the mechanisms of their action, especially the interorgan networks in which they participate, remain largely unclear.
Drosophila melanogaster has emerged as a useful model for studying organ-to-organ communication. In insects, many physiological processes are controlled by hormones. Early studies focusing on growth and metamorphosis used large insects. In these studies, the removal or transplantation of specific organs predicted the existence of inter-organ signaling molecules; later, Juvenile hormone (JH), Prothoracicotropic hormone (PTTH), and Ecdysone were biochemically purified3,4,5. A large family of intercellular signaling peptides is thought to be involved in various physiological events during the insect life cycle6,7. Most of these peptides act on G-protein-coupled receptors (GPCRs), although the specific GPCRs were initially difficult to identify using conventional approaches. The publication of the Drosophila whole genome sequence8 was the breakthrough that enabled the identification of Drosophila bioactive peptides based on their homology to those found in other insects. In addition, the receptors for several peptides were identified from GPCRs predicted in the genome using cell-culture-based GPCR-ligand binding assays. Next, expression analyses predicted the organ-to-organ pathways elicited by these peptides and receptors. Notably, many of the putative peptide receptors are expressed in the brain, suggesting that the brain is a major target of peptide hormones9. Moreover, the advanced genetic tools in Drosophila have contributed to the identification of physiological roles of peptide-GPCR combinations. For instance, the GAL4 and LexA-based binary transcriptional systems enable gene knockdown or overexpression in a spatially and temporally controlled manner. GAL4, a transcription factor identified in yeast, binds to a specific cis-regulatory sequence called Upstream Activation Sequence (UAS). In the GAL4/UAS system, the driver line provides tissue-specific or stage-specific GAL4 expression and the responder line carries UAS upstream of the gene of interest or the construct to drive shRNA expression. LexA/LexAop system is based on a similar mechanism. Phenotypic analyses of tissue-specific peptide-knockdown and tissue-specific GPCR-knockdown animals can reveal information about the peptide-GPCR signaling's mode and site of action. However, the conclusions that can be reached solely by genetic data are limited. On the other hand, once the putative target of a particular peptide hormone is narrowed down to a tissue or cell type, ex vivo calcium imaging in organ explants can be used to elucidate the organ-to-organ communication mediated by peptide-GPCR signaling. Upon the activation of a Gq-coupled GPCR, the intracellular Ca2+ concentration is increased due to the release of Ca2+ from the ER10. In the brain, neural activation also elevates the intracellular Ca2+ levels. Such Ca2+ increases can be detected by a calcium sensor, GCaMP, which undergoes conformational changes in the presence of Ca2+ resulting in fluorescence emission11.
In this article, a calcium imaging method using Drosophila brain explants is described. To test the ability of a peptide to activate specific neurons, a test peptide is applied to the GCaMP-expressing brain explant, and fluorescence changes are monitored by confocal microscopy. The involvement of the GPCR is then confirmed by performing the same assay using a mutant brain lacking the GPCR. This combination of imaging and genetics provides precise information about organ-to-organ communication by peptides-GPCRs. Possible modifications and applications of this protocol are also discussed.
Protocol
1. Preparation of Larval Brain Explants
- Make an imaging chamber. NOTE: Stable recording requires that the brain explants be tethered. Here, a low-cost, hand-made imaging chamber is used in the Ca2+ imaging system.
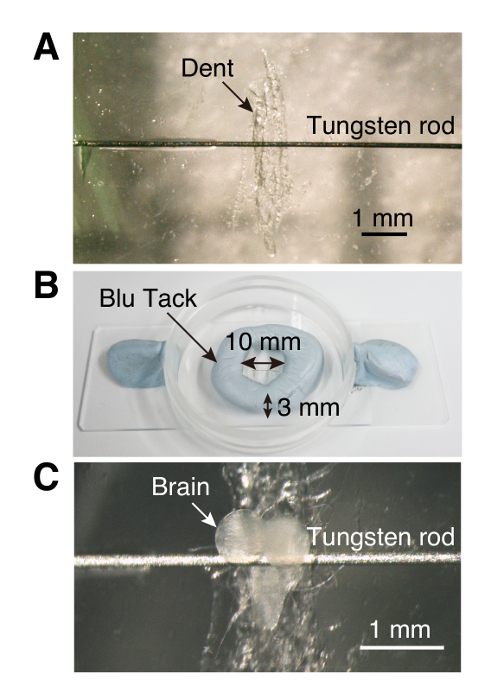
- Using forceps, scratch the bottom center of a plastic culture dish (35 mm x 10 mm) to create a dent for the ventral ganglion of the larval brain making mounting easier (step 1.3, Figure 1).
- With a toothpick, place a small drop of superglue on either side of the dent and attach a tungsten rod (0.125 mm diameter, 6 to 7 mm length) to the glue.
- Using a small piece of putty-like reusable adhesive, make a circular wall (10 mm in diameter and 3 mm high) surrounding the dent (Figure 1). NOTE: The space inside the wall will be later filled with phosphate buffered saline (PBS; 0.14 M NaCl, 0.0027 M KCl, 0.01 M PO43-, pH 7.4 ± 0.05 at 25 °C).
- Animal preparation NOTE: For calcium imaging, a calcium indicator, GCaMP6s, is expressed in the cell type of interest, which is commonly achieved using combinations of tissue-specific GAL4 and UAS-GCaMP6s. To ensure that the signal is mediated through the candidate receptor, GCaMP6s is also expressed in a mutant defective in the receptor.
- When experimental comparisons for different genotypes such as wild type and mutants are required, age match test larvae to avoid GCaMP6 expression level variations and physiological differences due to developmental stages.
- Keep parental flies (30 flies for each sex) in a culture vial containing standard fly food for 8 h to allow egg-laying on the surface of the food. Culture larvae in mass under a 12-h light-dark cycle at 25 °C and 60-70 % relative humidity.
- Dissection of the larval brain
- At 90-120 h after egg laying (AEL), collect larvae and wash them at least 3 times with distilled water to remove adherent food debris.
- Place a larva on a square 1.5-inch watch glass filled with ice cold PBS. NOTE: The next two steps are carried out in this watch glass under the dissecting microscope.
- Use forceps to gently grab the middle part of the larva. Use another forceps to grab the mouth hooks and pull the mouth hooks gently to separate the anterior part of the larva containing the brain from the rest of the body. NOTE: Alternatively, surgical scissors can be used.
- Hold the anterior tip by forceps and turn the larva inside out. Remove extraneous tissue attached to the brain, such as imaginal disks, fat bodies, and ring gland. Gently separate the brain from the mouthparts.
- Apply 200 µL of PBS to the inside of the ring made of the putty-like reusable adhesive in the imaging chamber. Gently suck the dissected brain with PBS into a Pasteur pipette, and then transfer it into the imaging chamber.
- Setting the brain into the imaging chamber
- Use forceps to grab muscle fibers extending from the ventral ganglia. Insert the brain gently into the dent underneath the tungsten wire.
- Using another forceps, pull the tungsten wire slightly up to place the brain at the correct position for imaging (Figure 1).
2. Acquisition of Ca2+ Fluorescence Images
NOTE: Brain explants immersed in PBS are imaged using a fluorescence microscope equipped with a 20X water-immersion objective lens (N.A. = 0.5). PBS does not activate cells in the brain. If Z-axis images are needed, the objective lens is mounted with a piezoelectric-activated lens mover. A spinning disc confocal head is used to enhance the time resolution. For low-light imaging, an electron multiplying charge-coupled device camera is mounted on the microscope. The imaging of GCaMP6s fluorescence (excitation/emission: 488/509 nm) requires a 488-nm laser for excitation with a dichroic beam splitter and an emission filter (e.g., 528 ± 38 nm band bandpass filter).
Place the imaging chamber containing the brain explant under the microscope.
Lower the objective lens until it touches the PBS. Under bright-field illumination, position the brain and bring it into focus. Switch to fluorescent light and adjust the focus on the GCaMP6s-labeled cells. NOTE: The basal green fluorescence of the GCaMP6s should be visible.
Start acquisition at 250 ms/frame at a resolution of 512 × 512 pixels in water-cooled mode using the appropriate acquisition software (e.g., µManager). Adjust the exposure time to obtain fluorescence values within the dynamic range of the CCD camera, but not lower than 1000 (arbitrary units), with 16-bit images (dynamic range 0-65,535). NOTE: A low exposure time should be used to minimize photo-bleaching of the GCaMP6s. Exposure time should be optimized in each experiment as it depends on the expression levels of GCaMP6 and the sensitivity of the detection system. It was 100 ms in our experiment using dilp2>GCaMP6s. When z-sections are required for a given imaging depth, several z-section images can be acquired depending on the exposure time and frame intervals. If the camera has enough spatial resolution, the binning size (number of registers on the chip that will be binned into one digital pixel) should be increased to reduce the exposure time.
Once imaging parameters are determined, take images for 1 min before peptide administration to detect baseline signal intensities (F0).
Apply the test peptide; for this, dissolve 100 µL of peptide solution in PBS and pipette it directly into the larval bath to yield an optimal concentration. NOTE: The peptide solution should be injected slowly (e.g., flow rate 20 µL/s) into the bath solution using a pipette. Unrelated synthetic peptide dissolved in PBS is used as a negative control.
Record the GCaMP6s emission for a few minutes.
3. Data Analysis
Note: Imaging data are analyzed offline with the ImageJ. During the time-lapse imaging, pipetting causes the larval brain to move. Therefore, positional aberrations of the serial images should be corrected before analysis. The correction can be performed using an ImageJ plug-in, TurboReg as follows.
Open the analysis software and use the first frame (before peptide application) as a reference image.
Select Plugins | Registration | TurboReg.
Choose the serial image file as the Source and the reference image as the Target.
Check Rigid Body (parallel displacement and rotation of images) and Accurate ("Fast" is coarse but fast analysis instead) for the processing method and quality, respectively.
Click Batch to start the image processing.
Select multiple regions of interest (ROIs) by using Analyze | Tools | ROI Manager in ImageJ.
Measure the pixel intensity by clicking More | Multi Measure | OK in the ROI Manager window.
Calculate ΔF/F0 = (F - F0)/F0, where F is the ROI intensity at the time of the stimulus, and F0 is the intensity at a time window immediately before a particular experimental event (e.g., 30 frames preceding the stimulus onset).
Repeat the experiment using multiple brain samples, conducting statistical processing to obtain the mean and standard error of the mean (SEM) at each time point.
Representative Results
Using this protocol, the activation of brain insulin-producing cells (IPCs) by the peptide hormone, CCHa2, was examined. IPCs of the wild-type brain were labeled with GCaMP6s using a combination of dilp2-GAL412 (a gift from Dr. Rulifson) and UAS-GCaMP6s13 (a gift from Dr. Kim). Larval brain explants were treated with a synthetic CCHa2 peptide, and the fluorescence from GCaMP6s was recorded in real time. In this experiment, images were obtained from a single focal plane. Each IPC cluster contains around 14 cells14, and signals from 3 to 6 cells were detected. The GCaMP6s signal intensity was dramatically increased upon the CCHa2 administration (Figure 2A, Movie 1). The wild-type brains did not show an obvious response to Ghrelin or Nociceptin (Movie 2 and 3, respectively), which are mammalian peptide hormones without a Drosophila homologue. These results indicate that the observed activation of IPCs by CCHa2 is a specific response to CCHa2. To clarify whether CCHa2 acts through CCHa2-R, the same analysis was conducted using CCHa2-R mutant brains. No such increase in the signal intensity was observed after CCHa2 administration in the mutant brains (Movie 4). Statistical analysis showed that the difference in signal intensity between the wild-type and CCHa2-R mutant brains became significant within 2 min after CCHa2 application and was maintained for at least 7 min (Figure 2B). These results indicate that IPCs are specifically activated by CCHa2 through CCHa2-R. All peptides used in this study were diluted by PBS to yield a final concentration of 10-9 M.
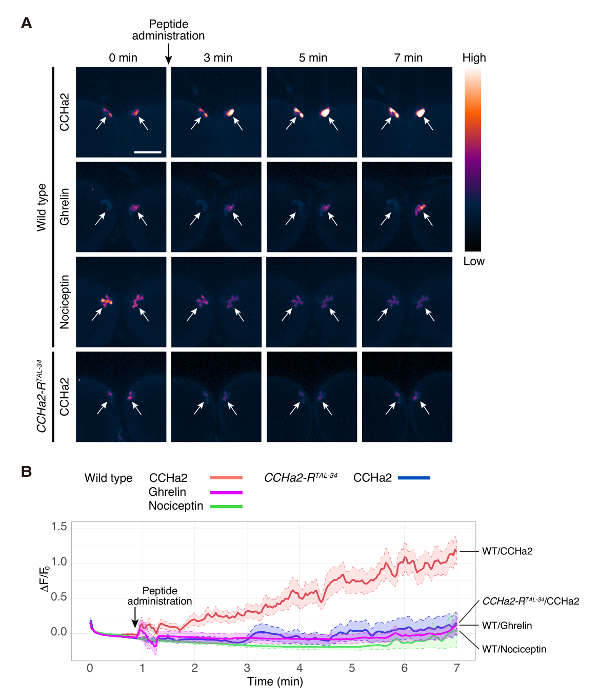
Figure 1. Imaging chamber (A) Culture dish (35 mm x 10 mm) with a small dent and tethering rod. (B) Ring-shaped wall made of putty-like reusable adhesives. (C) A larval brain explant inserted in the imaging chamber. Please click here to view a larger version of this figure.
Figure 2. Calcium imaging of the larval brain explant Wild-type (dilp2-Gal4/UAS-GCaMP6s) brain was exposed to CCHa2, Ghrelin, and Nociceptin. CCHa2-R mutant (dilp2-Gal4, CCHa2-RTAL-34/UAS-GCaMP6s, CCHa2-RTAL-34) brain was exposed to CCHa2. GCaMP6s signals in IPCs were detected by confocal microscopy at 250 ms/frame. Still images of selected time points are shown in (A). The images were pseudocolored to better visualize different signal intensities. ΔF/F0 at each time point was plotted in (B). ΔF/F0 was calculated from 5 to 10 different preparations. ROIs were set on cell bodies that were detected in a same focal plane. The solid lines indicate the mean of 5 to 10 samples, and the dotted lines mark upper and lower limits of the standard error of the mean. The shaded areas indicate the variation of the signals in the experiments. Scale bar indicates 50 µm. The figure is adapted from Sano et al.15. Please click here to view a larger version of this figure.
 Movie 1. Wild-type IPCs exposed to CCHa2
15
Please click here to view this video. (Right-click to download.)
Movie 1. Wild-type IPCs exposed to CCHa2
15
Please click here to view this video. (Right-click to download.)
 Movie 2. Wild-type IPCs exposed to Ghrelin
15
Please click here to view this video. (Right-click to download.)
Movie 2. Wild-type IPCs exposed to Ghrelin
15
Please click here to view this video. (Right-click to download.)
 Movie 3. Wild-type IPCs exposed to Nociceptin
15
Please click here to view this video. (Right-click to download.)
Movie 3. Wild-type IPCs exposed to Nociceptin
15
Please click here to view this video. (Right-click to download.)
 Movie 4.
CCHa2-R
mutant IPCs exposed to CCHa2
15
Please click here to view this video. (Right-click to download.)
Movie 4.
CCHa2-R
mutant IPCs exposed to CCHa2
15
Please click here to view this video. (Right-click to download.)
Discussion
The Ca2+ imaging method described here is a useful system for testing the function of hormones in the brain. In vivo, the brain receives hormones from various endocrine organs. In addition, the brain constantly perceives ascending sensory information, which causes spontaneous neuronal activation. This ex vivo system eliminates such noise and yields a better signal/noise ratio than in vivo imaging. In this system, molecules can be tested singly or in combination. In addition to peptide hormones, any water-soluble molecules, including natural or synthetic compounds, can be applied to the brain explant.
For Ca2+ imaging, brain explants need to be tethered. For this purpose, a gel matrix made of low-melting point agarose or fibrin has been used previously16,17. These gel matrixes can hold the sample stably, however, tissues are heated at above 30 °C. This temperature causes a heat-shock response in poikilothermic animals such as insects. To avoid heat stress, the brain could be tethered physically. The brain attached to the cuticle are fastened with insect pins18. Our method using a tungsten wire provides simple and stable holding of the sample. This system is easy to prepare and reusable, and allows stimulants to reach the target cells more easily compared to the gel embedding method. Thus, this will be useful for ligand screening.
The choice of fluorescent indicator depends on the type of target neurons, the type of receptors expressed in the neuron, and the biological questions being addressed. A series of GCaMP6 variants optimized for detecting various temporal Ca2+ dynamics has been generated13. In particular, GCaMP6 variants differ in their response kinetics: GCaMP6s (slow), GCaMP6m (medium), and GCaMP6f (fast). The decay times for GCaMP6s and 6m are relatively slow (i.e., τ1/2 after 10 action potentials in a hippocampal slice preparation) while that for GCaMP6f is fast (τ1/2 after 1 action potential). Other types of available fluorescent indicators for neural activity include EPAC-cAMP (cAMP), Synapto-pHluorin (synaptic release), and ArcLight (membrane potential)19,20,21. These genetically encoded fluorescent indicators can be expressed in specific cell types. Thus, the ex vivo imaging system can detect various responses in desired neurons.
There are several critical steps in this protocol. First, extraneous tissues must be removed from the brain to obtain a clear image. The use of sharpened forceps and surgical scissors eases the delicate work. Second, the dissected brain should be fixed into position. Occasionally, the brain sample slightly moves from pipetting (the range approximately 5 µm). To prevent the brain from moving, the height of the tungsten wire in the imaging chamber should be adjusted to the size of the brain sample. Alternatively, a gravity-feed perfusion system could be installed for gentle stimulation. Third, the results of the brain imaging should be interpreted with caution. The ligand applied to the brain might stimulate unexpected neurons, which in turn could activate the target neurons. To detect the direct effect of a hormone on specific neurons, the receptor should be knocked-down in the targetneurons. In this case, the expression of the fluorescent indicator can be combined with that of the receptor RNAi using the Gal4/UAS system.
A minor limitation of this protocol is that the duration of imaging is limited due to the eventual damage of the brain sample. In our hands, the Drosophila brain can be imaged for up to approximately 60 min using this protocol. Hemolymph-like solution, such as HL3.1 saline22, and laser intensities could enable long-term imaging. In addition, the current imaging setup is not suitable for repetitive stimulation of the same sample. For such experiments, a perfusion bath could be used.
Finally, this protocol has various applications. By modifying the imaging chamber, the adult brain and other Drosophila tissues or the tissues of other animals could be imaged. In non-model organisms, the lack of genetic tools has made it difficult to introduce fluorescent indicators into the genome. However, recently developed CRISPR/Cas9 genome editing techniques have opened the door to genetic engineering in these animals. Thus, this protocol could be used to study inter-organ signaling in a wide range of animals.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by Grants-in-Aid for Scientific Research (15K07147, 17K07419) from JSPS (to Hiroshi Ishimoto, Hiroko Sano), an Inamori Foundation Research Grant (to HI), and the Program of the Joint Usage/Research Center for Developmental Medicine, at the Institute of Molecular Embryology and Genetics, Kumamoto University (to HS). We thank Dr. Azusa Kamikouchi and Mr. Daichi Yamada for their help in using the imaging system.
References
- Morton GJ, Schwartz MW. Leptin and the central nervous system control of glucose metabolism. Physiological Reviews. 2011;91(2):389–411. doi: 10.1152/physrev.00007.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley S, Wynne K, McGowan B, Bloom S. Hormonal regulation of food intake. Physiological Reviews. 2005;85(4):1131–1158. doi: 10.1152/physrev.00015.2004. [DOI] [PubMed] [Google Scholar]
- Goodman WG, Cusson M. The juvenile hormones. In: Gilbert LI, editor. Insect Endocrinology. Academic Press; 2012. pp. 310–365. [Google Scholar]
- Lafont R, Dauphin-Villemant C, Warren JT, Rees H. Ecdysteroid chemistry and biochemistry. In: Gilbert LI, editor. Insect Endocrinology. Academic Press; 2012. pp. 106–176. [Google Scholar]
- Smith W, Rybczynski R. Protothoracicotropic hormone. In: Gilbert LI, editor. Insect Endocrinology. Academic Press; 2012. pp. 1–62. [Google Scholar]
- Claeys I, et al. Insect neuropeptide and peptide hormone receptors: current knowledge and future directions. Vitamins and Hormones. 2005;73:217–282. doi: 10.1016/S0083-6729(05)73007-7. [DOI] [PubMed] [Google Scholar]
- Gade G, Goldsworthy GJ. Insect peptide hormones: a selective review of their physiology and potential application for pest control. Pest Management Science. 2003;59(10):1063–1075. doi: 10.1002/ps.755. [DOI] [PubMed] [Google Scholar]
- Adams MD, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287(5461):2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- Park D, Veenstra JA, Park JH, Taghert PH. Mapping peptidergic cells in Drosophila: where DIMM fits in. PLoS One. 2008;3(3):1896. doi: 10.1371/journal.pone.0001896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361(6410):315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- Nakai J, Ohkura M, Imoto K. A high signal-to-noise Ca(2+) probe composed of a single green fluorescent protein. Nature Biotechnology. 2001;19(2):137–141. doi: 10.1038/84397. [DOI] [PubMed] [Google Scholar]
- Rulifson EJ, Kim SK, Nusse R. Ablation of insulin-producing neurons in flies: growth and diabetic phenotypes. Science. 2002;296(5570):1118–1120. doi: 10.1126/science.1070058. [DOI] [PubMed] [Google Scholar]
- Chen TW, et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature. 2013;499(7458):295–300. doi: 10.1038/nature12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassel DR, Kubrak OI, Liu Y, Luo J, Lushchak OV. Factors that regulate insulin producing cells and their output in Drosophila. Frontiers in Physiology. 2013;4:252. doi: 10.3389/fphys.2013.00252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano H, et al. The Nutrient-Responsive Hormone CCHamide-2 Controls Growth by Regulating Insulin-like Peptides in the Brain of Drosophila melanogaster. PLoS Genetics. 2015;11(5):1005209. doi: 10.1371/journal.pgen.1005209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao CK, Ku HY, Lee YM, Huang YF, Sun YH. Long Term Ex Vivo Culture and Live Imaging of Drosophila Larval Imaginal Discs. PLoS One. 2016;11(9):0163744. doi: 10.1371/journal.pone.0163744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabado VaN, Nagoshi E. Single-cell Resolution Fluorescence Live Imaging of Drosophila Circadian Clocks in Larval Brain Culture. Journal of Visualized Experiments. 2018;131:e57015. doi: 10.3791/57015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolopoulou AA, et al. Caffeine Taste Signaling in Drosophila Larvae. Frontiers in Cellular Neuroscience. 2016;10:193. doi: 10.3389/fncel.2016.00193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao G, et al. Genetically targeted optical electrophysiology in intact neural circuits. Cell. 2013;154(4):904–913. doi: 10.1016/j.cell.2013.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev VO, Bunemann M, Hein L, Hannawacker A, Lohse MJ. Novel single chain cAMP sensors for receptor-induced signal propagation. Journal of Biological Chemistry. 2004;279(36):37215–37218. doi: 10.1074/jbc.C400302200. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S, De Angelis D, Rothman JE, Ryan TA. The use of pHluorins for optical measurements of presynaptic activity. Biophysical Journal. 2000;79(4):2199–2208. doi: 10.1016/S0006-3495(00)76468-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Ueda A, Wu CF. A modified minimal hemolymph-like solution, HL3.1, for physiological recordings at the neuromuscular junctions of normal and mutant Drosophila larvae. Journal of Neurogenetics. 2004;18(2):377–402. doi: 10.1080/01677060490894522. [DOI] [PubMed] [Google Scholar]