

Abstract
Multiple enhancers often regulate a given gene, yet for most genes, it remains unclear which enhancers are necessary for gene expression, and how these enhancers combine to produce a transcriptional response. As millions of enhancers have been identified, high-throughput tools are needed to determine enhancer function on a genome-wide scale. Current methods for studying enhancer function include making genetic deletions using nuclease-proficient Cas9, but it is difficult to study the combinatorial effects of multiple enhancers using this technique, as multiple successive clonal cell lines must be generated. Here, we present Enhancer-i, a CRISPR interference-based method that allows for functional interrogation of multiple enhancers simultaneously at their endogenous loci. Enhancer-i makes use of two repressive domains fused to nuclease-deficient Cas9, SID and KRAB, to achieve enhancer deactivation via histone deacetylation at targeted loci. This protocol utilizes transient transfection of guide RNAs to enable transient inactivation of targeted regions and is particularly effective at blocking inducible transcriptional responses to stimuli in tissue culture settings. Enhancer-i is highly specific both in its genomic targeting and its effects on global gene expression. Results obtained from this protocol help to understand whether an enhancer is contributing to gene expression, the magnitude of the contribution, and how the contribution is affected by other nearby enhancers.
Keywords: Genetics, Issue 136, Gene regulation, CRISPR/Cas9, enhancer, epigenetics, functional genomics, transcription factors
Introduction
Large-scale sequencing projects such as ENCODE1, Roadmap Epigenomics2, and FANTOM3 have identified millions of putative enhancers within the human genome across hundreds of cell types. It is estimated that each promoter associates with an average of 4.9 enhancers and each enhancer contacts an average of 2.4 genes3, suggesting that gene expression is often the result of the integration of multiple distributed regulatory interactions. A significant remaining challenge is to define not only how individual enhancers contribute to gene expression, but how they combine to affect expression. Genetic approaches are commonly used to identify relationships between enhancers in model organisms from Drosophila4 to mice5. However, these experiments are time-consuming and low-throughput for the study of multiple enhancers at multiple genes.
One approach for studying enhancer function on a large scale involves massively parallel reporter assays. These assays allow for the simultaneous screening of thousands of DNA sequences for their ability to drive the expression of a reporter gene6. While these assays have shown that DNA sequence can alone be sufficient to convey gene regulation information7, they come with the caveats of being performed outside of the native chromatin context and with a heterologous promoter. In addition, the size of DNA sequence being analyzed in massively parallel reporter assays is usually less than 200 basepairs, which may exclude relevant surrounding sequence. Importantly, as reporter assays only measure the activity of one sequence at a time, they do not take into account the complex relationships that can exist between enhancers. Thus, while massively parallel reporter assays can be informative about the intrinsic activity of a DNA sequence, they do not necessarily inform us of the function of that DNA sequence in the context of the genome.
Recently developed CRISPR/Cas9 tools8 have facilitated the study of gene regulation as they allow for the deletion of enhancers at the endogenous locus. However, deleting multiple enhancers simultaneously may lead to genomic instability, and it is time consuming to generate successive enhancer deletions in a single cell line. In addition, new genomic sequence is created at the site of the deletion following repair, and this sequence may gain regulatory function. An alternative version of Cas9 has been developed specifically for modulating gene expression, relying on fusions of activating9,10 or repressing11,12 domains to the nuclease-deficient form of Cas9 (dCas9). These fusion proteins are ideal for studying multiple loci simultaneously as they do not physically alter the DNA sequence, and instead modulate epigenetics in order to interrogate a regulatory region. The most widely used repressive fusion is KRAB, which recruits the KAP1 co-repressor complex, promoting the deposition of the repression-associated histone H3 lysine 9 trimethylation (H3K9me3)13. dCas9-KRAB, also known as CRISPR interference14, has been used to target and screen individual enhancers for their contributions to gene expression15,16; however, it has not been optimized for targeting multiple regions simultaneously. One version of multiplex CRISPR interference for enhancers, Mosaic-seq17, uses single cell RNA-seq as a readout, but this technology is expensive and only suitable for the study of highly expressed genes due to the low sensitivity of single cell RNA-seq.
We sought to develop a CRISPR interference-based method for dissecting combinatorial enhancer function within the context of a transcriptional response to estrogen. About half of estrogen-responsive genes contain 2 or more enhancers bound by estrogen receptor alpha (ER) nearby18, suggesting that multiple enhancers may be participating in the estrogen response, and understanding the regulatory logic would require targeting multiple enhancers simultaneously. As initial studies using CRISPR interference at promoters suggested that not all promoters are equally responsive to KRAB-mediated repression19, we reasoned that the addition of a distinct repressive domain to dCas9 may facilitate the deactivation of diverse enhancers. We chose the Sin3a Interacting Domain of Mad1 (SID)20 as it leads to the recruitment of histone deacetylases21, which remove acetyl groups on histones that are associated with transcriptional activity. Importantly, the SID domain was effective at reducing gene expression when fused to dCas922 and TALEs23, and Sin3a has been shown to be a potent repressive co-factor in a variety of enhancer sequence contexts24. We used SID4x-dCas9-KRAB (Enhancer-i) to target 10 different enhancers bound by the ER, and identify ER binding sites (ERBS) that are necessary for the estrogen transcriptional response at 4 genes18. We also targeted the combinations of enhancers to identify the sites that cooperate in the production of the estrogen transcriptional response. We found that up to 50 sites can potentially be targeted simultaneously with detectable gene expression changes. Using ChIP-seq and RNA-seq, we demonstrated that Enhancer-i is a highly specific technique for studying multiple enhancers simultaneously.
In this protocol, we describe the steps involved in performing Enhancer-i, a flexible technique that enables the functional study of multiple enhancers simultaneously in a tissue culture setting. Enhancer-i is highly correlated with genetic deletion but provides transient deactivation that is dependent on histone deacetylases (HDACs). By delivering guide RNAs via transient transfection as opposed to stable integration via viral vectors, this protocol avoids deposition and potential spreading of H3K9me3. This protocol details guide RNA design and cloning via Gibson assembly, the transfection of guide RNAs using lipofection, and the analysis of resulting gene expression changes by qPCR. We also include the methods for evaluating the specificity of Enhancer-i targeting at the level of the genome and transcriptome. While this technique was developed to study gene regulation by ER bound enhancers in human cancer cell lines, it is applicable to the dissection of any mammalian enhancer.
Protocol
1. Generation of Cell Lines Stably Expressing SID4X-dCas9-KRAB
Note: The transfection conditions and drug concentrations presented here have been optimized for Ishikawa cells, an endometrial cancer cell line, grown in RPMI 1640 media supplemented with 10% FBS and 1% penicillin/streptomycin (complete RPMI). Other cell lines may require different transfection conditions and drug concentrations. Users can also perform transient transfection experiments in wild-type cells, instead of generating a stable cell line, with a plasmid expressing SID4X-dCas9-KRAB along with guide RNA expressing plasmids; however, results from transient transfections may be difficult to reproduce as SID4X-dCas9-KRAB levels may vary by transfection.
- Plate Ishikawa cells in at least 2 wells of a 6-well plate at 30 - 50% confluence (approximately 300,000 Ishikawa cells) in 3 mL of complete RPMI.
- Aspirate the media from the cells. Wash the cells once with 1x PBS (pH 7.4). Aspirate the PBS and add trypsin (4 mL for a 10 cm dish or 5 mL for a T-75 flask).
- Incubate the cells for ~5 min at 37 °C, checking every 2 min for detached cells and gently shaking the vessel.
- Once cells have detached, pipette trypsinized cells up and down a few times, and pipette gently down the side of the vessel to release any attached cells.
- Transfer the cells to a 15 mL conical tube and spin the cells down for 5 min at 250 x g.
- Aspirate the trypsin and resuspend the cells in 5 - 10 mL of media. Use a P1000 pipette to dissociate cell clumps if necessary.
- Count the cells and determine the volume needed to plate ~300,000 cells per well in a total volume of 3 mL. Add the cells to 2 separate wells of a 6-well plate. Fill each well to 3 mL with complete RPMI.
- Gently shake the plate every 5 min in the first 15 min after plating to ensure that the cells are evenly distributed on the plate. Use a microscope to ensure that the cells have dispersed from the middle of the well.
- Within 24 h of plating, perform the following transfections using an appropriate transfection reagent for the cell line of interest. For Ishikawa cells, use the procedure as described below. Note: This protocol assumes the use of cationic liposome-based transfection reagents. Electroporation provides an alternative method for cell types that are very sensitive to these reagents or that exhibit low transfection efficiency with lipofection. Transfection conditions must be optimized for the cell line of interest prior to attempting Enhancer-i experiments.
- In a 1.7 mL Eppendorf tube, dilute 2.5 μg of SID4X-dCas9-KRAB plasmid and 800 ng of plasmid expressing a fluorescent protein into serum-free media such that the final volume in the tube is 155 µL and the final concentration of plasmid is 0.020 µg/µL.
- In another tube, dilute 3.3 µg of a plasmid that does not contain a neomycin resistance cassette, such as pCMV-GFP, in serum-free media such that the final volume in the tube is 155 µL and the final concentration of plasmid is 0.020 µg/µL.
- Vortex each tube briefly and spin down using a microfuge.
- Add 9.9 µL of the transfection reagent (Table of Materials) to each tube. Mix by vortexing briefly at a low speed. Spin the tubes down with a microfuge.
- Incubate the tubes at room temperature for at least 5 min, but no more than 20 min.
- In the biosafety cabinet, add 150 µL of the prepared DNA:reagent mix dropwise to one well on the 6-well plate. Repeat for the other tube of prepared DNA:reagent mix. Mix the plates by swirling gently and return the plate to the incubator.
At day 2 post transfection, change the media and supplement with G418 to a final concentration of 600 ng/μL. This concentration may need to be optimized for the cell type.
Change complete RPMI media and supplement with G418 every other day for 2 - 4 weeks until the control transfected cells are dead and the wells containing SID4X-dCas9-KRAB become confluent. The exact amount of time needed for cells to recover will depend on the doubling time of the cells.
When the cells become confluent, passage to a T-25 or T-75 vessel in complete RPMI with a lower dose of G418 (300 ng/μL for Ishikawa cells). During this passage, make 2 aliquots of ~100,000 cells each (roughly 1/10th of a 6-well plate) into 2 separate 1.7 mL Eppendorf tubes for RNA and DNA isolation, respectively. Spin these tubes down (5 min, 250 x g), remove trypsin by pipetting, and freeze the tubes at -20 °C for future use.
Isolate genomic DNA using commercially available kits and perform PCR using the “pAC95_PCR” or “SID4X_PCR” primers (Table 1) to verify the presence of the fusion protein within in the cell line. Use genomic DNA extracted from the parental line as a negative control, and SID4x-dCas9-KRAB plasmid DNA as a positive control. Use a high-fidelity polymerase master mix with 50 - 100 ng of genomic DNA and the following cycling conditions: 98 °C for 30 s, 25 cycles of (98 °C for 10 s, 58 °C for 30 s, 72 °C for 2 min), 72 °C for 5 min, hold at 4 °C.
To verify the expression of the fusion protein at the RNA level, perform qPCR with RNA extracted from the cell line using commercially available kits. Use the “dCas9_qPCR” primers (Table 1), and the one-step qPCR protocol provided in step 6.3 of this protocol.
To verify the protein-level expression of the fusion, perform a Western blot on lysates from the cell line. Use either anti-FLAG or anti-HA antibodies to detect the fusion protein.
2. Guide RNA Design
Note: This protocol is designed for use with the U6 guide RNA cloning vector created by the Church lab and available on Addgene (Addgene 41824). To create a version of this vector containing puromycin resistance that allowed for the same cloning strategy as 41824, we moved the multiple cloning site from this vector into the pGL3-U6-sgRNA-PGK-puromycin vector (Addgene 51133). Either Addgene 41824 or our version with puromycin (Addgene 106404) are compatible with the cloning strategy outlined below.
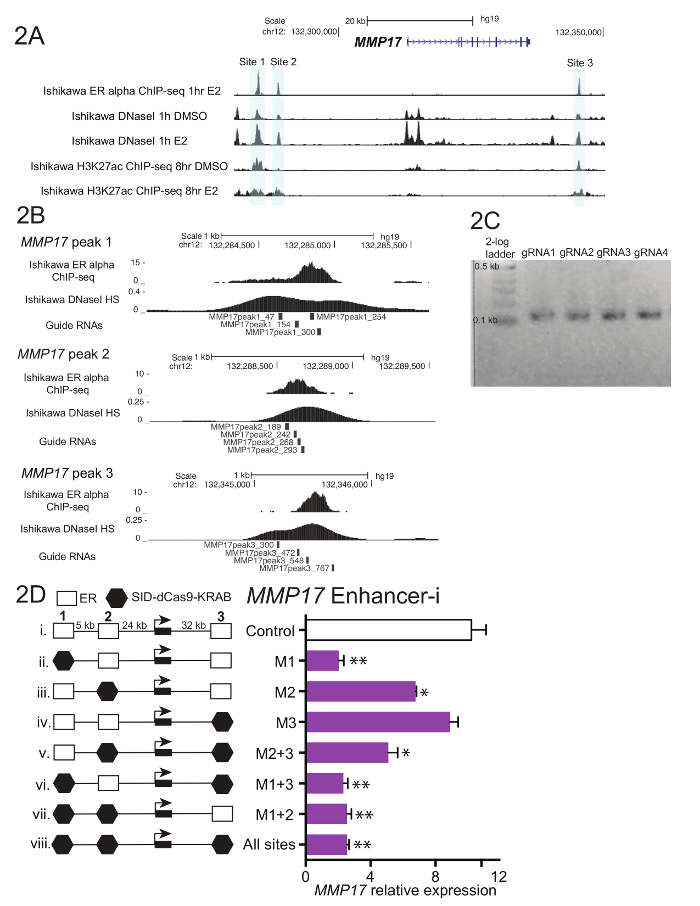
Obtain 600 - 900 basepairs of DNA sequence for each regulatory region of interest. Use transcription factor binding sites and/or chromatin accessibility for guidance as to where to define the region of interest (Figure 2A). Note: While the example in Figure 2A features upstream and downstream enhancers, it is also possible to target regulatory elements located within introns.
Place all sequences obtained in a single text file using the FASTA format.
Identify at least one negative control region that is not expected to change over experimental conditions, such as a promoter of a gene that is not expressed in the cell line of interest. Obtain DNA sequence for this region and add it to the text file in FASTA format. Note: We use guide RNAs targeting the IL1RN promoter25 as a negative control for all regions we target. Users can also select intergenic sequence near the region of interest that does not contain transcription factor binding sites as a negative control. However, if multiple loci are being targeted simultaneously, a single negative control region simplifies experimental design and interpretation of results. If the targeted enhancer is intronic, it may be useful to target an intronic region at the same locus that does not contain a putative regulatory element as an additional negative control, as the dCas9 fusion may interfere with transcription.
Identify positive control regions, such as the promoters that are the putative targets of the regulatory regions of interest, or promoters of genes that are highly transcribed in the cell line of interest. Obtain DNA sequence for these regions and add it to the text file in FASTA format.
- Use a program such as e-crisp26 (http://www.e-crisp.org/E-CRISP/) on the DNA sequences generated to find guide RNAs with low off-targets (ideally 0-3). Guide RNAs consist of 20 nucleotides upstream of a protospacer adjacent motif (PAM), which takes the form “NGG” for the dCas9 from S. pyogenes.
- On the e-crisp website, select the organism of interest using the drop-down menu. The genome assembly appears to the right of the species name.
- Select the Input is FASTA sequence radio button. Copy the FASTA sequences from above and paste them into the dialog box. Ensure that a FASTA header is included for each sequence. Note: Up to 50 sequences can be queried simultaneously.
- Select the Medium radio button and Single design in the drop-down menu.
- Click the button Start sgRNA search. A new browser tab will open, and results will be displayed. Download the candidate sequences by clicking the button Download an Excel formatted tabular report for all query sequences together.
- Open the tabular report using Excel or a text editing program.
- Use the UCSC genome browser to BLAT candidate full-length gRNA sequences (23 basepairs) to the genome.
- In a browser, navigate to the UCSC genome browser website (http://genome.ucsc.edu). Under the section Our tools, locate the word BLAT and click on it. The BLAT Search Tool will open.
- Use the drop-down menus located under the BLAT Search Genome text to select the organism and genome assembly of interest.
- Copy the guide RNA sequences from the tabular report generated by e-crisp and paste them into the dialog box. Ensure that each sequence has a unique FASTA header, then click Submit at the bottom of the dialog box. Note: Up to 25 sequences can be examined at once. On the BLAT Search Results page, alignments of each guide RNA sequence will appear, with each line representing an alignment. Ideally, there should be one alignment for each guide RNA, indicating the uniqueness of that guide RNA.
- Avoid the guides that align to multiple locations in the genome if possible.
- To examine guide RNA localization and distribution within the region of interest, click on the Browser link under the ACTIONS section for one of the queried guide RNAs. The Genome Browser will appear and will be centered on the selected guide RNA. Use the Zoom out buttons at the top of the page to visualize the distribution of other guide RNAs identified by e-crisp within the region of interest.
Select 4 preferably non-overlapping guide RNA sequences that are distributed throughout the region of interest (Figure 2B). If the region of interest exceeds 600 bp, consider adding 1 - 2 additional guides. Avoid guide RNAs with homopolymeric stretches and extreme GC content, as these features can hinder the guide RNA cloning process and reduce guide RNA targeting efficiency.
Once the guides have been selected, create a file containing the full guide RNA sequence (23 nucleotides) for each desired guide, and then remove the 5’ nucleotide as well as the PAM (NGG) from the 3’ end. This step facilitates oligo ordering.
Add the following sequence to the 5’ end of the oligonucleotide sequence: GTGGAAAGGACGAAACACCG.
Add the following sequence to the 3’ end of the oligonucleotide sequence: GTTTTAGAGCTAGAAATAGC. Note: The final sequence should be 59 nucleotides long and look like this: GTGGAAAGGACGAAACACCG-target (19 nt)-GTTTTAGAGCTAGAAATAGC.
Ensure that each regulatory element to be targeted with Enhancer-i has at least 4 unique oligonucleotides designed for it. Order these sequences along with the “U6_internal” primers listed in Table 1.
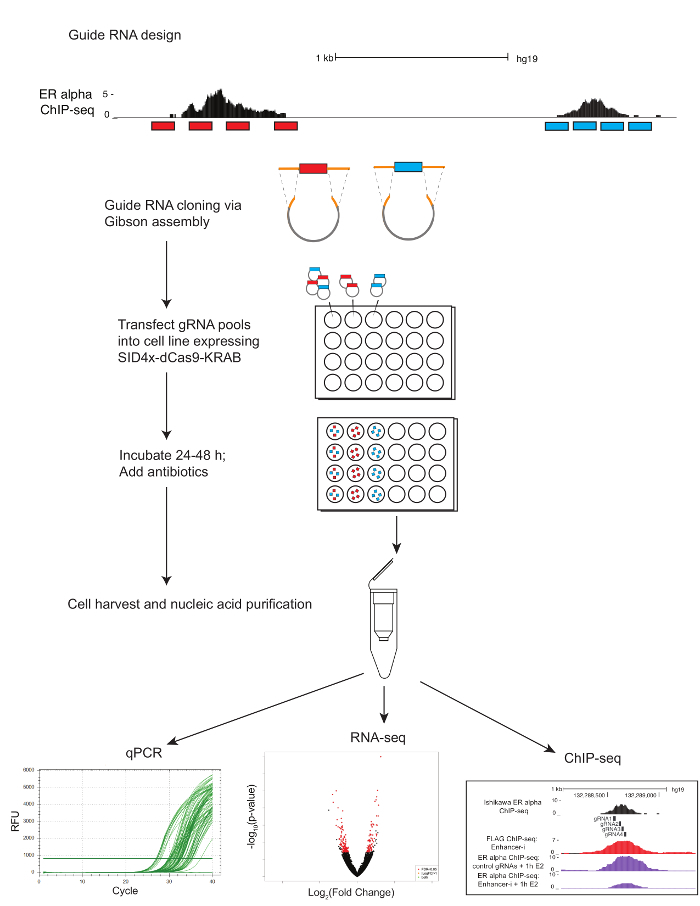
3. Guide RNA Cloning
Note: Guide RNA cloning via Gibson assembly has proven to be highly efficient in our hands, yielding hundreds of colonies per plate, with few if any colonies present in the vector only control. Such efficiency is critical for maintaining complexity during pooled cloning. Another advantage of Gibson assembly cloning is that users do not have to worry about the presence of a restriction enzyme cut site in the guide RNA they are trying to insert into the U6 cloning vector. Nonetheless, this protocol can be adapted for traditional restriction enzyme based cloning if desired.
Reconstitute guide RNA oligos at a final concentration of 100 μM in ultrapure water (RNase-free, DNase-free). There should be at least 4 separate guide RNA oligos for each region of interest.
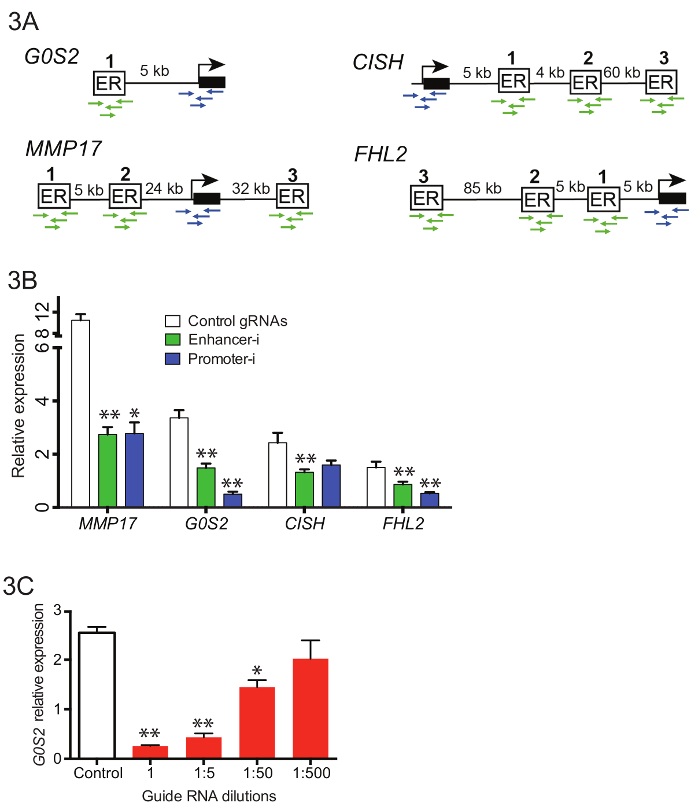
For each regulatory region of interest, create a pool of all the oligos corresponding to the region of interest. In an Eppendorf tube, combine 5 μL of each individual reconstituted guide RNA oligo for each region. Mix the pool well by vortexing, then remove 1 μL and dilute this aliquot 1:200 in ultrapure water. Note: If desired, these pools targeting individual regulatory regions can be further combined to generate a complex pool targeting multiple regions. Up to 50 regulatory regions may be targeted simultaneously in a single pool (Figure 3C).
- Perform a short PCR with the U6 primers to attach homology regions to the oligos prior to Gibson assembly. About 40 bases will be added to each oligo, yielding a ~100 bp product that contains sufficient homology to the U6 vector on both ends.
- For each guide RNA pool, set up a 20 μL PCR with a high-fidelity polymerase master mix and the following components: 1 μL of diluted oligo pool from step 3.2, 1 μL of U6 forward primer (10 μM), 1 μL of U6 reverse primer (10 μM), and water up to 20 μL.
- Incubate in a thermal cycler with the following conditions: 98 °C for 30 s, 10 cycles of (98 °C for 10 s, 55 °C for 30 s, 72 °C for 2 min), 72 °C for 5 min, and hold at 4 °C.
- Run 5 μL of the reaction on a 1 - 2% agarose gel with a low molecular weight ladder. The final product should be ~100 basepairs (Figure 2C).
- Clean up the extension reaction with a column-based DNA purification kit, and elute in 20 μL of elution buffer provided in the kit. Note: As the product is short, avoid using bead-based clean-ups, which are designed to exclude small fragments less than 100 bp.
- Quantify the purified DNA using a fluorometer or spectrophotometer (expected yield is 10-20 ng/μL). Guide RNA inserts can be stored at -20 °C, or can be used immediately in Gibson assembly with a linearized U6 vector.
- To prepare the recipient U6 cloning vector for Gibson assembly, set up a restriction enzyme digest. If many Gibson assembly reactions are to be performed, set up multiple digests to ensure sufficient yield of cut vector.
- Use 20 units of AflII enzyme and 1 μg of plasmid in a 20 μL reaction with the appropriate restriction enzyme buffer. Incubate at 37 °C for 1-2 h.
- Clean up the digest with beads or a column-based kit for DNA purification and elute in 20 μL of elution buffer. Quantify the purified DNA using a fluorometer or spectrophotometer. Samples can be frozen at -20 °C for later use.
- Perform Gibson assembly on the prepared vector and insert.
- Set up Gibson assembly reactions on ice. Use 50 ng of the vector and 7 ng of the insert in a 20 μL reaction. Dilute the inserts 1:10 in ultrapure water to facilitate pipetting. Set up a vector only Gibson assembly reaction, using 50 ng of the vector and replacing the insert with water.
- Incubate the Gibson assembly reactions for 15 min at 50 °C, followed by a hold at 4 °C.
- Transfer the assembled products to ice. Dilute the assembled products 1:4 in ultrapure water on ice. For example, add 5 μL of Gibson assembly product to 15 μL of ultrapure water.
- Transform the diluted Gibson assembly products.
- Thaw high efficiency competent cells on ice and make 25 μL aliquots for each transformation. If a complex pool targeting multiple sites is desired, thaw enough cells into different tubes to perform multiple independent transformations of the same complex gRNA pool.
- For each diluted product, add 1 μL of this dilution to a 1.7 mL Eppendorf tube containing 25 μL of competent cells. Mix by briefly flicking the tube. Incubate the tubes on ice for 30 min.
- Heat shock the cells for 30 s at 42 °C, then transfer immediately to ice for 2 min.
- Add 300 μL SOC media (2% tryptone, 0.5% yeast extract, 10 mM NaCl, 2.5 mM KCl, 10 mM MgCl2, 10 mM MgSO4, and 20 mM glucose) and let the cells recover for 1 h at 37 °C with shaking (300 rpm). During this time, warm the agar plates with ampicillin/carbenicillin to 37 °C in an incubator. Use one plate for each transformation.
- Plate 50 μL of cells and place the plates in a 37 °C incubator overnight. For the pools targeting individual sites, place directly in 3 - 5 mL of LB broth (Table of Materials) containing ampicillin/carbenicillin (1 mg/mL) and incubate overnight with shaking at 250 rpm at 37 °C for minipreps.
- Harvest the cells and isolate DNA.
- For large guide RNA libraries targeting multiple sites, use a plate scraper to collect all colonies from each individual plate into one maxiprep (150 mL liquid culture). This can be facilitated by pouring ~5 mL of LB with the appropriate antibiotic into a 50 mL falcon tube and scraping the colonies into the tube. For libraries targeting individual sites, scrape the plates into a miniprep (3 - 5 mL liquid culture).
- Incubate these cultures with the appropriate antibiotic for 3 - 5 h at 37 °C with shaking at 250 rpm.
- Perform DNA extraction using a kit that results in endotoxin-free preps.
- Quantify DNA using a fluorometer or spectrophotometer. Plasmids can be used immediately in transfection or stored at -20 °C for future use.
To confirm the presence of guide RNA sequence within the U6 vector for small pools targeting single sites, use Sanger sequencing on the prepared miniprep with the “U6_PCR_R” primer listed in Table 1. Due to pooling of guide RNAs, the 19 basepair gRNA target sequence will yield mixed bases, but the U6 promoter and guide RNA scaffold surrounding this sequence should be intact.
4. Transfection of Enhancer-i
Note: For the successful blockade of an estrogen response using Enhancer-i in Ishikawa cells, it is necessary to deprive the cells of estrogen for 5 - 7 days prior to transfection by maintaining them in phenol red free RPMI with 10% charcoal-stripped FBS and 1% penicillin/streptomycin. Cells should be cultured in this media during and after transfection if trying to block an estrogen response. We recommend the use of phenol red free trypsin for passage of cells in complete phenol red free RPMI.
The day before transfection, plate the cells (either wild-type or stably expressing SID4X-dCas9-KRAB) in a 24-well plate at 30 - 50% confluency (~60,000 cells per well for Ishikawa cells). Plate enough cells such that transfections can be performed in duplicate, and include wells to be transfected with control guide RNAs. Ensure that the cells are evenly distributed across the well by gently shaking the plate after cell plating as in step 1.1.7. Note: This protocol assumes the use of cationic liposome-based transfection reagents. Electroporation provides an alternative method for cell types that are very sensitive to these reagents. Transfection conditions must be optimized for the cell line of interest prior to attempting Enhancer-i experiments.
The following day, prepare transfections as per instructions of the transfection reagent of choice. For Ishikawa cells, use 550 ng of total plasmid for each well of a 24-well plate. Dilute the plasmids to a final concentration of 0.020 μg/μL in serum-free media (1.1 μg of DNA in 52 μL of total volume for transfecting 2 wells). Use 3 μL of transfection reagent for every 1 μg of DNA, vortex and incubate as described in step 1.2. Add 25 μL of the final mixture to each well. Note: To target combinations of sites, use the same weight of plasmid for each individual site, and then fill the leftover weight with a control plasmid (empty guide RNA cloning vector or guide RNAs targeting a negative control region such as the IL1RN promoter). For transient transfections, use a ratio of 3:2 Cas9 fusion:guide RNA plasmid. Plasmids containing fluorescent reporters may be added to monitor transfection efficiency.
At 36 h post transfection, change the media using phenol red free RPMI with 10% charcoal-stripped FBS and 1% penicillin/streptomycin (for Ishikawa cells) and supply puromycin (final concentration: 1 μg/mL) and neomycin (final concentration: 300 ng/mL). If cells are sensitive to transfection reagent, the media can be changed earlier, but antibiotics should be added no earlier than 24 h post transfection. Note: Wait at least 24 h after adding antibiotic before harvesting cells. Expression changes due to Enhancer-i can be detected as early as 48 h post transfection and up to 5 days post transfection. If working with Ishikawa cells that have been deprived of estrogen, perform an 8-h 10 nM 17β-estradiol (E2) induction the day after antibiotic treatment and then harvest cells immediately.
5. Cell Harvest and RNA Extraction
Prepare lysis buffer with 1% β-mercaptoethanol (BME). Ensure that there is enough lysis-BME mixture (300 μL for each well being harvested).
Aspirate the media using a vacuum aspirator.
Wash the cells once with an equal volume of 1x PBS (500 μL) and aspirate to remove as much PBS as possible.
Add 300 μL of lysis-BME solution to each well using a multichannel pipette. Pipette the lysis solution up and down 8 - 10 times, and transfer to a deep-well plate or 1.7 mL Eppendorf tubes on ice. RNA can be extracted immediately, or lysates can be frozen at -80 °C for future processing.
To extract RNA from lysates, use a commercially available kit that includes a DNase treatment. Elute in the smallest recommended volume of ultrapure water (RNase-free, DNase-free) or elution buffer and quantify RNA. For small numbers of samples, use a fluorometer or spectrophotometer. For large numbers of samples, use a fluorescent probe that detects RNA and measure on a plate reader. Samples can be frozen at -80 °C before or after quantification.
6. Quantifying Gene Expression Changes using One-step qPCR and RNA-seq
- Obtain qPCR primers for the genes of interest and for at least one housekeeping gene that is expressed near the level of targeted genes and does not change across experimental conditions. Ideally, these primers will span an exon-exon junction to avoid amplification of genomic DNA.
- Test these primers on RNA obtained from the cell line of interest. Use melt curve analysis to verify the production of a single product. If a single product is not produced, test additional primer pairs.
For each Enhancer-i and control guide RNA treated sample, identify how many genes must be assayed in that sample. This set of genes should include housekeeping genes, such as CTCF or GAPDH.
- Set up qPCR reactions.
- Dilute all samples to the same concentration in water, such that 50 ng of total RNA is easily pipetted, and there is enough diluted RNA for each reaction. For example, dilute RNA to ~16.6 ng/μL and use 3 μL of RNA in each reaction. Keep RNA on ice while setting up master mixes.
- Prepare separate master mixes for each gene to be measured using commercially available one-step qPCR kits. Use a 20 μL reaction volume with 1 μL of each primer (10 μM stock solution). Set up these reactions on ice.
- In a reaction plate that is appropriate for the thermal cycler, add RNA samples followed by master mixes. Seal with a plate sealer and mix gently by vortexing or pipetting. Briefly centrifuge the plate (140 x g for 60 s) to ensure that liquid is at the bottom of the wells.
- Incubate the plate in a thermal cycler as follows (or as kit instructs): 48 °C for 30 min, 95 °C for 10 min, 40 cycles of (95 °C for 15 s, 60 °C for 1 min).
- Obtain Ct values for each gene measured within each sample. Use the comparative Ct method to identify changes in gene expression.
- Subtract the Ct of the housekeeping gene from the Ct of each gene of interest for each sample to generate normalized Ct values.
- For the control treated samples, take an average of the normalized Ct values for each gene. Log base 2-scale fold repression can then be calculated by subtracting the normalized Enhancer-i treated sample Ct for each gene from the same value for the control treated sample for the corresponding gene.
To determine global changes in gene expression following Enhancer-i treatment, prepare the samples for RNA sequencing using a commercially available kit compatible with the user’s sequencing technology. Use ~500 ng of RNA for starting material and prepare libraries for at least 2 biological replicates.
7. Verification of Specific Genomic Targeting by SID4X-dCas9-KRAB Using ChIP-seq
Note: The SID4X-dCas9-KRAB fusion protein contains both a FLAG epitope tag and an HA epitope tag, but best results for ChIP-seq were obtained with anti-FLAG antibodies. If desired, the user can perform additional ChIP-seq experiments for transcription factors potentially affected by Enhancer-i, or for H3K27ac, a mark of enhancer activity that is decreased by Enhancer-i. However, each ChIP-seq experiment requires 10 x 106 cells, so plan accordingly.
- Transfect cells with Enhancer-i pools.
- Plate 10 x 106 cells in a 15 cm tissue culture dish. Each dish represents 1 ChIP-seq experiment for 1 factor of interest.
- The following day, transfect the cells using 20 μg of total DNA per dish. For transfections in cell lines stably expressing SID4X-dCas9-KRAB, the DNA should be a plasmid pool of guide RNAs targeting all sites of interest, and optionally a plasmid expressing fluorescent protein. For transient transfections, use a ratio of 3:2 dCas9 fusion protein:guide RNA pool. For ChIP-seq of other TFs or histone modifications, perform at least one additional control guide RNA transfection on another dish.
- Treat the dishes with puromycin (1 μg/mL) and neomycin (300 ng/mL) at 24 - 48 h post transfection. Wait at least 24 h before harvesting chromatin.
- Harvest chromatin from dishes. Note: To study the effects of Enhancer-i on ER genomic binding in Ishikawa cells, perform a 1 h 10 nM E2 treatment on dishes transfected with control guide RNAs and Enhancer-i prior to harvest. To study the effects of Enhancer-i on H3K27ac, perform an 8 h 10 nM E2 induction on dishes transfected with control guide RNAs and Enhancer-i prior to harvest.
- Apply 500 μL of 37% formaldehyde to each dish (final concentration of 1%). Swirl the plates briefly. Let the plates sit at room temperature for 10 min.
- Add 1 mL of 2.5 M glycine (final concentration of 125 mM). Swirl the plates briefly.
- Pour off the media with formaldehyde and glycine. Add an equal volume (~20 mL) of cold 1x PBS.
- Pour off the PBS. Aspirate with a vacuum aspirator to remove as much PBS as possible. Place the dishes on ice.
- Add 3 - 5 mL of cold 1x PBS or Farnham lysis buffer (5mM PIPES pH 8.0, 85 mM KCl, 0.5% NP-40) with 1x protease inhibitor (added just before use) to each plate. Scrape the dish with a plate scraper and transfer the solution to a 15 mL conical tube on ice.
- Pellet chromatin by spinning down tubes in a centrifuge for 5 min at 4 °C at 1000 x g. Discard the supernatant and store the pellets at -80 °C for future use, or proceed with the ChIP-seq protocol of choice using an anti-FLAG antibody or antibodies targeting other transcription factors or histone modifications of interest (H3K27ac, H3K9me3).
Representative Results
Figure 1 shows a schematic of the workflow described in the protocol. To determine the contributions of ER-bound enhancers near the estrogen-regulated gene MMP17, which has 3 binding sites nearby as defined by ChIP-seq (Figure 2A), guide RNAs were designed for each region. To design guide RNAs, a 600 - 900 bp window of sequence surrounding each ER binding site of interest was selected and put into a guide RNA design program. Resulting guide RNA sequences with 0-2 predicted off target sites were aligned to the human genome using BLAT. Four non-overlapping guide RNAs that spanned the region defined by ChIP-seq and DNaseI hypersensitivity were chosen for targeting (Figure 2B). Additional sequence (Table 1) was added to each end to facilitate downstream cloning and the resulting 59 nucleotide fragments were ordered. Upon arrival, guide RNAs were diluted and pooled by site, and a short PCR was performed to add homology regions prior to Gibson assembly. Figure 2C shows the expected guide RNA product after a short PCR using the "U6_internal" primers (Table 1), which will add 20 basepairs of sequence to each end of the 59 basepair guide RNA fragment, resulting in a ~100 basepair sequence. Following Gibson assembly, these guide RNA pools were transformed into bacteria and plasmid minipreps were prepared the following day. Figure 2D shows results from an enhancer dissection experiment, where multiple enhancers nearby MMP17 are targeted alone and in combination using Enhancer-i. Sites targeted by Enhancer-i are indicated with a black hexagon. Guide RNA plasmids targeting the indicated sites were transfected into an estrogen-deprived Ishikawa cell line stably expressing SID4X-dCas9-KRAB. Two days later, the media was changed and puromycin was added to enrich for transfected cells. The following day, the cells were harvested following an 8 h 10 nM estradiol treatment. RNA was isolated, and a one-step qPCR was performed. In this example, sites 1 and 2 are necessary for a complete estrogenic response of MMP17, while site 3 does not contribute under these conditions (Figure 2D, lanes ii-iv). When only sites 2 or 3 are active (vi and vii), the estrogen response is similar to when no sites are active (viii), suggesting that these sites cannot contribute independently. Site 1 can contribute some expression by itself (v), but the greatest activity is seen when sites 1 and 2 are active (iv).
To manipulate 10 enhancers near 4 different genes simultaneously (Figure 3A), complex pools of guide RNAs were generated containing 42 enhancer guides and 16 promoter guides. Guide RNA oligos were pooled before the initial guide RNA extension PCR (Step 3.3), and resulting PCR products were purified and combined with the empty puromycin U6 cloning vector using Gibson assembly. Following the Gibson assembly, multiple independent transformations were performed and plated. The plates were scraped into LB and allowed to grow out for 2 - 4 h prior to maxiprep. Figure 3B shows representative reductions in gene expression by qPCR when these guide RNA pools were transfected into an estrogen-deprived Ishikawa cell line stably expressing SID4X-dCas9-KRAB and treated as described above (Figure 2D). Reductions from Enhancer-i are similar to those obtained by targeting the promoter of the putative target gene. Figure 3C shows the effects of dilution of guide RNAs on reduction of the estrogen response using Enhancer-i. A 1:50 dilution of a guide RNA pool targeting the enhancer near G0S2 still yields significant reduction in gene expression, suggesting that Enhancer-i can be used to target up to 50 sites at once. However, the deactivation can be diluted out, indicating that hundreds of sites cannot be targeted simultaneously unless more sensitive detection methods are employed.
Figure 1. Protocol schematic for multiplex enhancer dissection using Enhancer-i. Guide RNAs (red and blue) are designed using e-crisp and selected using the UCSC genome browser. Four guide RNAs are chosen that span the regions of interest (transcription factor binding sites as defined by ChIP-seq). Guide RNA oligonucleotides that have been pooled by the region of interest (red and blue) undergo a PCR to add homology regions (orange) prior to the Gibson assembly and transformation. Resulting plasmid pools are transfected via lipofection into cell lines stably expressing SID4X-dCas9-KRAB or into wild-type cells in conjunction with SID4X-dCas9-KRAB plasmid. Guide RNA plasmid pools can be transfected individually to target one site at a time, or in combination to target multiple sites simultaneously. Transfected cells are treated with antibiotics to enrich for cells containing guide RNAs. At ~72 h post transfection, the cells are harvested. Nucleic acids can be extracted for qPCR, RNA-seq, or ChIP-seq. Please click here to view a larger version of this figure.
Figure 2. Guide RNA design and enhancer dissection for MMP17.(A) Genome browser screenshot of the ER alpha-bound enhancers (gray) to be targeted near MMP17. This figure has been modified from Carleton, et al.18. (B) Guide RNA designs for the 3 binding sites18. The binding site for ER as defined by ChIP-seq is the target, and the 4 guide RNAs tile across this region. The DNaseI sensitivity signal, which spans the binding site, can also be used to define target sequence for guide RNA design. Both ChIP-seq and DNaseI HS data were obtained from Ishikawa cells treated with 10 nM estradiol for 1 h. (C) Representative guide RNA sequences that are ready for Gibson assembly, having undergone a short PCR to add homology regions. (D) Relative expression of MMP17 measured via qPCR following targeting of specific regions with Enhancer-i and an 8-h10 nM estradiol treatment. Expression is relative to CTCF and expression level of MMP17 in cells not treated with estradiol. Control guide RNAs target the promoter of IL1RN. All error bars represent SEM, double asterisks indicate p <0.01 and single asterisks indicate p <0.05 in a paired t-test. This figure has been modified from Carleton, et al.18. Please click here to view a larger version of this figure.
Figure 3. Targeting multiple enhancers near different genes simultaneously with pooled Enhancer-i.(A) Schematic of the binding sites and promoters to be targeted in pooled Enhancer-i. (B) The effects on expression as measured by qPCR after E2 treatment on Ishikawa cells transfected with Enhancer-i plasmid pool (green), Promoter-i plasmid pool (blue) or control gRNAs (white)18. A significant reduction at all genes is observed with Enhancer-i. This figure was modified from Carleton, et al.18. (C) The effects on G0S2 expression levels after E2 treatment on Ishikawa cells transfected with different amounts of guide RNAs targeting G0S2. A significant reduction can be seen even with small amounts of guide RNA (1:50 dilution), suggesting that up to 50 sites may targeted simultaneously. All error bars represent SEM, double asterisks indicate p <0.01 and single asterisks indicate p <0.05 in a paired t-test. Please click here to view a larger version of this figure.
| Name | Sequence |
| U6_internal_F | TTTCTTGGCTTTATATATCTTGTGGAAAGGACGAAACACCG |
| U6_internal_R | GACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAAC |
| U6_PCR_F | CCAATTCAGTCGACTGGATCCGGTA |
| U6_PCR_R | AAAAAAAGCACCGACTCGGTGCCA |
| gRNA_qPCR_F | GCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCG |
| gRNA_qPCR_R | AAAAAGCACCGACTCGGTGCC |
| dCas9_qPCR_F | GTGACCGAGGGAATGAGAAA |
| dCas9_qPCR_R | AGCTGCTTCACGGTCACTTT |
| pAC95_PCR_F | AGAAGAGAAAGGTGGAGGCC |
| pAC95_PCR_R | CGTCACCGCATGTTAGAAGG |
| SID4X_PCR_F | CAATAGAAACTGGGCTTGTCG |
| SID4X_PCR_R | TCGTGCTTCTTATCCTCTTCC |
Table 1. Primers used for guide RNA extension and sequencing, qPCR, and detection of the fusion protein.
Discussion
This protocol describes a simple and flexible method for dissecting enhancer function at the endogenous genomic locus without physically altering the DNA sequence. While similar in concept to previously published CRISPR interference protocols using dCas9-KRAB27, Enhancer-i differs from these protocols in 3 main ways. First, Enhancer-i utilizes the SIN3A interacting domain of MAD120 to achieve enhancer deactivation. Enhancer deactivation can be rescued using HDAC inhibitors, suggesting that the primary mechanism of deactivation is HDAC dependent. Unlike CRISPR interference with dCas9-KRAB, Enhancer-i does not lead to the deposition of H3K9me3. This is likely due to the fact that Enhancer-i relies on transient introduction of guide RNAs, with the cells being harvested at 3 days post transfection. In CRISPR interference, an increase in H3K9me3 is observed at 7 days post transduction12. Finally, the Enhancer-i protocol provides a strategy to target multiple sites simultaneously and monitor the efficiency of targeting. In Mosaic-seq17, dCas9-KRAB is used to target multiple enhancers simultaneously, but this technique relies on single-cell RNA sequencing to identify expression changes, and many genes (such as estrogen-responsive genes) go undetected due to the low sensitivity of single-cell RNA-seq. Enhancer-i provides a reliable method to study enhancers individually and in combination for any gene.
The most critical step of Enhancer-i is transfection, which should be optimized for the cell line of interest. This protocol relies on puromycin treatment to enrich for transfected cells, but it is possible that co-transfecting guide RNAs with a fluorescent protein and sorting for fluorescent cells using flow cytometry may prove to be a better enrichment method for some cell types. We recommend monitoring the expression level of guide RNAs and SID4x-dCas9-KRAB by qPCR to troubleshoot and confirm transfection. If guide RNA levels are low (cycle threshold >30), users may also consider alternative guide RNA production strategies such as in vitro transcription28. It is also possible that despite high gRNA levels, guide RNA targeting of the SID4x-dCas9-KRAB protein is inefficient, in which case selecting different guide RNA sequences may be necessary. By performing ChIP-seq on the fusion protein with chromatin from Enhancer-i treated cells, the efficiency of targeting can be monitored. If there is high signal of SID4x-dCas9-KRAB at the region of interest, and no expression changes in its putative target gene are detected, then the region likely does not contribute to the expression of that gene under the conditions studied.
One potential limitation of Enhancer-i is that off-target effects may accumulate if too many sites are targeted simultaneously. Nonetheless, CRISPR interference strategies for knockdown have fewer off target effects than RNAi29, particularly when a polyclonal cell line expressing dCas9-KRAB is used. While we have seen off-target genomic binding of SID4X-dCas9-KRAB when targeting 10 sites simultaneously, we have not identified gene expression changes as a result of those binding events. As some enhancers may contact multiple promoters and/or other enhancers, it is possible that many genes may change expression upon targeting of a single enhancer, though it is unclear if this form of gene regulation is common. To confirm that the expression changes observed are due to targeting a specific enhancer, and not off-target effects, users can perform Enhancer-i with two distinct sets of non-overlapping guide RNAs targeting the same region. In addition, the genetic deletion of the region using nuclease-competent Cas9 can further confirm its effects on gene expression.
As Enhancer-i functions through histone deacetylation, it is possible that its deactivation abilities are limited to enhancers that have appreciable levels of histone acetylation. There are a variety of alternative repressive fusions that may be more effective at targeting specific enhancers. DNA methyltransferase fusions to dCas9 can be used to reduce gene expression when targeted to distal enhancers30, but this repression is often not transient. Another repressive fusion uses the Friend of GATA1 (FOG1) domain, which leads to histone H3 lysine 27 trimethylation and represses gene expression at levels similar to dCas9-KRAB across a variety of cell lines and promoters31. Interestingly, adding more copies of FOG1 to dCas9 reduced the repressive potential at promoters, suggesting that a single copy of the SID domain may provide more enhancer deactivation than the 4 copies currently used in Enhancer-i. It is possible that some loci may benefit from dual targeting by different combinations of the above dCas9 fusions. For example, stable long-term repression can be achieved by simultaneous transduction of dCas9-DNMT3a and dCas9-KRAB32. Most of these repressive fusions have only been targeted to a single locus at a time, and it remains unclear which is most effective at manipulating multiple enhancers simultaneously.
Enhancer-i, while a suitable method for studying combinations of enhancers for a handful of genes, is still somewhat limited in throughput if the user wishes to study putative enhancers for hundreds of genes. Future applications of this technique will incorporate imaging-based technologies to quantify multiple genes in multiple samples simultaneously. Importantly, these technologies are compatible with direct detection of RNA molecules from lysate, eliminating the need for time-consuming RNA isolation. These adaptations will facilitate the interrogation of larger sets of enhancers.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by NIH/NHGRI R00 HG006922 and NIH/NHGRI R01 HG008974 to J.G., and the Huntsman Cancer Institute. J.B.C. was supported by NIH Training Program in Genetics T32GM007464.
References
- Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenomics C, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518(7539):317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson R, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507(7493):455–461. doi: 10.1038/nature12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry MW, Boettiger AN, Bothma JP, Levine M. Shadow enhancers foster robustness of Drosophila gastrulation. Curr Biol. 2010;20(17):1562–1567. doi: 10.1016/j.cub.2010.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam DD, et al. Partially redundant enhancers cooperatively maintain Mammalian pomc expression above a critical functional threshold. PLoS Genet. 2015;11(2):e1004935. doi: 10.1371/journal.pgen.1004935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwardhan RP, et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat Biotechnol. 2012;30(3):265–270. doi: 10.1038/nbt.2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savic D, et al. Promoter-distal RNA polymerase II binding discriminates active from inactive CCAAT/ enhancer-binding protein beta binding sites. Genome Res. 2015;25(12):1791–1800. doi: 10.1101/gr.191593.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339(6121):823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng AW, et al. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 2013;23(10):1163–1171. doi: 10.1038/cr.2013.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton IB, et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol. 2015. [DOI] [PMC free article] [PubMed]
- Kearns NA, et al. Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat Methods. 2015;12(5):401–403. doi: 10.1038/nmeth.3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakore PI, et al. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat Methods. 2015;12(12):1143–1149. doi: 10.1038/nmeth.3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groner AC, et al. KRAB-zinc finger proteins and KAP1 can mediate long-range transcriptional repression through heterochromatin spreading. PLoS Genet. 2010;6(3):e1000869. doi: 10.1371/journal.pgen.1000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi LS, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152(5):1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulco CP, et al. Systematic mapping of functional enhancer-promoter connections with CRISPR interference. Science. 2016;354(6313):769–773. doi: 10.1126/science.aag2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo JY, Schaukowitch K, Farbiak L, Kilaru G, Kim TK. Stimulus-specific combinatorial functionality of neuronal c-fos enhancers. Nat Neurosci. 2016;19(1):75–83. doi: 10.1038/nn.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie S, Duan J, Li B, Zhou P, Hon GC. Multiplexed Engineering and Analysis of Combinatorial Enhancer Activity in Single Cells. Mol Cell. 2017;66(2):285–299. doi: 10.1016/j.molcel.2017.03.007. [DOI] [PubMed] [Google Scholar]
- Carleton JB, Berrett KC, Gertz J. Multiplex Enhancer Interference Reveals Collaborative Control of Gene Regulation by Estrogen Receptor alpha-Bound Enhancers. Cell Syst. 2017;5(4):333–344. doi: 10.1016/j.cels.2017.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154(2):442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayer DE, Laherty CD, Lawrence QA, Armstrong AP, Eisenman RN. Mad proteins contain a dominant transcription repression domain. Mol Cell Biol. 1996;16(10):5772–5781. doi: 10.1128/mcb.16.10.5772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alland L, et al. Role for N-CoR and histone deacetylase in Sin3-mediated transcriptional repression. Nature. 1997;387(6628):49–55. doi: 10.1038/387049a0. [DOI] [PubMed] [Google Scholar]
- Konermann S, et al. Optical control of mammalian endogenous transcription and epigenetic states. Nature. 2013;500(7463):472–476. doi: 10.1038/nature12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennoll SA, Scott SA, Yochum GS. Targeted repression of AXIN2 and MYC gene expression using designer TALEs. Biochem Biophys Res Commun. 2014;446(4):1120–1125. doi: 10.1016/j.bbrc.2014.03.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stampfel G, et al. Transcriptional regulators form diverse groups with context-dependent regulatory functions. Nature. 2015;528(7580):147–151. doi: 10.1038/nature15545. [DOI] [PubMed] [Google Scholar]
- Perez-Pinera P, et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat Methods. 2013;10(10):973–976. doi: 10.1038/nmeth.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heigwer F, Kerr G, Boutros M. E-CRISP: fast CRISPR target site identification. Nat Methods. 2014;11(2):122–123. doi: 10.1038/nmeth.2812. [DOI] [PubMed] [Google Scholar]
- Parsi KM, Hennessy E, Kearns N, Maehr R. Using an Inducible CRISPR-dCas9-KRAB Effector System to Dissect Transcriptional Regulation in Human Embryonic Stem Cells. Methods Mol Biol. 2017. pp. 221–233. [DOI] [PubMed]
- Romanienko PJ, et al. A Vector with a Single Promoter for In Vitro Transcription and Mammalian Cell Expression of CRISPR gRNAs. PLoS One. 2016;11(2):e0148362. doi: 10.1371/journal.pone.0148362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith I, et al. Evaluation of RNAi and CRISPR technologies by large-scale gene expression profiling in the Connectivity Map. PLoS Biol. 2017;15(11):e2003213. doi: 10.1371/journal.pbio.2003213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XS, et al. Editing DNA Methylation in the Mammalian Genome. Cell. 2016;167(1):233–247. doi: 10.1016/j.cell.2016.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Geen H, et al. dCas9-based epigenome editing suggests acquisition of histone methylation is not sufficient for target gene repression. Nucleic Acids Res. 2017;45(17):9901–9916. doi: 10.1093/nar/gkx578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amabile A, et al. Inheritable Silencing of Endogenous Genes by Hit-and-Run Targeted Epigenetic Editing. Cell. 2016;167(1):219–232. doi: 10.1016/j.cell.2016.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]