

Abstract
The study of microbe domestication has witnessed major advances that contribute to a better understanding of the emergence of artificially selected phenotypes and set the foundations of their rational improvement for biotechnology. Several features make Saccharomyces cerevisiae an ideal model for such a study, notably the availability of a catalogue of signatures of artificial selection and the extensive knowledge available on its biological processes. Here, we investigate with population and comparative genomics a set of strains used for cachaça fermentation, a Brazilian beverage based on the fermentation of sugar cane juice. We ask if the selective pressures posed by this fermentation have given rise to a domesticated lineage distinct from the ones already known, like wine, beer, bread, and sake yeasts. Our results show that cachaça yeasts derive from wine yeasts that have undergone an additional round of domestication, which we define as secondary domestication. As a consequence, cachaça strains combine features of wine yeasts, such as the presence of genes relevant for wine fermentation and advantageous gene inactivations, with features of beer yeasts like resistance to the effects of inhibitory compounds present in molasses. For other markers like those related to sulfite resistance and biotin metabolism our analyses revealed distributions more complex than previously reported that support the secondary domestication hypothesis. We propose a multilayered microbe domestication model encompassing not only transitions from wild to primarily domesticated populations, as in the case of wine yeasts, but also secondary domestications like those of cachaça yeasts.
Keywords: microbe population genomics, microbe domestication, yeast evolutionary biology, domestication traits, comparative and population genomics, cachaça fermentation
Introduction
Several features make the yeast Saccharomyces cerevisiae an ideal model for the study of the mechanisms of microbe domestication. It shows clear signatures of artificial selection (Fay and Benavides 2005; Legras et al. 2007; Liti et al. 2009; Novo et al. 2009) and the extensive knowledge available on its biological processes facilitates the interpretation of new data. The study of the genomic, genetic and phenotypic transitions associated with microbe domestication has witnessed recent advances in the case of two major yeast products: ale beer and wine. In the first case, ale-type beer yeasts were found to be essentially distinct from other industrially relevant lineages, including wine yeasts, and different phylogenetic subgroups of beer yeasts could be associated with distinct beer types (Gallone et al. 2016; Gonçalves et al. 2016). In the second case, the closest wild relatives of wine yeasts were discovered in the Mediterranean region and in association with oak trees (Almeida et al. 2015). The detection of this unique lineage allowed the disclosure of the predomesticated version of the genome of wine strains (Almeida et al. 2015). This enabled a direct comparison of wine yeasts with their wild relatives that revealed widespread genome-wide divergence, particularly at noncoding sites and in transacting DNA binding proteins, thus implicating transcriptional regulation as a driver of divergence between these two groups (Almeida et al. 2017). These and other advances contribute to uncover a complex scenario involving the emergence of various domesticated lineages.
Given the worldwide ubiquity of fermented foods and beverages that rely on the conversion of simple sugars to ethanol implicating S. cerevisiae as the dominant microorganism (Romano et al. 2006; Legras et al. 2017), it can be hypothesized that this yeast was coopted multiple times by humans. If those putative domestication events involved the artificial selection of local wild S. cerevisiae lineages or the utilization of a globally dispersed domesticated lineage is still an open question. The case of sake yeasts, that are pylogenetically distinct from wine yeasts (Fay and Benavides 2005; Liti et al. 2009), argues in favor of independent domestication. However, the global dispersion of the domesticated wine yeast lineage (Borneman et al. 2016) and the widespread mosaicism, that is, interpopulation recombination detected in S. cerevisiae (Tilakaratna and Bensasson 2017) make it possible that a single “proto”-domesticated lineage was globally dispersed and further and independently domesticated, locally. The assessment of these alternative hypotheses and even the accommodation of an intermediate scenario that accepts both independently domesticated lineages and cases of multiple rounds of domestication of a single lineage, requires a detailed knowledge of additional domestication events. Here, we use a population and phylogenomics approach to investigate the domestication of S. cerevisiae strains employed in the fermentation of cachaça, a Brazilian distilled spirit based on the conversion of sugar cane juice.
Sugar cane cultivation and cachaça production were introduced by Portuguese settlers in the XVI century. Originally consumed only by slaves working on sugar cane plantations, its production was gradually improved and by early XIX century it was considered a typical Brazilian beverage (Badotti et al. 2012). Nowadays cachaça is the third most popular distilled beverage in the world and 1.3 billion liters are produced annually (Badotti et al. 2012). Almost one billion liters are annually produced industrially in stainless steel distillation columns. Another 300 million liters are produced by approximately 35,000 rural producers through the distillation of the sugar cane wine (must) in copper alembics, the traditional production method (Rosa et al. 2009). Industrial cachaça producers employ baker's yeast or active dried yeast used for ethanol production as the starter for the fermentation process, whereas traditional producers use natural ferments (spontaneous fermentation) cultured by various methods (Rosa et al. 2009). The fermentation employs a dilution of the original sugar cane juice that reduces sucrose concentration from around 20% to 16–14% w/v, and a starter culture from a previous fermentation that normally represents 20% of the total volume of the fermentation vat. At the beginning of the season a natural inoculum is prepared in traditional distilleries by mashing rice, maize flour, and salt biscuits with undiluted cane juice and lemon or orange juice to lower the pH. Typically the cachaça fermentation stage lasts 18–30 h and proceeds at elevated temperatures that in some regions can reach 41 °C (Vianna et al. 2008). At the end of fermentation ethanol concentration is around 8% and products with organoleptic relevance are ethyl esters, aldehydes, and organic acids (Cardoso et al. 2004). At this stage four fifths of the fermented must is distilled and fresh sugar cane juice is added to start a new fermentation cycle. Although cachaça fermentation resembles grape wine fermentation in that simple sugars are directly fermented, cachaça possesses some unique features such as a short fermentative cycle with daily additions of fresh sugar cane juice and high fermentation temperatures. Some studies have aimed at understanding the genetic diversity and specific attributes of S. cerevisiae cachaça strains associated with traditional fermentations. Using PCR-fingerprinting, Guerra et al. (2001) revealed a considerable genetic diversity during a single fermentation that was attributed to the spontaneous nature of the process. Moreover, different cachaça strains exhibited different properties in terms of growth rates and concentration of fermentation products (Campos et al. 2010). More recently, multilocus sequencing was employed to analyze cachaça strains from traditional distilleries of different regions in Brazil that were resolved in two main populations, one corresponding to the wine group and the other corresponding to “native strains” (Badotti et al. 2014). According to these authors, a third group of cachaça strains corresponded to hybrids of the two groups mentioned above.
In this study, we employed whole genome data to try to understand in more detail the relationships of cachaça strains among themselves and with representatives of well-established populations of S. cerevisiae, in particular to the wine group. More specifically, we wanted to know if cachaça fermentation selected for typical wine strains, for modified wine strains further adapted to this niche, or, instead, for strains unrelated to the wine group. We also wanted to assess the degree of genetic variability within the cachaça group. Also, taking advantage of the availability of complete genome data we wanted to know if domestication signatures typical of wine strains were present in cachaça yeasts or if distinct signatures could be detected. Moreover, given the recent identification of wild populations of S. cerevisiae in Brazil (Barbosa et al. 2016), their relationship with cachaça yeasts and the evaluation of possible genetic contact between these two groups were also investigated.
Materials and Methods
Genome Sequencing, Read Alignment, and Genotype Calling
Whole-genome Illumina MiSeq paired-end reads (500 cycles) were obtained for monosporic or single cell derivatives. Single-cell derivatives were used when cultures did not form ascospores (supplementary table S1, Supplementary Material online). Genome data of strains not sequenced in this study was retrieved from public databases (supplementary table S1, Supplementary Material online). Where only finished genome sequences were available, the corresponding error-free Illumina reads were simulated using dwgsim (https://github.com/nh13/DWGSIM). Reads for each isolate were mapped to S. cerevisiae reference genome (UCSC version sacCer3) using SMALT v0.7.5 aligner (https://www.sanger.ac.uk/science/tools/smalt-0). The reference index was built with a word length of 13 and a sampling step size of 2 (–k 13 –s 2). An exhaustive search for alignments (–x) was performed during the mapping step with the random assignment of ambiguous alignments switched off (–r –1) and the base quality threshold for the look-up of the hash index set to 10 (–q 10). With these settings, SMALT v0.7.5 only reports the best unique gapped alignment for each read. For paired-end information, the insert size distribution was inferred with the “sample” command of SMALT prior to mapping. Conversion of SAM format to BAM, sorting, indexing, several mapping statistics, and consensus genotype calling were performed using the tools available in the SAMtools package v1.18 (Li et al. 2009) as described previously (Almeida et al. 2014). Multiple sequence alignments for each reference chromosome were generated from the resulting fasta files. For downstream analysis, all bases with Phred quality score below Q40 (equivalent to a 99.99% base call accuracy) or ambiguous base calls were converted to an “N.” All strains with >20,000 heterozygous sites with a Phred quality score above Q40 were selected for phasing. The BAM file of each strain with the paired-end read sequences mapped to the reference genome was analyzed with the phase command of SAMtools to infer both phases, thus solving the heterozygous SNPs. This algorithm uses the pair-end read information and attempts to find the best phase using the Minimum Error Correction method, solving each local haplotype with the highest probability based on the observed reads and the reference genome. The –F option was used to exclude errors from unmapped or misaligned sequences. Two haplotypes were obtained for each strain, each with reduced levels of heterozygosis. One haplotype per strain was randomly chosen and used in subsequent analyses.
Phylogeny and Population Structure
Chromosomal single nucleotide polymorphisms (SNPs) were extracted from multiple sequence alignments only if the evaluated site was represented by unambiguous high-confidence alleles in at least 85% of the isolates. SNPs were then concatenated to generate a whole-genome SNP alignment. A Neighbor-Joining phylogeny was estimated using the p-distance model as implemented in MEGA 6. Single gene phylogenies were prepared with MEGA 6, employed sequence alignments obtained with Muscle and were constructed using the Neighbor-Joining method and Tamura's 3-parameter model. Population structure was explored using the model-based Bayesian clustering method implemented in STRUCTURE v2.3.4 (Falush et al. 2003) and using SNPs present in all the sequences, not allowing for gaps. STRUCTURE was run with a subset of approximately 10,000 equally spaced parsimony informative sites. The number of Markov chain Monte Carlo (MCMC) iterations was set to an initial burn-in period of 100,000 iterations, followed by 100,000 iterations of sampling. The ancestry model allowed for admixture and allele frequencies were assumed to be correlated among populations. Five independent simulations were run for each value of K, varying from K = 1 to K = 15, and stability was assessed by monitoring the standard deviation between simulations.
Survey of Specific Genes
We performed de novo genome assemblies using SPAdes v.3.1.0 (Bankevich et al. 2012). Prior to assembly, reads were processed with Trimmomatic (Bolger et al. 2014) based on a quality score threshold of 20 for windowed trimming, discarding reads <100 bp in length or harboring ambiguities. To retrieve genes of interest, a local BLAST database was set up for each genome and ORFS were searched by BLASTN (1e−4 E-value cutoff), using as queries sequences of YPS 163 for AQY1 and AQY2 (Will et al. 2010), sequences of EC1118 for regions A, B, and C (Novo et al. 2009; Marsit et al. 2015), sequences of CEN.PK13 for RTM1, BIO1, and BIO6 (Nijkamp et al. 2012) and SGD sequences for PAD1 and FDC1 (Mukai et al. 2014). Blast hits were retained if sequence identity was above 90% and if the sequence was aligned to at least 10% of the query.
Polymorphism and Divergence Analyses
Whole-genome levels of polymorphism and divergence were estimated using Variscan v2.0 (Hutter et al. 2006). Only sites with valid high quality alleles (Q > 40) in at least 75% of ingroup sequences were used in calculations. Sites with more than this threshold were randomly subsampled to 75% of the total number of sequences (defined with the NumNuc parameter together with CompleteDeletion = 0 and FixNum = 1). For divergence estimates between populations, only positions with valid alleles in at least four ingroup individuals were used for calculations (defined with the NumNuc parameter together with CompleteDeletion = 0 and FixNum = 0).
Analysis of Introgressions from S. paradoxus
We searched for evidence of introgressions from other Saccharomyces species by mapping the reads to a combined reference that includes all the available annotated coding sequences of Saccharomyces species. Reads were mapped to this combined reference using BWA v0.6.2 (Li and Durbin 2009) with default parameters but setting the quality threshold to 10 (–q 10). SAMtools v1.1852 (Li et al. 2009) was used for the manipulation of the resulting BAM files. Only genes with orthologs unambiguously annotated in all six species were analyzed. An ORF was considered to have a foreign origin to S. cerevisiae if its coverage was at least higher than 1/4 of the median whole-genome coverage for the analyzed strain. The ORF coverage was defined as the product of the total number of mapped reads to the orthologous ORFs by the read size, dividing by the sum of the length of each ORF, considering only the ones with >25% of reads mapped (relative to the orthologous ORF with the highest number of reads) to control for spurious alignment counts. This coverage threshold allowed for some heterogeneity in the read counts and for the eventual presence of a foreign ORF together with the native S. cerevisiae ORF.
Pairwise divergence between S. paradoxus (strain YPS 138) and S. cerevisiae (strain S288c) was used as a proxy to search for evidence of DNA segments of S. paradoxus in the genomes of S. cerevisiae strains. Divergence per site, k, (with Jukes–Cantor correction) was calculated using a nonoverlapping sliding window of 10,000 sites, using Variscan v2.0 (Hutter et al. 2006). Using de novo genome assemblies a local BLAST database was set up for each genome in order retrieve the introgressed genes. The introgressed ORFS were searched by BLASTN, using the correspondent S. cerevisiae ORF sequences available at SGD as queries.
GO Analysis
The Standard GO term discovery was performed with the GO Term Finder tool, available at SGD.
PCR Detection of SSU1 Translocations
Primers and PCR conditions followed those of Pérez-Ortín et al. (2002) and Zimmer et al. (2014). In the case of the detection of the translocation first observed by Pérez-Ortín et al. (2002), we modified the original primer sequences and the primers used to amplifly SSU1 were NOG1_FW (5′-GAATCTGATAGACACAATGC-3′) and GLR1_RV (5′-TACTCTAGTAGCGAGGTC-3′), whereas the primers used to amplifly SSU1R were YHL044W_FW (5′-CAAGTACTGGGAGGATAAG-3′) and GLR1_RV (5′-TACTCTAGTAGCGAGGTC-3′). All essays employed DNA obtained from parental strains.
Results
Complete Genome Sequences of Brazilian S. cerevisiae Strains
In this study, we obtained the complete genome sequences of 26 Brazilian S. cerevisiae strains—21 strains from cachaça, three strains from jabuticaba wine, and two strains from grape wine (supplementary table S1, Supplementary Material online). Jabuticaba wine results from the fermentation of the fruits of a tree of the Myrtaceae (Jabuticaba, Plinia cauliflora) that is native to Brazil, Argentina, Paraguay, Peru, and Bolivia. The cachaça strains had distinct geographical provenances and were obtained in six Brazilian states. Three additional genome sequences from bioethanol producing strains available in public databases were also used (CBS 7960, JAY 291, and BG1). Finally, a group of 26 wild Brazilian strains studied recently by Barbosa et al. (2016) was also included in our study, so that a total of 55 genomes of Brazilian S. cerevisiae strains were investigated. For comparison and as indicated in supplementary table S1, Supplementary Material online, we used representatives of all the known S. cerevisiae populations for which genomic data is available: wild lineages found in Brazil and in oak trees in the Mediterranean region and in North America–Japan (NA–JPN), and representatives of the Malaysian, Philippine, and West African populations, together with members of domesticated lineages (Wine, Beer, Bread, and Sake). This group on non-Brazilian representatives encompassed 135 genomes.
Cachaça Strains Are Polyphyletic
Similarly to the beer and bread strains that had a high number of heterozygous sites, indicative of ploidy levels higher than 2n (Gonçalves et al. 2016; supplementary table S1, Supplementary Material online), 10 out of the 21 cachaça strains studied had also >20,000 heterozygous sites (supplementary table S1, Supplementary Material online) and were phased prior to further analysis. Since a preliminary analysis showed that the vast majority of phased cachaça haplotypes were phylogenetically very close to each other (supplementary fig. S1, Supplementary Material online), one haplotype per strain was randomly chosen and used in subsequent analyses. For strain UFMG-CM-Y623 both phased haplotypes, that showed some divergence, were retained in the final phylogenetic analysis. Among the wild strains we detected a single Brazilian strain, UFMG-CM-Y456, with an elevated number of heterozygous sites. In this case, the two phased haplotypes were phylogenetically distinct and were also retained in the final phylogenetic analysis.
The phylogenetic relationships of cachaça strains were analyzed based on 1,108,048 high quality polymorphic sites as shown in figure 1. Instead of forming a single group, cachaça strains were markedly polyphyletic. Two clades containing mostly cachaça strains were placed at the base of the Wine + Mediterranean oak population (MO) + Beer 2 clades. One, that we designated C1 was composed of eight strains and the other (C2) contained six strains, but two strains were isolated from Tapirira guianensis, a candidate Brazilian natural habitat for S. cerevisiae (Barbosa et al. 2016). The group of three Brazilian strains from sugar cane bioethanol was also placed at the base of the Wine + MO + Beer 2 clades. Whereas group C2 contained only strains from the Tocantins state (cachaça strains and wild strains), group C1 contained only cachaça strains but from four Brazilian states, none of them being the Tocantins state (supplementary table S1, Supplementary Material online). Other cachaça strains were included in the Wine, Bread and Wild Brazilian B1 clades (one, three, and one strains, respectively). Finally, four cachaça strains (UFMG-CM-Y623, UFMG-CM-Y628, UFMG-CM-Y637, and UFMG-CM-Y638) were placed in isolated positions outside any known clade. Interestingly, the Brazilian strains from grape and jabuticaba wine clustered within the wine group (fig. 1). We note that the Brazilian wine strains were placed very close to commercial wine strains, being therefore possible that they are descendants of starter cultures, instead of typically autochthonous wine strains.
Fig. 1.

—Phylogenetic placement of cachaça strains among the known lineages of Saccharomyces cerevisiae. Whole-genome phylogenetic tree of 188 sequences, inferred from 1,108,048 SNPs using the Neighbor-Joining method and the p-distance model of sequence evolution. The tree was rooted with S. paradoxus, branch lengths correspond to the expected number of substitutions per site and black dots depict bootstrap support values above 90% (100 replicates). Lineages of cachaça or bioethanol strains are highlighted in red. For phased sequences (those with heterozygous sites >20,000) a single phase is depicted except when the two phases are phylogenetically distinct (two exceptions with strain designations indicated in red). Abbreviations of populations: MO, Mediterranean oaks; MY, Malaysia; NA–JPN, North America–Japan; PHL, Philippines; WA, West Africa.
Population Structure
Using a selection of eight cachaça/bioethanol strains and also representatives of all populations of S. cerevisiae known so far (supplementary table S1, Supplementary Material online), we analyzed the population structure of this data set using STRUCTURE (Falush et al. 2003) and testing from 2 to 15 ancestral (K) clusters. A comprehensive representation of sequence ancestry was achieved with K = 12 (fig. 2a) and analyses using higher K values did not reveal new meaningful clusters. Similarly to other recent studies (e.g., Almeida et al. 2015; Barbosa et al. 2016) our analysis recovered the main groups of industrial variants or geographically delimited populations such as 1) Wine—Mediterranean oak, 2) Beer, 3) Sake, 4) Philippines—North American—Japanese populations, 5) West Africa, 6) Wild Brazilian B1, and 7) Wild Brazilian B3. In this comprehensive analysis involving a wide representation of populations of S. cerevisiae, the cachaça strains shared most of their ancestry with the wine group. However, they did not appear to be identical to the wine strains but seemed rather to present admixture between wine ancestry and a hitherto unknown genetic cluster (cluster 8). Interestingly, the two wild Brazilian populations recently described (Barbosa et al. 2016) appear not to have relevant contributions to the genetic composition of cachaça strains (fig. 2). In order to confirm these results, we performed a second analysis in STRUCTURE eliminating some representatives of populations that did not show an association with cachaça strains (e.g., West Africa, Philippines, and Malaysia) and increasing the proportion of cachaça and wild Brazilian strains in the data set. Overall, the results of this second study were consistent with those of the previous analysis and the correspondent K = 12 population representations are shown in figure 2b. This analysis evidenced the important role of wine ancestry for other domesticated groups besides the wine group, such as the main clade of ale-type beer strains (Beer 1), bread strains, the so-called Beer 2 clade, recently revealed and composed of Belgian ale beer strains and other beer strains from high ethanol beers (Gallone et al. 2016), and for the cachaça strains analyzed in this study. Except for the wine strains, all the other groups have clear signs of admixture (fig. 2). The resemblance between JAY 291, a strain used for the production of bioethanol, and cachaça strains is worth noting. Cluster 8, the hallmark of cachaça strains, was present in distinct proportions in different subgroups of cachaça strains. Strains of group C1 had a higher proportion of wine ancestry and a correspondingly lower proportion of Cluster 8 (85 –90% Wine; 10–15% Cluster 8), whereas for group C2 the proportion of cluster 8 tended to be higher (45–80% Wine; 20–30% Cluster 8). For the cachaça strains not assigned to the two main cachaça groups (C1 and C2), nor to the Wine, Bread and B1 clades, more complex admixture patterns were detected and the proportion of cluster 8 was even lower than that observed for cachaça C1 group (UFMG-CM-Y628 10.1%, UFMG-CM-Y637 5.8%, and UFMG-CM-Y623 5.7%). One cachaça strain, UFMG-CM-Y638, that occupied an isolated position in the phylogeny of figure 1 together with UFMG-CM-Y639, a Brazilian wild strain isolated from Tapirira guianensis, lost its genetic ancestry in cluster 8 with K = 12 (but not with lower K values, similarly with UFMG-CM-Y639). The K = 12 analysis of the second STRUCTURE run indicated that a new genetic cluster (cluster 9) explains most of the genetic ancestry of these two strains, being also a minor fraction of a few other cachaça strains, especially those with more admixed genomes.
Fig. 2.

—Population structure of Saccharomyces cerevisiae. (a) STRUCTURE plot (run 1) that depicts the analysis of 47 strains from all major populations and from the cachaça lineages that emerged from the phylogeny depicted in figure 1. The plot is based on a subset of 10,445 parsimony informative sites for K = 12. Numbers from 1 to 8 represent the different clusters that capture the maximum representation of population ancestry. The phylogenetic groups inferred in figure 1 are color-coded at the bottom of the plot. (b) STRUCTURE plot (run 2) that depicts the analysis of 61 strains, excluding representatives of less relevant populations as inferred from run 1 and including all cachaça strains. The plot is based on a subset of 10,308 parsimony informative sites for K = 12. Numbers up to 10 represent the different clusters that capture the maximum representation of population ancestry.
Domestication Signatures—Regions A, B, and C
One of the hallmarks of wine yeast domestication is the acquisition of three genomic regions (named A, B, and C), independently and through horizontal gene transfer from non-Saccharomyces yeasts. These regions encompass 39 genes potentially relevant for the winemaking process (Novo et al. 2009; Galeote et al. 2011; Marsit et al. 2015). In line with these findings, regions A, B, and C can be found in wine yeasts, but not in beer or sake yeasts (Gonçalves et al. 2016). A fourth domesticated lineage, that of bread yeasts, is mostly admixed and combines wine, beer and sake ancestries, so that regions A, B, and C are found in bread strains too (Gonçalves et al. 2016). Given that our phylogenetic analysis pointed to a closeness of cachaça strains of clades C1 and C2 to wine strains (fig. 1), and that cachaça strains share with wine strains most of their genetic ancestry (fig. 2), we surveyed the genomes of cachaça strains for the presence of regions A, B, and C. In the eight strains of the C1 clade, six strains had at least one of these regions and the same happened with four of the six strains of C2 clade (supplementary table S1, Supplementary Material online). Therefore, for these two clades, and considering only strains isolated from cachaça, one of these regions was present in 67% of the strains. For comparison, among the wine strains included in this study (Brazilian wine strains excluded) at least one of the regions A, B, or C was present in 71% of the strains. Moreover, both the Brazilian grape and jabuticaba wine strains had at least one of these regions (supplementary table S1, Supplementary Material online). As mentioned before, two strains of the C2 cachaça clade were firstly regarded as wild because they were isolated from a natural substrate, the tree T. guianensis (Barbosa et al. 2016). However, these strains not only resembled cachaça strains in the phylogenetic and population structure analyses but harbored the complete region B, thus suggesting that these are feral strains, that is, domesticated cachaça strains that have escaped their original environment and have colonized natural environments. Since Brazilian jabuticaba wine and grape wine strains from Brazil were assigned to the wine group (fig. 1), it is not surprising that they harbor these regions. Similarly, the cachaça strains assigned to the bread group possessed these regions as it normally occurs for bread strains. Conversely, the Brazilian wild strains of clades B1 and B3 did not harbor any of these regions, as already observed for other wild populations (e.g., MO and NA–JPN) (Almeida et al. 2015).
Borneman et al. (2011) and Galeote et al. (2011) studied in more detail S. cerevisiae strains that harbored region B, that corresponds to a cluster of five ORFs, and reported several variants concerning copy number, synteny and chromosome location. Contrasting with the reference wine strain, EC 1118, that had three copies in chromosomes X (sinteny “e-a-b-c-d-e”), XII (sinteny “a-b-c-d-e”), and XIV (sinteny “a-b-c-d-e-a”) (fig. 3a), the bioethanol strain JAY 291 had a single copy of region B in chromosome XI with a distinct synteny (Galeote et al. 2011). The alternative synteny observed for region B is suggestive of a distinct mode of linearization of this region prior to its integration in the chromosome from a putative circular precursor (Borneman et al. 2011; Galeote et al. 2011). Interestingly, the distinctive features previously observed for JAY 291 were present in most of the cachaça strains that harbored region B, in Brazilian jabuticaba wine strains and in L1374, a wine strain from Chile (sinteny “d-e-a-b-c”, fig. 3b, supplementary table S1, Supplementary Material online). Moreover, other cachaça strains or other South American wine strains had organizations of this region that match those of seen in wine strains other than EC 1118 (sinteny “e-a-b-c-d”, fig. 3c and d). Taken together, our results suggest that South American wine—bioethanol—cachaça strains are similar in terms of sinteny of region B.
Fig. 3.

—Similar localization and organization of region B in cachaça and other South American domesticated strains. (a) Organization of region B in EC 1118 as reported by Galeote et al. (2011). Colored arrows represent syntenic ORFs designated as in Novo et al. (2009). (b) Most cachaça strains that have region B share with bioethanol strain JAY 291 and with South American wine strains its organization and location in chromosome XI. (c) Alternative organization of region B in cachaça strains (chromosome location was not possible to determine). (d) third organization type of region B, detected in chromosome IV in South American wine strains.
Domestication Signatures—Inactivation of Aquaporin Genes
The domestication of wine strains and the consequent adaptation to sugar-rich and high osmolarity environments involved the adaptive loss of water channels encoded in the aquaporin genes AQY1 and AQY2 (Will et al. 2010). Several frame-shifting deletions or mutations giving rise to premature stop codons have been identified in the aquaporin genes of wine strains. Ale-type beer also has inactive aquaporin genes as well as other strains obtained from sugar rich environments (Gonçalves et al. 2016). All wine strains included in our study had at least one inactive aquaporin gene and 62% had the two genes inactive. For the six Brazilian strains clustering in the wine clade (one strain from cachaça, two strains from grape wine and three strains from jabuticaba wine), the inactivation of AQY genes was detected in all strains (supplementary table S1, Supplementary Material online). All but one of the 14 strains in the cachaça clades C1 and C2 had at least one aquaporin gene inactivated and most (10 out of 14) had the two genes inactivated. The single strain that had both genes functional was UFMG-CM-Y260 (cachaça C2), a strain not isolated from cachaça and a putative feral strain. For AQY1, the typical inactivation of wine strains, an adenine deletion at base position 881 that renders AQY1 inactive (A881 deletion), was also observed in cachaça strains of C1 and C2 clades (8 out of 12 cases of inactivation). The remaining four cases revealed a new type of inactivation caused by a thymine deletion at position 498 (supplementary table S1, Supplementary Material online). This deletion is unique of cachaça strains and has not been reported before. For AQY2 the typical 11 bp deletion of wine strains was detected in C1 and C2 strains. For a single cachaça strain, the AQY2 inactivation was caused by a guanine deletion at position 25, which is a typical sake strain inactivation. For the cachaça strains falling outside the C1 and C2 clades, the deeply admixed strain UFMG-CM-Y268 had both aquaporin genes inactivated but, interestingly, AQY1 was inactivated through a new kind of mutation, detected only in that strain (A817del). The cachaça strains belonging to the wine and bread groups had the inactivations typical of those groups whereas Brazilian wild strains had functional aquaporin genes (supplementary table S1, Supplementary Material online). For the bioethanol strains, the inactivations typical of the wine group were observed, similarly to what was seen for cachaça strains. The exception was a unique 20 bp insertion at position 326 of AQY2 detected for strain BG1.
Domestication Signatures—RTM1
RTM1 provides resistance to the effects of inhibitory compounds present in molasses and is a member of a three-gene cluster that also includes SUC telomeric genes (Ness and Aigle 1995). RTM1 can be viewed as a domestication signature of beer yeasts since it is consistently present in the Beer 1 clade and absent in wine strains (Gonçalves et al. 2016). This gene is also present in other domesticated groups like the Sake clade, albeit infrequently, and in this study we observed that it is consistently present in the Bread and Beer 2 clades (supplementary table S1, Supplementary Material online). Interestingly, RTM1 was present in cachaça strains of clades C1 (infrequently) and C2 (frequently) and one to three copies of this gene were detected in cachaça strains. We suggest that the RTM1 donors of cachaça, bread and beer strains of the Beer 2 clade belong to the Beer 1 clade. In spite of being present in several domesticated groups, RTM1 is also present consistently in wild Malaysian strains associated with sugary substrates (supplementary table S1, Supplementary Material online) and in West African strains. RTM1 was also detected, infrequently, in the Philippine and wild Brazilian B1 populations (supplementary table S1, Supplementary Material online).
Domestication Signatures—FZF1 and SSU1
FZF1 regulates the transcription of SSU1, a sulphite efflux pump conferring resistance to sulphite (Avram et al. 1999; Park and Bakalinsky 2000) and is a target of recent adaptive evolution in S. cerevisiae and S. paradoxus (Engle and Fay 2012). Moreover, most of the representatives of the wild population associated with Mediterranean oaks harbor a introgressed version of the allele found in the European population of S. paradoxus (Almeida et al. 2017). We compared the FZF1 sequences of Brazilian strains belonging to wild populations, to cachaça, and to grape and jabuticaba wine with sequences from representatives of all other populations. With the exception of introgressed alleles from S. paradoxus, the diversity of native S. cerevisiae sequences could be resolved in two basic alleles that we designated the Cosmopolitan allele (C) and the Wine allele (W) (fig. 4). Whereas the W allele was detected in the Wine, Beer 2, and Bread clades (although together with the C allele in some heterozygous bread strains), the C allele was found in most of the remaining populations, namely Beer 1, Sake, West Africa, NA–JPN, Philippines, Malaysia, and Brazil 1 (supplementary table S1 and fig. S2, Supplementary Material online). Interestingly, the cachaça clades C1 and C2 presented a unique situation because they included the W and C allele in roughly equal frequencies. Two of the seven strains from clades C1 and C2 that had an increased number of heterozygozyties, suggestive of a ploidy higher than 2n, had the C and W alleles (supplementary table S1 and fig. S2, Supplementary Material online), whereas the remaining were homozygous for this gene. Similarly with the cachaça strains, the group of bioethanol strains contained also the two FZF1 alleles. It thus appears that the presence of the C allele differentiates, at a population level, cachaça and wine strains. Another interesting observation concerned the wild Brazilian population B3. Among the studied strains of this population, the complex and probably tetraploid genome of UFMG-CM-Y456 had two copies of FZF1, one corresponding to allele C and the other corresponding to the FZF1 allele present in the North American population of S. paradoxus (fig. 4a andsupplementary fig. S2, Supplementary Material online). The other two representatives of population B3 (UFMG-CM-Y641 and UFMG-CM-Y642) had S. cerevisiae x S. paradoxus recombinant sequences as illustrated for the first strain in figure 4b. Therefore, in addition to the already described introgression of the European FZF1 allele of S. paradoxus into the population of S. cerevisiae associated with Mediterranean oaks (Almeida et al. 2017), we observed here a second case of introgression implicating the North American FZF1 allele of S. paradoxus.
Fig. 4.

—Phylogenetic analysis of FZF1 and BIO1 in Saccharomyces cerevisiae. (a) Phylogenetic tree based on a FZF1 alignment showing the cosmopolitan (C) and the wine (W) allele. The C allele is present in Beer 1, Brazil 1, Malaysia, North America–Japan, Philippines, Sake and West Africa populations, and also on bioethanol and cachaça strains, whereas the W allele is present in Wine, Bread and Beer 2 populations, and also on bioethanol and cachaça strains. Populations are color-coded and sequences from cachaça/bioethanol strains are highlighted by a red border. The phylogenetic analyses employed the Neighbor-Joining method and Tamura's 3-parameter model with bootstrap values > 90% indicated by black circles (1000 replicates). The tree also includes FZF1 sequences of different populations of S. paradoxus (EUR, Europe—SGD sequence; FE, Far East; NA, North America), and cases of introgression into the wild Brazilian B3 population, and was rotted with FZF1 (SGD) sequences of S. mikatae and S. uvarum (not included in the image). (b) Divergence plot of recombinant (North American S. paradoxus X S. cerevisiae) FZF1 sequence of UFMG-CM-Y641. (c) Phylogenetic tree of BIO1 from S. cerevisiae and S. paradoxus showing the origin of BIO1 in cachaça strains. The allele present in wild Brazilian populations and in cachaça and beer strains of BEER 2 clade is the North American S. paradoxus allele. One cachaça strain has the S. cerevisiae SAKE allele. The phylogeny was constructed as in (a) and was rooted with S. arboricola.
Two chromosomal translocations upstream of SSU1, a gene that codes for a sulfite efflux pump, have been implicated in sulfite resistance in wine strains because they lead to the overexpression of this gene (Pérez-Ortín et al. 2002; Zimmer et al. 2014). We compared wine strains and cachaça strains using PCR and primers designed to detect the native SSU1 version (chromosome XVI) or a translocated and more sulfite-resistant allele (SSU1-R, chromosome VIII or SSU1-R2, chromosome XV). Among 24 wine strains analyzed, we detected SSU1 in 20 occasions and SSU1-R in 10 occasions (in some cases both SSU1 and SSU1-R were detected in the same genome; supplementary table S1, Supplementary Material online). Interestingly all of the 19 cachaça strains analyzed (clades C1, C2, and mosaic strains) were positive for SSU1 and negative for SSU1-R and for SSU1-R2. These results suggest that sulfite resistance is not a trait selected for during cachaça fermentation, which is line with the absence of this compound during the fermentation process.
Domestication Signatures—Presence of BIO1/BIO6
BIO1 and BIO6 encode enzymes involved in the synthesis of biotin, a pathway that also includes enzymes coded by four additional genes (BIO2–BIO5). Whereas strains used in sake fermentation have the complete genetic makeup necessary to synthesize biotin de novo, the remaining industrially used strains like wine, bread, and beer yeasts are auxotrophic for biotin and lack functional versions of BIO1 and BIO6 (Hall and Dietrich 2007; Borneman and Pretorius 2015; Gonçalves et al. 2016). Moreover, the biotin pathway appears to have been lost in the Saccharomycotina and subsequently rebuilt by a combination of horizontal gene transfer and gene duplication followed by neofunctionalization (Hall and Dietrich 2007). Our survey for the presence or absence of BIO1/BIO6 in cachaça strains and remaining populations yielded interesting results. First, we detected that these two genes are present in the Brazilian wild populations B1 and B3 and also in the Philippine population (supplementary table S1, Supplementary Material online). Confirming earlier reports (Borneman and Pretorius 2015), we also recorded the presence of BIO1/BIO6 in sake strains and in the Malaysian and West African populations. These findings challenge the notion that in S. cerevisiae only sake strains are able to synthesize biotin. Moreover, a phylogenetic analysis of BIO1 revealed two clades, one containing native S. cerevisiae alleles from Sake, Philippine, Malaysian, and West African populations and the other corresponding to the North American S. paradoxus allele but including also the Brazilian wild populations, thus suggesting that BIO1 from S. paradoxus was introgressed in these S. cerevisiae populations (fig. 4c). The same pattern was observed for BIO6 (supplementary fig. S3, Supplementary Material online). We also observed that, contrary to wine strains, some cachaça strains also harbored BIO1 and BIO6. Most of the alleles found in cachaça strains could be assigned to the introgression from North American S. paradoxus observed in Brazilian wild strains. However, in one cachaça strain we detected the sake alleles of BIO1 and BIO6 (fig. 4c andsupplementary fig. S3, Supplementary Material online). Surprisingly, the introgressed S. paradoxus alleles of BIO1 and BIO6 were also detected in some beer strains of the Beer 2 clade (fig. 4c andsupplementary fig. S3, Supplementary Material online). It is not evident at this stage how this allele was acquired in beer strains.
Introgressions from S. paradoxus
We have previously reported a widespread dissemination of introgressions from the North American S. paradoxus population into the wild populations (B1 and B3) of Brazilian S. cerevisiae (Barbosa et al. 2016). Although the pattern of distribution of introgressions was not uniform among strains, overall distinct patterns could be discerned for populations B1 and B3. Also, the presence of these introgressions in Brazilian wild populations contrasted with their absence in other populations. Here we used the same approach to analyze cachaça strains. Introgressions from S. paradoxus (but not from other Saccharomyces species) were also detected but, globally, they were distinct from those previously found in Brazilian wild strains (fig. 5). In spite of some heterogeneity in their introgression patterns, all but one of the cachaça strains of clades C1 and C2 had introgressions. Moreover, except for a single ORF in chromosome XV, none of the introgressions detected in cachaça strains was detected in the Wine group. The introgressions of cachaça strains involved single ORFs or larger regions encompassing up to four ORFs. In total 20 ORFs and five chromosomes were implicated, less than those reported by Barbosa et al. (2016) for wild Brazilian strains (62 introgressed ORFs in 13 chromosomes). Remarkably, only three ORFs were simultaneously found in cachaça and wild Brazilian strains (fig. 5), thus pointing to independent origins in these two groups. We could determine that the majority of introgressions originated from the most widespread North American population of S. paradoxus, normally designated as population B, thus pointing to the occurrence of hybridization events in the American continent, rather than in Europe. However, an exception to this pattern was recorded for cachaça strains and encompassed a considerable number of introgressions from the European population of S. paradoxus, all located in chromosome III (fig. 5). As indicated above, some cachaça strains belong to the Wine and Bread groups, but these strains did not harbor the S. paradoxus introgressions detected for the C1 and C2 clades (supplementary fig. S4, Supplementary Material online). However, other cachaça strains with complex admixture patterns discussed above like UFMG-CM-Y623 and UFMG-CM-Y628 also had typical cachaça introgressions (supplementary fig. S4, Supplementary Material online). Interestingly, the bioethanol strains shared with cachaça strains five introgressions and had also two introgressions found in Brazilian wild strains but not in cachaça strains (supplementary fig. S4, Supplementary Material online). A Gene Ontology (GO) analysis of the complete set of introgressed genes detected in cachaça strains revealed that it was significantly enriched in genes encoding secondary active transmembrane transporters. Three genes were implicated: STL1, a glycerol proton symporter; SMF1, a broad specificity divalent and trivalent metal ion transporter; and OPT1, a proton-coupled oligopeptide transporter. Notably, these last two genes belong to the restricted group of three introgressed genes found simultaneously in cachaça and Brazilian wild strains (fig. 5). They belong also to a group of introgressed and significantly enriched genes coding for secondary active transmembrane transporters in wild Brazilian strains (Barbosa et al. 2016). For cachaça strains, the introgressed region in chromosome IV containing STL1, includes also PAD1 and FDC1 (fig. 5 and supplementary table S1, Supplementary Material online), two genes responsible for the decarboxylation of aromatic acids like ferulic and cinnamic acids, thus rendering them less toxic. In an attempt to understand if the introgressed genes improved resistance to ferulic acid in acidic conditions (pH 4.5), we compared the phenotypes of cachaça strains harboring the native and the introgressed alleles (eight strains in each group). Cachaça strains harboring the introgressed genes were slightly more resistant to 0.2% ferulic acid but without statistical significance since 50% of cachaça strains harboring the introgressed genes had intermediate resistance, whereas 25% of cachaça strains with native PAD1/FDC1 had the same resistance and all other strains had low resistance (supplementary table S2, Supplementary Material online).
Fig. 5.

—Distinct introgressions of Saccharomyces paradoxus in the genomes of Brazilian S. cerevisiae cachaça (C1 and C2) and wild (B1 and B3) populations and comparison with the Wine group. Introgressed ORFs are indicated in the first rows of the two panels and introgressions common to the cachaça and wild populations are highlighted in purple. (a) chromosome I–VII; (b) chromosome VIII–XVI. The presence of introgressions is color coded and refers to the donor population of S. paradoxus: European (light green), North American population B (dark green), unknown population or recombinant sequences (brown). The frequency (%) of each introgressed ORF in each population is indicated by numerals. The common names of the genes discussed in the text are indicated.
Diversity, Divergence, and Fixed Differences
Nucleotide diversity (pairwise differences, π*100) of the Cachaça population for which a more representative number of isolates is available (C1) was 0.0018% (table 1). This value is slightly higher than that of the wine population (0.0011%). We also measured the mean pairwise divergence between two alleles drawn from two populations (πB, estimated per site from pairwise comparisons across the total length of the genome). As expected, nucleotide divergence (πB*100) between cachaça C1 and the wine group was lower (0.176) that the divergence between C1 and other populations (0.223–0.595, divergence to MO and West Africa populations, respectively). We also observed that C1 and Wine had the highest number of shared polymorphisms and the lowest number of fixed differences in pairwise comparisons of the various populations with the cachaça C1 group (fig. 6).
Table 1.
Whole-Genome Diversity of Cachaça Strains and Comparison with Other Populations of Saccharomyces cerevisiae
| No. of Strains | Analyzed Sites | Segregating Sites | π | |
|---|---|---|---|---|
| Cachaça (C1) | 8 | 10,930,605 | 46,633 | 0.0018302 |
| Cachaça (C2) | 6 | 11,275,315 | 60,642 | 0.0029448 |
| Brazil (B1)a | 17 | 11,075,830 | 70,292 | 0.002169 |
| North America–Japanb | 42 | 11,348,218 | 1,19,184 | 0.002560 |
| Mediterranean oaksb | 31 | 11286153 | 56053 | 0.0009901 |
| Wineb | 19 | 11216288 | 56367 | 0.0011166 |
Note.—Diversity values are per site estimates calculated for the total length of the genome.
Taken from Barbosa et al. (2016).
Taken from Almeida et al. (2015).
Fig. 6.
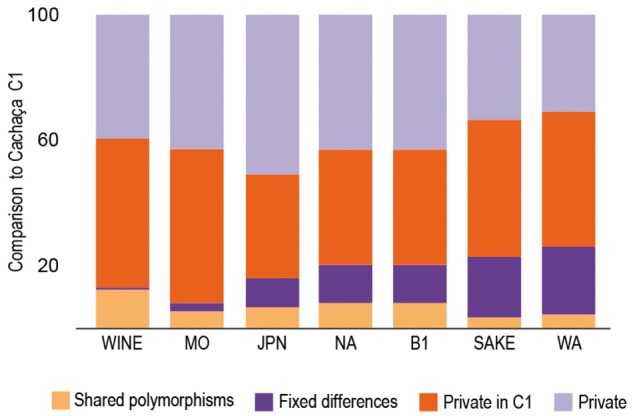
—Proportion of shared polymorphisms and fixed differences in pairwise comparisons of the cachaça C1 group with other populations of S. cerevisiae (B1, Wild Brazil, JPN, Japan; MO, Mediterranean oak, NA, North America; WA, West Africa).
Cachaça Feral Strains and Wild Brazilian Strains in Cachaça Fermentations
In three cases, strains with cachaça genotypes (UFMG-CM-Y260, UFMG-CM-Y636, and UFMG-CM-Y639, figs.1 and 2) were found in a wild environment, the tree Tapirira guianensis (supplementary table S1, Supplementary Material online). Therefore, we regard these strains as feral and as evidence that domesticated strains can disperse into wild environments. Evidence of gene flow from domesticated lineages into Brazilian wild populations had already been reported (Barbosa et al. 2016) and here we also observed partial wine ancestry in the population structure analysis of the Brazilian wild population B3 (fig. 2). The reverse situation, that is, the occurrence of wild Brazilian strains in cachaça fermentations was also observed, but in a single occasion (UFMG-CM-Y648, Brazilian wild population B1). Overall, the limited number of cases of transitions from the original niche observed suggests moderate genetic contact. An apparent consequence of this is the absence of genetic homogeneity at a regional scale, which might also be due to distinct selective pressures and a strong selective effect in anthropic and wild environments.
Discussion
Cachaça and Wine Strains—Similar but Not Identical
Here, we analyzed a comprehensive group of S. cerevisiae strains used in the fermentation of sugar cane juice, the first step of cachaça production. Most strains were isolated from spontaneous fermentations in Brazil but we also surveyed commercial cachaça strains and strains used for bioethanol production from sugar cane juice and molasses. Our analysis included, for comparison, representatives of the main domesticated and wild populations of S. cerevisiae known so far. The hypothesis that the yeasts that ferment cachaça are, or derive from, wine yeasts makes sense from an historical and technological perspective. Alternative hypotheses worth considering are that the genetic stock of cachaça yeasts derives mostly from local (Brazilian) wild populations or that cachaça yeasts form a distinct and unique domesticated group not related to any of the known populations.
Our genome-based phylogenetic analysis represents the first population genomics study of cachaça strains and provides two important findings. First, cachaça strains are distinct from the main groups of domesticated strains (Wine, Beer 1, Beer 2, Bread, and Sake). Secondly, and contrary to those domesticated groups (except Beer 1), cachaça strains do not cluster in a single clade and are therefore not monophyletic. Indeed most cachaça strains belong to two clades (C1 and C2) and a third clade comprehends the bioethanol strains. Moreover, a population structure analysis indicated that cachaça strains share most of their genetic ancestry with wine yeasts. It is therefore conceivable, both from the historical and from the population genomics perspective, that the early stock of cachaça yeasts corresponded to wine yeasts transported from Europe by Portuguese settlers. This means that cachaça yeasts are modified, that is, further domesticated, wine yeasts. In addition, because Brazilian bioethanol yeasts closely resemble cachaça yeasts, our conclusion can also be extended to this technologically important group. We note that the divergence of cachaça yeasts from wine yeasts cannot be explained simply by a geographic effect and by genetic isolation of South American strains. On the contrary, the genomes of Brazilian grape and jabuticaba wine strains suggest that typical wine yeasts belonging to the global population of wine strains occur in Brazil and not exhibit the differences observed in cachaça yeasts. An earlier study analyzed cachaça strains based on the sequences of four genes (Badotti et al. 2014) and recognized three groups: wine, “native” and hybrids between those two allelic variants. The higher resolution of our analysis confirmed the closeness between cachaça and wine strains but failed to reveal a substantial presence of native alleles in cachaça strains.
Combining the phylogenetic and the technological perspectives, three domesticated main groups emerge: the Wine-super group, including particular cases of beer yeasts (Beer 2 clade), cachaça and bioethanol strains; The Bread–Beer group (Beer 1, main group of ale-type beer strains); and the Sake group. With respect to cachaça yeasts, we find it relevant that their polyphyletic nature contrasts with the monophyletic nature of wine and sake yeasts. We also find it relevant that wine yeasts are typically diploid whereas cachaça yeasts are a mixture of diploid and probably tetraploid strains. These and other observations of discontinuities among domesticated lineages of S. cerevisiae (e.g., Gallone et al. 2016; Gonçalves et al. 2016) suggest that multiple domestication events have occurred in this species and also that fundamentally distinct patterns of adaptation to anthropic environments have unfolded. Based on such observations we propose here a multilayered domestication model, encompassing primary and secondary domestications with wine and cachaça as two epitomes of primary and secondary domestications, respectively (fig. 7).
Fig. 7.

—A model of secondary domestication in Saccharomyces cerevisiae. (a) Primarily domesticated lineages, WINE, BEER 1 (BE1) and SAKE, contribute to the secondarily domesticated genomes of CACHAÇA (C1, C2), BIOETHANOL (BETH), BREAD (BRD) and BEER 2 (BE2) yeasts. Populations including simultaneously wild and domesticated strains are placed at the borderline between wild and primarily domesticated populations. Hypothetical genetic transfers are marked with a question mark (abbreviations of other populations: B1 and B3, WILD BRAZIL B1 and B3; MO, MEDITERRANEAN OAK; MY, MALAYSIA; NA–JPN, NORTH AMERICA–JAPAN; PHL, PHILIPPINES). (b) Schematic representation of the phylogenetic relationships of wild and domesticated populations of S. cerevisiae. Stars depict the main domesticated groups.
The Case of Secondary Domestications
Cachaça yeasts are likely to represent a case of a second round of domestication of primarily domesticated wine yeasts because their genomes exhibit not only signatures of wine yeast domestication but also other markers that we postulate were acquired more recently (fig. 7). Signatures of primary (wine yeast) domestication are the presence of regions B and C and some of the types of inactivating mutations of aquaporin genes that were detected simultaneously in wine and cachaça yeasts. The signatures of secondary domestication are additional kinds of inactivation of aquaporin genes not found in wine yeasts or in any other group and that are therefore specific of cachaça yeasts. Another evidence of secondary domestication concerns the presence of the RTM1 gene in cachaça strains. Since this cluster provides resistance to inhibitory compounds that occur in molasses, its acquisition by cachaça yeasts might have enhanced adaptation to sugar cane juice. It is likely that this cluster was acquired from beer yeasts (Beer 1 clade) given its prevalence in this clade. Reacquisition of biotin prototrophy through cooptation of BIO1/BIO6 from S. paradoxus (or from Brazilian wild populations that originally acquired these genes from S. paradoxus) and, albeit less frequently, from Sake strains, also configures another set of secondary modifications. The acquisition of the FZF1 C allele and the apparent loss of SSU1-R, that enhances resistance to sulfite, are also two additional secondary modifications from a hypothetical ancestral wine genome. Besides a primary level of discontinuities detected between wine and cachaça yeasts at a phylogenetic and population level and a secondary category of differences that concerns gene-level domestication signatures, a third category of changes corresponds to the introgressions from S. paradoxus. We detected the introgression in cachaça strains, but not in wine strains, of eight ORFs from the North American population of S. paradoxus. As discussed earlier it is likely that this population is present also in South America and therefore these introgressions were acquired after the introduction of wine yeasts in South America.
We propose a three-step model for the domestication of S. cerevisiae. From wild populations, some of which known only from wild representatives (e.g., North-America Japan or Malaysian populations) whereas others include strains from both wild and anthropic environments (e.g., Philippines population), primarily domesticated populations have evolved. Wine and sake yeasts are the best examples of such events and beer yeasts of Beer 1 clade might also represent a primary domestication event (fig. 7). We tentatively place the Beer 1 clade in an intermediary position between primarily and secondarily domesticated lineages mostly because the genomes of beer yeasts are tetraploid, contrary to the genomes of wine and sake yeasts that are diploid. It was recently shown that polyploid S. cerevisiae cells explore more genotypic and phenotypic space than lower ploidy cells (Scott et al. 2017). According to our model, >2n genomes are common in secondarily domesticated populations, a situation that can relate to more derived states in yeast domestication trajectories (fig. 7). The Bread, Beer 2, and Cachaça populations would represent cases of secondary domestications where the genome of a wine yeast acquired some additional domestication-related features (e.g., RTM1 and BIO1/BIO6) while other features were lost (e.g., region A and SSU1-R in cachaça yeasts) (fig. 7).
Recent studies on the domestication of S. cerevisiae not only clarified the acquisition of several domestication-relevant traits (Novo et al. 2009; Marsit et al. 2015; Coi et al. 2017) and structural rearrangements (Borneman et al. 2012), but also unveiled unanticipated levels of complexity. For example, they showed that beer yeasts are genetically very diverse and contrary to wine yeasts do not have an obvious wild ancestor (Sampaio et al. 2017). Their ploidy is higher than 2n, a trait shared with bread yeasts, but the exact causes that elicited this change in some anthropic environments but not in others are unknown. Furthermore, it is possible that the evolution of their genomes included multiple interpopulation hybridizations, thus making it challenging to trace back their deep roots in one (or more) wild lineage. Also, Asian wild yeasts are not related to domesticated sake yeasts, thus rendering the origins of this domesticated group also obscure. These and other questions call for a better understanding of the domestication trajectories of S. cerevisiae which in turn will help ascertain the weight of independent domestications versus the role of the wine group in the global pattern of S. cerevisiae domestication. The results presented here reinforce this last aspect because cachaça yeasts clearly emerge as secondarily domesticated wine yeasts. The relevance of meta-domestication of wine yeasts is strengthened by our observation that bread and beer yeasts of the Beer 2 clade are also additional cases of secondary domestications of wine yeasts (fig. 7).
What Is a Cachaça Yeast?
In spite of being used worldwide, wine yeasts are better circumscribed phylogenetically than cachaça yeasts. The latter are not monophyletic as clades C1 and C2 demonstrate, and can even include strongly admixed strains as well as strains from other industrial groups as wine and bread strains. It might be speculated that because of its recent origins, cachaça fermentations have not yet shaped into a single and typical population, contrary to what is observed for the historically much older wine fermentations. Here, we have shown that wine yeasts constituted the main genetic source of cachaça strains. However multiple additional contributions originating in other domesticated populations and possibly also in native wild lineages or even in the sister species S. paradoxus also contributed to shape the unique genomes of cachaça yeasts. Therefore, from the three hypothesis stated earlier, the first one stating that cachaça yeasts are, or derive from, wine yeasts gets stronger support.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
This work was supported by Fundação para a Ciência e a Tecnologia (Portugal) grants PTDC/AGR-ALI/118590/2010 and PTDC/BIA-EVF/118618/2010 (J.P.S., P.G.), and UID/Multi/04378/2013 (A.P., J.P.S., P.G.). This work was supported by Conselho Nacional de Desenvolvimento Cientifico e Tecnológico (CNPq—Brazil, process numbers 407415/2013 and 0457499/2014-1, C.A.R.), Fundação do Amparo a Pesquisa do Estado de Minas Gerais (FAPEMIG, process number APQ-01525-14, C.A.R.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Literature Cited
- Almeida P, Barbosa R, Bensasson D, Gonçalves P, Sampaio JP.. 2017. Adaptive divergence in wine yeasts and their wild relatives suggests a prominent role for introgressions and rapid evolution at noncoding sites. Mol Ecol. 26(7):2167–2182. [DOI] [PubMed] [Google Scholar]
- Almeida P, et al. 2015. A population genomics insight into the Mediterranean origins of wine yeast domestication. Mol Ecol. 24(21):5412–5427. [DOI] [PubMed] [Google Scholar]
- Almeida P, et al. 2014. A Gondwanan imprint on global diversity and domestication of wine and cider yeast Saccharomyces uvarum. Nat Commun. 5:4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avram D, Leid M, Bakalinsky AT.. 1999. Fzf1p of Saccharomyces cerevisiae is a positive regulator of SSU1 transcription and its first zinc finger region is required for DNA binding. Yeast 15(6):473–480. [DOI] [PubMed] [Google Scholar]
- Badotti F, Gomes FCO, Rosa CA.. 2012. 38 Brazilian cachaça: fermentation and production In: Hui Y, editor. Handbook of plant-based fermented food and beverage technology. 2nd ed Boca Raton, London, New York: CRC Press; p. 639–648. [Google Scholar]
- Badotti F, Vilaça ST, Arias A, Rosa CA, Barrio E.. 2014. Two interbreeding populations of Saccharomyces cerevisiae strains coexist in cachaça fermentations from Brazil. FEMS Yeast Res. 14(2):289–301. [DOI] [PubMed] [Google Scholar]
- Bankevich A, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa R, et al. 2016. Evidence of natural hybridization in Brazilian wild lineages of Saccharomyces cerevisiae. Genome Biol Evol. 8(2):317–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B.. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borneman AR, et al. 2011. Whole-genome comparison reveals novel genetic elements that characterize the genome of industrial strains of Saccharomyces cerevisiae. PLoS Genet. 7(2):e1001287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borneman AR, et al. 2012. The genome sequence of the wine yeast VIN7 reveals an allotriploid hybrid genome with Saccharomyces cerevisiae and Saccharomyces kudriavzevii origins. FEMS Yeast Res. 12(1):88–96. [DOI] [PubMed] [Google Scholar]
- Borneman AR, Forgan AH, Kolouchova R, Fraser JA, Schmidt SA.. 2016. Whole genome comparison reveals high levels of inbreeding and strain redundancy across the spectrum of commercial wine strains of Saccharomyces cerevisiae. G3 (Bethesda) 6(4):957–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borneman AR, Pretorius IS.. 2015. Genomic insights into the Saccharomyces sensu stricto complex. Genetics 199(2):281–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos CR, et al. 2010. Features of Saccharomyces cerevisiae as a culture starter for the production of the distilled sugar cane beverage, cachaça in Brazil. J Appl Microbiol. 108(6):1871–1879. [DOI] [PubMed] [Google Scholar]
- Cardoso DR, et al. 2004. Comparison between cachaça and rum using pattern recognition methods. J Agric Food Chem. 52:3429–3433. [DOI] [PubMed] [Google Scholar]
- Coi AL, et al. 2017. Genomic signatures of adaptation to wine biological ageing conditions in biofilm-forming flor yeasts. Mol Ecol. 26(7):2150–2166. [DOI] [PubMed] [Google Scholar]
- Engle EK, Fay JC.. 2012. Divergence of the yeast transcription factor FZF1 affects sulfite resistance. PLoS Genet. 8(6):e1002763.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falush D, Stephens M, Pritchard JK.. 2003. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164(4):1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay JC, Benavides JA.. 2005. Evidence for domesticated and wild populations of Saccharomyces cerevisiae. PLoS Genet. 1(1):e5.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galeote V, et al. 2011. Amplification of a Zygosaccharomyces bailii DNA segment in wine yeast genomes by extrachromosomal circular DNA formation. PLoS One 6(3):e17872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallone B, et al. 2016. Domestication and divergence of Saccharomyces cerevisiae beer yeasts. Cell 166(6):1397–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves M, et al. 2016. Distinct domestication trajectories in top-fermenting beer yeasts and wine yeasts. Curr Biol. 26(20):2750–2761. [DOI] [PubMed] [Google Scholar]
- Guerra JB, et al. 2001. Genetic diversity of Saccharomyces cerevisiae strains during the 24 h fermentative cycle for the production of the artisanal Brazilian cachaça. Lett Appl Microbiol. 33(2):106–111. [DOI] [PubMed] [Google Scholar]
- Hall C, Dietrich FS.. 2007. The reacquisition of biotin prototrophy in Saccharomyces cerevisiae involved horizontal gene transfer, gene duplication and gene clustering. Genetics 177(4):2293–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutter S, Vilella AJ, Rozas J.. 2006. Genome-wide DNA polymorphism analyses using VariScan. BMC Bioinformatics 7:409.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legras J-L, Galeote V, Camarasa C, Blondin B, Dequin S.. 2017. Ecology, diversity and applications of Saccharomyces yeasts in food and beverages In: Satyanarayana T, Kunze G, editors. Yeast diversity in human welfare. Singapore: Springer Singapore; p. 283–321. [Google Scholar]
- Legras JL, Merdinoglu D, Cornuet JM, Karst F.. 2007. Bread, beer and wine: saccharomyces cerevisiae diversity reflects human history. Mol Ecol. 16(10):2091–2102. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R.. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14):1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25(16):2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liti G, et al. 2009. Population genomics of domestic and wild yeasts. Nature 458(7236):337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsit S, et al. 2015. Evolutionary advantage conferred by an eukaryote-to-eukaryote gene transfer event in wine yeasts. Mol Biol Evol. 32(7):1695–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai N, Masaki K, Fujii T, Iefuji H.. 2014. Single nucleotide polymorphisms of PAD1 and FDC1 show a positive relationship with ferulic acid decarboxylation ability among industrial yeasts used in alcoholic beverage production. J Biosci Bioeng. 118(1):50–55. [DOI] [PubMed] [Google Scholar]
- Ness F, Aigle M.. 1995. RTM1: a member of a new family of telomeric repeated genes in yeast. Genetics 140(3):945–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijkamp JF, et al. 2012. De novo sequencing, assembly and analysis of the genome of the laboratory strain Saccharomyces cerevisiae CEN.PK113-7D, a model for modern industrial biotechnology. Microb Cell Fact. 11(1):36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novo M, et al. 2009. Eukaryote-to-eukaryote gene transfer events revealed by the genome sequence of the wine yeast Saccharomyces cerevisiae EC1118. Proc Natl Acad Sci USA. 106(38):16333–16338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Bakalinsky AT.. 2000. SSU1 mediates sulphite efflux in Saccharomyces cerevisiae. Yeast 16(10):881–888. [DOI] [PubMed] [Google Scholar]
- Pérez-Ortín JE, Querol A, Puig S, Barrio E.. 2002. Molecular characterization of a chromosomal rearrangement involved in the adaptive evolution of yeast strains. Genome Res. 12(10):1533–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano P, Capece A, Jespersen L.. 2006. Taxonomic and ecological diversity of food and beverage yeasts In: Querol A, Fleet G, editors. The Yeast Handbook, volume 2, Yeasts in food and beverages. Berlin, Heidelberg: Springer-Verlag; p. 13–53. [Google Scholar]
- Rosa C, AM S, Faria J.. 2009. Cachaça production In: Ingledew WM, Kersall DR, Austin G., Kluhsipies C, editors. The alcohol textbook. 5th ed Nottingham: Nottingham University Press; p. 481–490. [Google Scholar]
- Sampaio JP, Pontes A, Libkind D, Hutzler M.. 2017. Taxonomy, diversity, and typing of brewing yeasts In: Bokulich NA, Bamforth CW, editors. Brewing microbiology: current research, omics and microbial ecology. UK: Caister Academic Press; p. 86–117. [Google Scholar]
- Scott AL, Richmond PA, Dowell RD, Selmecki AM.. 2017. The Influence of polyploidy on the evolution of yeast grown in a sub-optimal carbon source. Mol Biol Evol. 34(10):2690–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilakaratna V, Bensasson D.. 2017. Habitat predicts levels of genetic admixture in Saccharomyces cerevisiae. G3 (Bethesda) 7(9):2919–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vianna CR, Silva CLC, Neves MJ, Rosa CA.. 2008. Saccharomyces cerevisiae strains from traditional fermentations of Brazilian cachaça: trehalose metabolism, heat and ethanol resistance. Antonie Van Leeuwenhoek 93(1–2):205–217. [DOI] [PubMed] [Google Scholar]
- Will JL, et al. 2010. Incipient balancing selection through adaptive loss of aquaporins in natural Saccharomyces cerevisiae populations. PLoS Genet. 6(4):e1000893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer A, et al. 2014. QTL dissection of lag phase in wine fermentation reveals a new translocation responsible for Saccharomyces cerevisiae adaptation to sulfite. PLoS One 9(1):e86298. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.