


Abstract
The first nucleosomes in the downstream of transcription starting sites are called +1 nucleosomes, which are expected to be readily unwrapped for DNA transcription. To investigate DNA accessibility in +1 nucleosomes, MNase-seq experiments were carried out with 20 reconstituted +1 nucleosomes of budding yeast. Although MNase has been known for its sequence preference in DNA digestions, we confirmed that this sequence preference is overwhelmed by DNA accessibility by identifying the sequence-driven and accessibility-driven cleavages. Specifically, we find that sequences favoured by MNase at the end regions such as TA dinucleotide are prohibited from cleavage at the internal sites in the early stage of digestion. Nevertheless, sequences less favoured by MNase at the end regions such as AA/TT dinucleotide are predominantly cleaved at the internal sites in the early stage of digestion. Since AA/TT is known as a rigid dinucleotide step resistant to DNA bending, these internal cleavages reflect the local site exposures induced by DNA mechanics. As the DNA entry site of +1 nucleosomes in yeast is found AA/TT-rich, this sequence element may play a role in gene activation by reducing DNA–histone affinities along the direction of DNA transcription.
INTRODUCTION
Eukaryotic genes are organized in arrays of nucleosomes initiating from their transcription starting sites (TSS). The first nucleosomes in these arrays, termed as +1 nucleosomes, are highly positioned, leading the downstream nucleosomes (i.e. +2, +3 …) in a gradually decreased positioning (1). Such a pattern of nucleosome organization of genes has been found in many species, though the reason behind is still under investigation (2).
As the first hurdle to DNA transcription, +1 nucleosomes have been found to play critical roles in gene regulation. It has been reported that +1 nucleosomes interfere in transcription elongation by enhancing RNA Polymerase II (Pol II) pausing at the promoter-proximal region (3). The stall enhanced by +1 nucleosomes can serve as a break to recruit factors for transcription elongation and pre-mRNA processing. Furthermore, genome-wide analyses have shown that +1 nucleosomes especially of active genes are largely subjected to histone variant substitutions and histone modifications (4,5). Histone variant H2A.Z is found prevalent in the +1 nucleosomes of yeast; by replacing the canonical histone H2A, it can stimulate histone turnover and destabilize +1 nucleosomes for gene activation (6,7). On the other hand, histone modifications such as acetylation, methylation and phosphorylation have been extensively studied on their effects to perturb nucleosome structure and/or regulate molecular recognition by transcription factors and chromatin remodellers (8,9). The frequent presence of these modifications in the +1 nucleosomes is believed to play an important role in modulating DNA accessibility in nucleosomes and facilitating the sequential binding events for Pol II transit. Nevertheless, spontaneous site exposures of nucleosomes have also been observed at physiological conditions in vitro (10,11), indicating the inherent dynamic nature of nucleosomes. DNA sequence, which can encode DNA–histone interactions and DNA bending mechanics, should also play a non-negligible role. In this work, we examined the role of DNA sequence on DNA accessibility in the +1 nucleosomes of budding yeast by using Micrococcal Nuclease (MNase).
MNase is an endo- and exo-nuclease, which can specifically break down the phosphodiester bond between adjacent nucleotides at the presence of calcium ions and in a pH range from 8.6 to 10.3 (12). In the crystal structure of MNase complexed with a 3′, 5′-biphosphate thymidine (13), it shows that MNase engulfs the thymine base into a binding pocket and exposes the leaving phosphate group towards a calcium ion. It is clear that MNase cleaves DNA in a single-stranded manner. When the substrate is double helix, the original DNA base pairing and base stacking must be interrupted so that a single DNA base can insert into the binding pocket, forming a proper binding complex for cleavage. Since the dissociation of base pairing, deformation of base stacking and accommodation of a DNA base at the binding pocket are apparently sequence-dependent, MNase exhibits evident sequence preference as reported earlier (14–17). Despite this, MNase has been successfully used as a probe to detect DNA accessibility in DNA foot-printing assays. Not only histones but also other DNA-binding proteins have demonstrated their ability to shield DNA from MNase digestions (18–20).
In this study, we used MNase to probe the +1 nucleosomes of yeast in vitro, to investigate the correlation between DNA sequence and nucleosome site exposures mirrored by MNase digestions. Specifically, we looked into the cleavages made by MNase on the nucleosome core structures. This topic may be overlooked in studies where MNase has been used for chromatin digestions whilst only the mononucleosomal-sized DNA fragments were sifted for nucleosome positioning mapping. One of the direct consequences is exemplified by the observed discrepancy of nucleosome positioning near the 5′ nucleosome free region (NFR) at very low and high MNase concentrations (21,22). Since cleavages made on the nucleosome core structures could lead to an underestimated positioning, features of nucleosomes which can be both well-positioned and dynamically site-exposed may not be fully recognized. In short, our results depict how MNase cleaves nucleosomal DNA at a base pair resolution. We find that the digestion is dictated by MNase sequence preference at the end regions whereas driven by a bending-resistant sequence element at the internal sites in the early stage of digestion. This sequence element observed in the +1 nucleosomes of yeast and examined along the direction of DNA transcription is found to associate with gene transcription profile of the yeast genome (23), suggesting the comprehensive role of DNA sequence in gene regulation.
MATERIALS AND METHODS
Extracting DNA sequences of +1 nucleosomes
The reference genome and TSS of Saccharomyces cerevisiae were obtained from http://www.yeastgenome.org and http://cosbi3.ee.ncku.edu.tw/yna/statistic, respectively. Nucleosome dyad positions were obtained from (24). Along the direction of DNA transcription, nucleosomes with their dyad positions within 73 bp in the downstream of TSS were defined as +1 nucleosomes in this study. Hence, 3043 +1 nucleosomes were identified and their sequences were extracted.
Frequencies of occurrence of DNA dinucleotides
The +1 nucleosomal DNA sequences were aligned from the DNA entry site to the exit site, i.e. the direction of DNA transcription. Frequencies of occurrence were calculated for each type of DNA dinucleotide steps at each position in a nucleosome based on the 3043 +1 nucleosomes.
MNase-seq experiments
Selection of 20 +1 nucleosomal DNA sequences
The +1 nucleosomes with a nucleosome centre positioning score-to-noise ratio higher than 2.0 (24) were selected and sorted by the ratio of adenine contents in the entry-to-dyad region and exit-to-dyad region of the DNA sense/+ strand in a descending order. The top 10 +1 nucleosomes with the highest ratios of adenine content and another 10 with a ratio of 1.0 were selected (Supplementary Table S1).
Nucleosome reconstitution assays
Chemically synthesized 147 bp DNAs (Fasmac) were used for the nucleosome reconstitution. The human histone proteins (H2A, H2B, H3.1 and H4) were expressed and purified as described previously (25,26). To reconstitute the histone octamer, freeze-dried H2A, H2B, H3.1 and H4 were mixed in 20 mM Tris–HCl (pH 7.5) buffer, containing 20 mM 2-mercaptoethanol and 7 M guanidine hydrochloride. The sample was dialyzed against 10 mM Tris–HCl (pH 7.5) buffer, containing 2 M NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA) and 5 mM 2-mercaptoethanol. Reconstituted histone octamer was purified by superdex200 gel filtration column chromatography in 10 mM Tris–HCl (pH 7.5) buffer, containing 2 M NaCl, 1 mM EDTA and 5 mM 2-mercaptoethanol. The nucleosomes were reconstituted by the previously described method with modification (27). The DNAs were mixed with the histone octamer in 17 mM Tris–HCl (pH 7.5) buffer, containing 0.4 mM EDTA, 0.7 M NaCl, 1.3 M KCl, 1.7 mM 2-mercaptoethanol and 0.3 mM DTT. The samples were diluted with 20 mM Tris–HCl (pH 7.5) buffer, containing 1 mM DTT in six steps: sum of the NaCl and KCl concentrations was decreased in each step to 2, 1.33, 1, 0.8, 0.57 and 0.25 M. Reconstituted nucleosomes were incubated at 55°C for 1 h.
MNase digestion assays
The nucleosome mixture (94 nM), containing all types of nucleosomes, was incubated with 5.5 units/ml of MNase (Takara) at 37°C for 1, 3, 6 and 10 min, in 50 mM Tris–HCl (pH 8.0) buffer, containing 2.5 mM CaCl2, 69 mM NaCl, 81 mM KCl and 1.9 mM dithiothreitol. The reaction was stopped by adding half volume of deproteinization solution (20 mM Tris–HCl (pH 8.0), 20 mM EDTA, 0.5 mg/ml proteinase K (Roche) and 0.1% sodium dodecyl sulphate). The DNA fragments were purified by using Wizard SV Gel and PCR Clean-Up System (Promega). For the MNase digestion experiments with nuc19 and its mutants (Supplementary Table S1), each nucleosome (94 nM) was incubated with 5.5 units/ml of MNase (Takara) at 37°C for 1 and 3 min under the same conditions as described above. The DNA fragments were extracted by phenol/chloroform/isoamyl alcohol and precipitated by ethanol. For the MNase digestion experiments with free DNAs, DNA mixture (94 nM) was incubated with 0.5 units/ml of MNase (Takara) at 37°C for 1 and 3 min under the same conditions as nucleosome digestion experiments. The DNA fragments were purified by phenol/chloroform/isoamyl alcohol extraction and ethanol precipitation.
Next-generation sequencing
The libraries were prepared by using the NEBNext Ultra DNA Library Prep Kit (New England Biolabs). The optimal cycle number was determined by using the KAPA Real-time Library Amplification Kit (KAPA Biosystems). The libraries for the digestion assays of the 20 +1 nucleosomes were subjected to single-end sequencing for 50 cycles on an Illumina HiSeq 1500. The libraries for the digestion assays of free DNAs of the 20 +1 nucleosomes and the mutation experiments were subjected to paired-end sequencing for 50 cycles on an Illumina MiSeq.
Sequence alignments with Bowtie
After sequencing, DNA reads were mapped to the original DNA sequences of the 20 +1 nucleosomes with Bowtie. Default parameters for the ‘-n’ alignment mode in Bowtie were used, so that up to two mismatches were allowed in the seed region of a length of 28 bp and the sum of quality values at all mismatched positions would not exceed 70.
Read frequency and cut frequency
A read frequency is defined as the ratio of the number of reads starting from position ‘p’ on a strand of nucleosome and the total number of reads aligned on that strand, as follows:
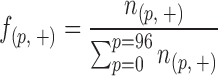 |
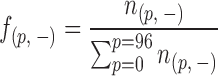 |
where ‘+’ and ‘−’ denote the sense and antisense strands, respectively. Since the minimum length of a DNA fragment sequenced is 51 bp, the last read start position is 96. A cut frequency describing the frequency of cleavage between position ‘p’ and ‘p+1’, denoted as 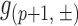 , is equivalent to the read frequency at position ‘p+1’,
, is equivalent to the read frequency at position ‘p+1’, 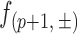 .
.
Non-parametric statistic tests
To compare the cut frequencies of dinucleotides at the specified region, one-sided Mann–Whitney U-test was used at a significance level of 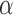 = 0.05. The sample to be compared in the tests is a group of cut frequencies for each dinucleotide at the end or internal regions, respectively. The end region is defined from position 1 to 7, which is around the contacting site of the first super helical turn on nucleosome core structure. At the end region, U-tests were carried out with the alternative hypothesis defined as H1: AA < TX (X = A, C, G). For the internal region, a tentative region is defined from ‘p’ to 73 (dyad), where ‘p’ varies from 1 to 73. For each tentative region, a U-test between AA and TA dinucleotide was carried out with H1: AA > TA. It is found that when p is in the range from 37 to 42, the one-sided P-value is uniformly <0.05, with p = 38 showing the highest significance (P-value = 0.01). The internal region is thus defined from 38 to 73. At the internal region, U-tests were carried out with H1: AA > TX (X = A, C, G).
= 0.05. The sample to be compared in the tests is a group of cut frequencies for each dinucleotide at the end or internal regions, respectively. The end region is defined from position 1 to 7, which is around the contacting site of the first super helical turn on nucleosome core structure. At the end region, U-tests were carried out with the alternative hypothesis defined as H1: AA < TX (X = A, C, G). For the internal region, a tentative region is defined from ‘p’ to 73 (dyad), where ‘p’ varies from 1 to 73. For each tentative region, a U-test between AA and TA dinucleotide was carried out with H1: AA > TA. It is found that when p is in the range from 37 to 42, the one-sided P-value is uniformly <0.05, with p = 38 showing the highest significance (P-value = 0.01). The internal region is thus defined from 38 to 73. At the internal region, U-tests were carried out with H1: AA > TX (X = A, C, G).
Correlation between AA/TT content and MNase digestion by comparing the two ends of a nucleosome
The amount of digestion at one end of nucleosome is defined as the sum of read frequencies from position 2 to ‘p’, where ‘p’ varies from 2 to 73. Position 1 was not included because of the patent MNase sequence specificity at this position. Similarly, the AA/TT contents of one end of nucleosome were determined accordingly. Then, the ratio of digestions and the ratio of AA/TT contents (entry versus exit) were calculated for each 2 to ‘p’ region of each nucleosome. The ratios of AAAA/TTTT contents were similarly determined for each 3 to ‘p’ region of each nucleosome.
RESULTS
DNA entry site of +1 nucleosomes in yeast is AA/TT-rich
Genome-wide nucleosome positioning map of S. cerevisiae has been determined at a base-pair resolution by Brogaard et al. in 2012 (24), which enabled us to extract the DNA sequences of +1 nucleosomes in budding yeast for this study.
In our initial analyses on the +1 nucleosomal DNA sequences, we found that there are more AA/TT dinucleotide steps positioned near the DNA entry site than the exit site and such a positioning diminishes gradually from the entry site to the dyad (Figure 1A). However, we did not observe any featured patterns for the other types of dinucleotide steps. In the downstream nucleosomes (+2, +3 and +4), it is only shown that AA/TT is more abundant than the other types of dinucleotide steps as in the +1 nucleosomes; whereas there is no difference in AA/TT content between the entry and exit sites (Supplementary Figure S1). This finding has also been reported in an earlier study in which the nucleosome positions of yeast were mapped with a different method (2). Since AA/TT has been characterized as a bending-resistant dinucleotide step in previous computational and experimental studies (28–30), we asked if this sequence element is utilized in yeast to assist their +1 nucleosomal DNA unwrapping for transcription.
Figure 1.

Frequencies of occurrence of DNA dinucleotide steps in the +1 nucleosomes of yeast and the sketch of MNase-seq experiments. (A) Frequencies of occurrence of dinucleotide steps at each position in the +1 nucleosomes of yeast were plotted. The DNA sequences were aligned from the DNA entry to exit site. It is shown that the frequencies of AA/TT dinucleotide step are distinctively higher than those of the other dinucleotide steps at all positions and that the DNA entry site of +1 nucleosomes in yeast is AA/TT-rich. (B) Schematic illustration of MNase-seq experiments carried out in this study is shown.
To investigate this, MNase-seq experiments were carried out as sketched in Figure 1B. Firstly, 20 +1 nucleosomal DNA sequences (147 bp) were selected to reconstitute mononucleosomes with human histones. The gel mobility shift assay revealed that all the reconstituted nucleosomes migrated similarly on a native polyacrylamide gel as single sharp bands (Supplementary Figure S2). This result suggests that all DNAs were wrapped properly around histone octamers within the nucleosomes. It is possible that the reconstituted nucleosomes exhibit local structural defects (1 or 2 bp) due to the spontaneous rotational repositioning of nucleosome. However, at a length of 147 bp and without the presence of chromatin remodellers or other chromatin regulators, nucleosomal DNAs in the reconstituted nucleosomes would not undergo the long-range rotational repositioning in super-helical turns. The reconstituted nucleosomes were then incubated with MNase for 1, 3, 6 and 10 min at 37°C. After digestions, DNA fragments on nucleosomes were released from histone cores and sequenced via single-end sequencing approach. Each experiment was repeated three times.
Digestion profile changes with incubation time
After sequencing, DNA reads were mapped to the original DNA sequences of the 20 +1 nucleosomes with Bowtie. In our sequencing processes, each DNA fragment was sequenced in 50 cycles; therefore, each read originated from a DNA fragment of a minimum length of 51 bp from the starting position to the 3′ end. Read frequencies were calculated for each position on each DNA strand of a nucleosome, averaged over the three parallel experiments for each incubation time. A read frequency, denoted as 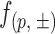 , is the frequency of fragments starting from position ‘p’ on the DNA sense (+) or antisense (−) strand of a nucleosome. To index DNA position ‘p’, the 5′ terminal nucleotides of both DNA strands were defined from ‘0’.
, is the frequency of fragments starting from position ‘p’ on the DNA sense (+) or antisense (−) strand of a nucleosome. To index DNA position ‘p’, the 5′ terminal nucleotides of both DNA strands were defined from ‘0’.
Firstly, read frequencies at positions 0(+) and 0(−) and positions inward are shown in Supplementary Figure S3 and Figure 2A–B, respectively. The read frequencies at 0(+) and 0(−) can approximately reflect the extent of digestions in the assays, suggesting that around 40–60% of nucleosomes were digested amongst different incubation times. However, these frequencies (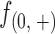 and
and 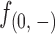 ) are not precisely equivalent to the amount of undigested nucleosomal DNAs. Since single-end sequencing approach was used in this set of experiments, we could not differentiate DNA fragments starting from 0(+) or 0(−) by their lengths (≥51 bp) to calculate the exact amount of intact nucleosomal DNAs (147 bp). However, because the read frequencies were normalized on a strand basis and the cleavages on nucleosomes were measured by read frequencies from 1(+) and 1(−) inward, we do not think the use of single-end sequencing approach here would bias our further analyses. By using paired-end sequencing approach, however, the information of where a DNA fragment terminates is provided and therefore the frequencies of fragments terminating at each position on each DNA strand of a nucleosome can be determined. In our additional experiments, paired-end sequencing approach was used.
) are not precisely equivalent to the amount of undigested nucleosomal DNAs. Since single-end sequencing approach was used in this set of experiments, we could not differentiate DNA fragments starting from 0(+) or 0(−) by their lengths (≥51 bp) to calculate the exact amount of intact nucleosomal DNAs (147 bp). However, because the read frequencies were normalized on a strand basis and the cleavages on nucleosomes were measured by read frequencies from 1(+) and 1(−) inward, we do not think the use of single-end sequencing approach here would bias our further analyses. By using paired-end sequencing approach, however, the information of where a DNA fragment terminates is provided and therefore the frequencies of fragments terminating at each position on each DNA strand of a nucleosome can be determined. In our additional experiments, paired-end sequencing approach was used.
Figure 2.

Read frequency as a function of read start position and overall digestion profile as a function of time. Read frequencies of the 20 +1 nucleosomes as a function of read start position for the 1-min and 3-min assays are shown in (A) and (B), respectively. The 5′ terminal nucleotides of both DNA strands are indexed from ‘0’. Read frequencies of the DNA sense/+ and antisense/− strands are shown in the upper and lower panels in the figures, respectively. DNA base compositions for each read start position are also indicated. (C) Cumulative read frequency as a function of read start position at different incubation times. Cumulative read frequencies were calculated from position 1 (instead of 0), averaged over the 40 strands of the 20 +1 nucleosomes. (D) Differentials of cumulative read frequencies between incubation times are shown.
Next, we compared the read frequencies from 1(+) and 1(−) to the dyad between the 1- and 3-min assays, shown in Figure 2A and B, respectively. We found that read frequencies at regions from 7(+) and 7(−) to the 5′ ends are higher in the 3-min assay, indicating that the end regions were subjected to more digestions in the longer assay. However, there is a decrease in read frequency at regions from 20(+) and 20(−) toward the dyad in 3 min, revealing that the 1-min assay is characteristic of more internal cleavages.
To further compare digestions at the end and internal regions between incubation times, cumulative read frequencies were calculated from position 1 averaged over the 40 strands of the 20 +1 nucleosomes, as shown in Figure 2C. It is clear that at the region from 1 to ∼20, digestions were uniformly enhanced with time. However, at the internal region from ∼20 to ∼50, there were more digestions resulted in the 1-min assay than the longer assays, indicated by the steepest increment of cumulative read frequencies in 1 min. More details can be obtained from the differentials of cumulative read frequencies between times as shown in Figure 2D. Compared with 1 min (3 versus 1 min, 6 versus 1 min and 10 versus 1 min), digestions at positions from 1 to ∼20 were gradually intensified from 3, 6 to 10 min. However, from ∼20 to ∼50, cleavages at each position of this region in 1 min were much more frequent, so that the differentials decrease evidently. Moreover, if compared between the longer assays (6 versus 3 min, 10 versus 3 min and 10 versus 6 min), the differentials first increase as a result of enhanced cleavages at the end regions and then reach a slowly growing plateau indicating that the internal cleavages were slightly increased with time in these assays.
In summary, the overall digestion profile reflects the general accessibility of DNA in nucleosomes, i.e. the end regions are more accessible than the internal sites. More importantly, the outstanding internal cleavages in the 1-min assay reveal an evolution of substrate for digestion with time. In 1 min, the initial substrates were intact nucleosomes; therefore, MNase would cleave the relatively accessible end regions and the internally exposed sites. However, with time going on, substrates in the 3, 6 and 10 min assays became diversified, including both intact nucleosomes and diversely cleaved intermediates. Hence, although MNase would continue cleaving the internally exposed sites in the longer assays, digestions were much more frequent at the end regions of previously cleaved nucleosomes.
MNase digestion on nucleosomal DNA involves sequence-driven and accessibility-driven cleavages
To understand how MNase cleaves nucleosomal DNA in a sequence-dependent manner, we correlate the read frequencies with sequence information of the 20 +1 nucleosomes as shown in Supplementary Appendix I. From these data, we identified two distinct digestion patterns on the TA- and AA-repeated regions.
Digestion of TA-repeated region is hindered by DNA accessibility
Amongst the 20 +1 nucleosomes, there are three TA-repeated regions located at different sites in nuc01, nuc02 and nuc10, respectively (Figure 3A, left panel). In each of these regions, read frequencies are gradually going up from the upstream to the downstream since 1 min. This pattern features a fast digestion of MNase on TA dinucleotides in the exo-cleaved manner, so that the upstream is substantially digested, contributing to the gradually ascending frequencies in the downstream. Moreover, if compared between the three TA-repeated regions, digestions are markedly dependent on the distance to the 5′ end of DNA. This position-dependent manner suggests that TA dinucleotides are generally well wrapped on histones, therefore MNase is prohibited from accessing the inward TAs if the upstream has yet been digested.
Figure 3.

MNase digestions on TA- and AA-repeated regions. (A) Read frequencies of TA-repeated regions from the sense/+ strand of nuc01, nuc02 and nuc10 are shown as a function of incubation time and DNA position. The digestions of nucleosomal DNAs (left panel) are compared with the digestions of free DNAs (right panel). It shows that although TAs are favourably cleaved in free DNA, they are generally well wrapped on histones and cleavages on nucleosomal TAs are suspended by the upstream. Therefore, MNase cleaves TAs in nucleosomes from the 5′ end of DNA as an exonuclease. (B) Read frequencies of AA-repeated regions from the antisense/− strand of nuc01, nuc03 and nuc07 are shown as a function of incubation time and DNA position. The digestions of nucleosomal DNAs (left panel) are compared with the digestions of free DNAs (right panel). It shows that at the inward sites of nucleosomes, digestions of AAs are allowed via nucleosome site exposures. The evenly distributed read frequencies in AA-repeated regions suggest that MNase cleaves AAs as an endonuclease in the early stage of digestion.
Further evidence is provided by the digestion results on free DNA of the same sequence (Figure 3A, right panel) (full data of free DNA digestions shown in Supplementary Appendix II). In the form of free DNA, all of the TAs within the same TA-repeated regions were substantially digested, regardless of the distance to the 5′ end of DNA; as a result, there were barely reads starting from thymine and nearly all of the reads were starting from adenine. However, in the form of nucleosomal DNA, reads starting from thymine were still present in the digestion products, suggesting that digestions on these TAs were prohibited by histone binding. This result provides the evidence that histone binding can shield nucleosomal DNA from MNase digestion, even if the DNA sequence is favoured by MNase. It is worth mentioning that if compared between the three sequences in free DNA, digestions were also weakened from the end to the inward sites. This is because digestions on TAs in free DNA proceed in an exo- and endo-manner; therefore, regions near the 5′ end would be more digested due to the exo-cleavages.
Digestion of AA-repeated region is allowed by nucleosome site exposure
Digestions of the AA-repeated regions from nuc01, nuc03 and nuc07 are presented in Figure 3B (left panel). It is shown that AA dinucleotides in each of these regions were evenly cleaved by MNase in the 1-min assay, nearly independent of the distance to the 5′ end of DNA. This pattern suggests that MNase can evenly cleave each dinucleotide in an AA-repeated region as an endonuclease. To result such a simultaneous accessibility for MNase digestion, exposure of the AA-repeated region is expected. If these regions were well protected by histone binding, MNase could only cleave from the 5′ end of DNA to result the wedge-shaped read frequencies as observed in TAs. However, with digestion going on, the evenly cleaved products were further digested by MNase as an exo-nuclease; as a result, the ascending read frequencies are observed in the longer assays.
Digestions of AA-repeated regions in free DNA are shown in Figure 3B (right panel). No clear differences are observed for AA digestions in the form of free and nucleosomal DNA, as AAs in nucleosomes can also be accessible to MNase via nucleosome site exposure. Nevertheless, AAs in free DNA are digested in the exo- and endo-manner; and the exo-cleavages can lead to the wedge-shaped read frequencies as observed in the free DNA of nuc07 since 1 min. Differently, AAs in nucleosomes are mainly cleaved in the endo-manner in 1 min, indicated by the evenly distributed frequencies.
Observations in TA- and AA-repeated regions are confirmed by mutation experiments
To confirm the observations in TA- and AA-repeated regions, mutation experiments were performed. ‘nuc19’ was selected as the wild-type since it does not have these sequence features and did not exhibit characteristic digestion patterns in the digestion assays of the 20 +1 nucleosomes. Four mutants were designed by replacing the end (position: 0–15) and a further inward site (position: 14–29) on the ‘+’ strand of nuc19 with TA-repeated and imperfect poly-A sequences, respectively (Supplementary Table S1). The wild-type and four mutants were incubated with MNase individually for 1 and 3 min, and the digestion products were sequenced via paired-end sequencing approach.
As shown in Figure 4 (upper half) (full data shown in Supplementary Appendix III), TAs at the end region (mut_1) were dramatically digested from the 5′ end of DNA in the exo-manner in 1 min; whilst at the internal site (mut_3), digestions on TAs were clearly hindered by DNA accessibility. Differently, AAs at the internal site (mut_4) were still evenly cleaved due to nucleosome site exposure, and the digestions in this region were clearly more frequent than the digestions of the same region in the wild-type sequence.
Figure 4.

MNase digestions on nuc19 and its four mutants. Read frequencies of nuc19 and its four mutants from 1-min digestion assays are shown. The digestion products were sequenced via paired-end sequencing approach. In ‘mut_1’ and ‘mut_2’, positions 0–15 on the ‘+’ strand of nuc19 were replaced with the TA-repeated and imperfect poly-A sequences, respectively. In ‘mut_3’ and ‘mut_4’, positions 14–29 on the ‘+’ strand of nuc19 were replaced with the TA-repeated and imperfect poly-A sequences, respectively. The mutated regions are shaded in blue. The frequencies of reads starting from each position in the range from 0 to 40 on the ‘+’ strand are shown in the upper panels. The frequencies of reads terminating at each position on the opposite strand (‘−’) of the mutated regions are shown in the lower panels. Note that frequencies at positions 0(+) and the paired positions on the opposite strand (‘−’) of nuc19, mut_2, mut_3 and mut_4 are above 0.08 and therefore not shown.
What is more, since paired-end sequencing approach was used in the mutation experiments, frequencies of DNA fragments terminating at each position on each DNA strand of a nucleosome can be determined. In Figure 4 (lower half), we plotted the frequencies of fragments terminating at positions on the opposite strand of the mutated regions. It is shown that both DNA strands were nearly symmetrically digested. As MNase cleaves DNA in a single-stranded manner, our results suggest that when MNase nicks one strand of DNA at a certain position, it will also cleave the same or neighbouring positions on the opposite strand.
MNase sequence preference and DNA accessibility dominate DNA digestions in nucleosome at different regions
We have demonstrated that the sequences favoured by MNase are well wrapped on histones, therefore, at the end regions where MNase can easily access from the 5′ end of DNA, digestions could be largely affected by MNase sequence preference. Whilst at the internal sites where DNA is supposed to be inaccessible to MNase, digestions should be mainly attributed to nucleosome site exposures. To verify this assumption, we analysed the frequencies of dinucleotide cleavages at the end and internal regions, respectively, based on the digestion results of the 20 +1 nucleosomes. Cut frequency is used hereafter to describe the frequency of cleavage on a DNA dinucleotide composed of positions ‘p’ and ‘p+1’ (see ‘Materials and Methods’ section).
MNase sequence preference biases digestions at the end region
There are three types of dinucleotides standing out with the highest cut frequencies at position = 1; they are TA, TC and TG (Figure 5A). Compared with others, these dinucleotides show extensive digestions at this presumably accessible region, indicating that they are the preferential sequences for MNase. Referring to the cleaving mechanism of MNase revealed by the X-ray crystal structural analysis (13), it is implied that these three dinucleotides are prone to deform their base pairing and base stacking to be favourably cleaved by MNase, which is supported by earlier studies on DNA mechanics (28,29,31). More importantly, when extending inward, i.e. from position 1 to 7 which is around the contacting site of the first super helical turn on nucleosome core structure, cut frequencies of these preferential sequences decrease with distance, consistent with our earlier observations in the TA-repeated regions. Whereas AA dinucleotide behaves in a different manner that the cut frequencies are modest at position = 1 and nearly independent of the distance to the 5′ end of DNA from position 2 to 7 in 1 min.
Figure 5.

Frequencies of dinucleotide cleavage at the end and internal regions. (A) Cut frequencies of dinucleotides AA, TA, TC, TG and TT at the end region are shown in boxplots for different incubation times. The end region was defined from dinucleotide position 1 to 7 and the frequencies were transformed in the log10 scale. One-sided Mann–Whitney U-tests were carried out to compare the digestions at the end region between AA/TT and TX (X = A, C, G) at a significance level of 0.05. The alternative hypotheses were defined as H1: AA < TX and H1: TT < TX, respectively. If the one-sided P-value is lower than 0.05, the null hypothesis is rejected in favour of the alternative hypothesis. The symbolic indications of statistical significance are as follows: (i) *: P-value ≤ 0.05; (ii) **: P-value ≤ 0.01; (iii) ***: P-value ≤ 0.001; (iv) ****: P-value ≤ 0.0001 (same below). (B) Cut frequencies of dinucleotides AA, TA, TC, TG and TT at the internal region are shown in boxplots for different incubation times. The internal region was defined from dinucleotide position 38 to 73 and the frequencies were transformed in the log10 scale. One-sided Mann–Whitney U-tests were carried out to compare the digestions at the internal region between AA/TT and TX (X = A, C, G) at a significance level of 0.05. The alternative hypotheses were defined as H1: AA > TX and H1: TT > TX, respectively. Here, the alternative hypotheses were reversed from those defined for the end region so that the expected decisions are always indicated by P-value ≤ 0.05.
Next, we did statistic tests to verify the significance of differences observed in the dinucleotide digestions. We incorporated the cut frequencies of each dinucleotide at the specified region into a group and compared the groups of cut frequencies between dinucleotides by using the one-sided Mann–Whitney U-test (α = 0.05). U-test compares the medians of two samples and does not require the samples to be normally distributed as t-test, which meet our needs. Because the frequencies are position-dependent and the occurrences of different dinucleotides at the same position given by the 20 +1 nucleosomes are not even, which lead to the non-normal distributions of cut frequencies.
As shown in Figure 5A, although the preferential sequences show extensive digestions at position = 1, the differences between AA and TX (X = A, C, G) at the end region (from position 1 to 7) turn out to be insignificant in 1 min. This is because as mentioned earlier the preferential sequences are of a higher affinity to histones, as a result, in the early stage cleavages on these sequences are suspended by the upstream. However, in 3 and 6 min, the preference of MNase for TA and TC over AA is shown to be significant. Whilst in 10 min, digestions at the end region have been exhaustive, therefore the significance of differences vanishes. As for TG, although it shows a higher cut frequency at position = 1 than AA as well as a higher median in the 6 and 10 min assays, the differences are suggested to be insignificant.
DNA accessibility dominates digestions of inward DNA in the early stage
Initially we did not know from which starting position to the dyad can be defined as an internal region to account for nucleosome site exposure. Thus, we varied the starting position from 1 to 73 (dyad) and did U-tests for each tentative internal region between AA and TA dinucleotides for the 1-min assay. The result suggests that when the region is defined from 37∼42 to the dyad, AA is significantly more cleaved than TA, with 38 showing the highest significance (P-value = 0.01) (Supplementary Figure S4). Such a range (37∼42) for the starting position of internal region is expected, because MNase sequence preference affects the digestions in the upstream towards the 5′ end and the digestions in the downstream close to the dyad are too weak to observe a significant difference. Thus, we defined the internal region from 38 to 73.
The results for the internal region are shown in Figure 5B. Firstly, digestions of each dinucleotide in 1 min are shown to diminish towards the dyad where DNA–histone interactions have been predicted as the strongest (32). More importantly, as expected, AA dinucleotide is significantly more cleaved than all the identified preferential sequences in 1 min, confirmed by the U-tests (Figure 5B). However, in 3 min, TA is quickly more digested than AA, due to the increased accessibility of TA to MNase with digestion going on. Whilst for TC and TG, it is only in the 10-min assay that the preference of MNase starts to effectively bias the digestions.
Thus, we conclude that it is DNA accessibility rather than MNase sequence preference that dominates digestions of inward DNA in the early stage and that AA dinucleotide is the site-exposure sequence, less favoured by MNase compared with the preferential sequences.
Successive rather than isolated AA/TT steps are responsible for site exposure
Since AA and TT refer to the same dinucleotide step, they should share the same mechanics to induce site exposure and be detected by MNase. However, we did observe discrepancies between the two dinucleotides in the U-tests. It is shown that at the end region the median of TT is generally lower than those of the preferential sequences in all assays, whereas a significant difference is only observed in 10 min against TC (Figure 5A). As for the internal region in 1 min, TT is only shown to be significantly more cleaved than TG (Figure 5B). If TT is also responsible for site exposure as AA, overwhelming digestions on TT should be expected when compared with TA and TC as well.
To find out the reason, we examined the frequencies of occurrence of AA and TT in the 20 +1 nucleosomes. We find that there are 49% of AA dinucleotides residing in a AA-repeated region of a minimum length of 4 bp (i.e. an A-tetramer) in the regions from 0 to 73 of both DNA strands. However, this value for TT is only 26%, indicating that most TTs are in the same region as isolated dinucleotides. This difference may explain our concern above, as successive AA/TT dinucleotide steps should be able to induce site exposures whereas the bending rigidity of a single AA/TT step may not.
In addition, since single-end sequencing approach was used to sequence the digestion products of the 20 +1 nucleosomes, we could not determine the frequencies of cleavage on the opposite strand of AA-repeated regions (i.e. TT-repeated regions). However, in our mutation experiments where paired-end sequencing approach was used, we do observe the comparable digestions of TT-repeated regions on the opposite strand (Figure 4).
Besides the dinucleotides discussed so far, we also examined digestions on the other dinucleotides (Supplementary Figures S5 and 6). For the preferential sequences, we did not identify dinucleotides other than those mentioned above for which MNase shows a preference (Supplementary Figure S7). However, for the site-exposure sequences, we did observe others which have also been recognized as rigid dinucleotides in previous studies (28,29), showing significant cleavages in 1 min, such as AC, AG and AT (Supplementary Figure S8). We believe that these dinucleotides could also induce nucleosome site exposures at sites in genomes where they are abundant.
Correlation between MNase digestions and contents of site-exposure sequence
Next, we compared the two ends of a nucleosome to investigate the correlation between MNase digestion and AA/TT content. The two ends refer to the DNA entry and exit site of a nucleosome. By extending from the entry and exit site to the dyad one position at a time, we calculated the ratio of digestions (sum of read frequencies) between the two ends (entry versus exit) and the corresponding ratio of AA/TT contents.
Firstly, we classified the 20 +1 nucleosomes into two groups based on the numbers of site exposure sequence elements (denoted as ‘SESEs’ and defined as discrete AAAA or TTTT segments) in their sequences. Specifically, if there are three or more SESEs in the region from 0 to 73 on either strand of a nucleosome, that nucleosome is grouped into the class of nucleosomes with SESEs; on the other hand, nucleosomes with two or fewer SESEs in the same region on each strand are grouped into the class of nucleosomes with no or fewer SESEs.
Correlations of the two classes of nucleosomes from the 1- and 3-min assays are shown in Figure 6A. The rectangle regions shaded in red and blue indicate that the entry site of a nucleosome with more AA/TTs is more digested and that the exit site with more AA/TTs is more digested, respectively. It is shown that data points of the class of nucleosomes with SESEs are well confined in the shaded rectangle regions, with a correlation coefficient of 0.35 and 0.63 for the 1- and 3-min assays, respectively. This result reveals a positive correlation between AA/TT content and the tendency of one end of nucleosome to dissociate from histones driven by the AA/TT-dependent site exposures and reflected by MNase digestions.
Figure 6.

Correlation between MNase digestions and contents of site-exposure sequence by comparing the two ends of a nucleosome. Correlations between MNase digestions and AA/TT contents in the 1- and 3-min assays are shown on the left and right panels of (A), respectively. Similarly, (B) shows the correlations of MNase digestion versus AAAA/TTTT content from the 1- and 3-min assays. The shaded rectangle regions in red indicate that the entry site of a nucleosome with more AA/TTs or AAAA/TTTTs gets more digested; the regions in blue indicate that the exit site with more AA/TTs or AAAA/TTTTs gets more digested. The 20 +1 nucleosomes are divided into two groups based on the numbers of site exposure sequence elements (SESEs, defined as discrete AAAA or TTTT segments) in their sequences. Specifically, nucleosomes with SESEs (coloured in black) are those with three or more SESEs on either strand of the nucleosomes, including nuc01, nuc02, nuc03, nuc05, nuc07, nuc10 and nuc20. Oppositely, nucleosomes with no or fewer SESEs (coloured in orange) consisting of the rest of the +1 nucleosomes, are those with two or fewer SESEs on each strand. Correlation coefficients for each class of nucleosomes under each incubation time are also indicated.
However, for nucleosomes with no or fewer SESEs, data points are largely scattered outside of the shaded regions and show nearly no correlations, as indicated by the correlation coefficients of 0.07 and 0.00 for the 1- and 3-min assays, respectively. This result is expected that not all of the 20 +1 nucleosomes show positive correlations between MNase digestions and AA/TT contents. Because MNase digestion is not exclusively on AA/TT dinucleotides, substantial digestions can also occur on other types of dinucleotides if they are favoured by MNase especially at the end regions where DNA is presumably accessible. For nucleosomes with no or fewer SESEs, AA/TT dinucleotides in these nucleosomes are mostly as isolated dinucleotides which cannot provoke nucleosome site exposures, therefore digestions were mainly proceeded from the 5′ end of DNA and fluctuated by the sensitivity of MNase to different types of dinucleotides encountered. Hence, for nucleosomes with no or fewer SESEs, MNase digestion is not AA/TT-dependent.
In addition, we have also examined the correlations between MNase digestions and AAAA/TTTT contents (Figure 6B). Compared with the correlations examined on the AA/TT content, higher correlation coefficients were observed, especially for the class of nucleosomes with no or fewer SESEs (0.19 and 0.33 for the 1- and 3-min assays, respectively). This result suggests that as long as there are SESEs in the sequence which can induce nucleosome site exposures to promote MNase digestions, MNase digestion is positively correlated with AAAA/TTTT content and that AAAA/TTTT is more robust than AA/TT when referred as a site-exposure sequence.
AA/TT contents in the +1 nucleosomes of yeast are associated with gene transcription levels
Since AA/TT is abundant at the entry site of +1 nucleosomes in yeast, we propose that this sequence element might be utilized in gene regulation to facilitate DNA unwrapping for transcription. Here, we associate the transcription levels of yeast genes reported in an earlier study (23) with the AA/TT contents in their +1 nucleosomes.
The results are shown in Table 1. For the low transcribed genes, for example, there are 368 of them associated with a +1 nucleosome of a greater AA/TT content in the entry-to-dyad region (than the exit-to-dyad region). On the other hand, there are 257 low transcribed genes with more AA/TTs in the exit-to-dyad region in their +1 nucleosomes. This difference in the numbers of genes gives a ratio of 1.43. It is found that when the transcription level advances, this ratio also increases. For the highly transcribed genes, this ratio reaches 1.99, indicating that nearly two thirds of the highly transcribed genes are coupled with a +1 nucleosome with more AA/TTs in the entry-to-dyad region. The significance of the ratios is indicated by the P-values from Chi-squared tests shown in the parentheses. Similar results were obtained for the AAAA/TTTT element.
Table 1.
AA/TT contents and gene transcription levels
| AA/TT content | AAAA/TTTT content | ||||||
|---|---|---|---|---|---|---|---|
| Level | Range of level (in log2) | Entry-to-dyad | Exit-to-dyad | Ratio | Entry-to-dyad | Exit-to-dyad | Ratio |
| No_trans | (0, 2] | 81 | 67 | 1.21 (0.3) | 68 | 66 | 1.03 (0.93) |
| Low | (2, 4] | 368 | 257 | 1.43 (2.6 × 10−2) | 322 | 212 | 1.52 (1.7 × 10−2) |
| Medium | (4, 6] | 531 | 348 | 1.53 (2.0 × 10−3) | 457 | 299 | 1.53 (4.1 × 10−3) |
| High | (6, +∞) | 287 | 144 | 1.99 (5.7 × 10−4) | 245 | 131 | 1.87 (3.3 × 10−3) |
The transcription levels of yeast genes have been measured in a previous study via RNA-seq approach (23) and classified into four categories as listed above. For each transcription level, the numbers of genes associated with a +1 nucleosome of a greater AA/TT content in the entry-to-dyad region, in the exit-to-dyad region and the ratio between the two are shown, respectively. Results examined for the AAAA/TTTT element are also shown. P-values from Chi-squared tests are shown in the parentheses. In the Chi-squared tests, the null hypothesis H0 is defined as the ratio = 1.
This analysis unveils a coincidence in the yeast genome that the highly transcribed genes are more likely to position more AA/TTs in the entry-to-dyad region than the exit-to-dyad region in their +1 nucleosomes. As AA/TT is able to induce nucleosome site exposure, this positioning could lower the overall DNA–histone affinities in the entry-to-dyad region of +1 nucleosomes to facilitate DNA dissociation for Pol II transit.
However, it is worth mentioning that we did not observe a simple linear correlation between the absolute amount of AA/TTs and transcription levels, i.e. genes with their +1 nucleosomes possessing more AA/TTs in the entry-to-dyad region than the other genes do not turn out to be more transcribed. Since transcription is a complex process involving a variety of regulating factors; DNA sequence, as one of them, may help in nucleosome unwrapping whereas it is not likely to interfere with gene transcription levels.
What is more, the transcription levels of yeast genes we referenced in our study were measured under the vegetative growth. It should be noted that the transcription levels of genes can vary, dependent on the growth conditions, cellular states, etc. Hence, it is worth of efforts in future to investigate how the correlation evolves under different growth conditions and cellular states.
DISCUSSION
DNA sequence plays a non-negligible role in regulating DNA accessibility in nucleosomes
Existing studies have elaborated that access to DNA in nucleosomes can be regulated via different mechanisms, e.g. chromatin remodelling (33–35), histone modifications (36,37), histone variants (5,6) and chaperones (38–40). These mechanisms differ in their ways and outcomes of triggering nucleosome destabilization, but ultimately, they all destabilize nucleosomes by altering DNA–histone interactions. It has been known that the DNA–histone interactions to promote and sustain the nucleosome core structure are barely sequence-dependent, evidenced by the reconstituted nucleosomes with different DNA sequences. However, upon formation, nucleosomes can be highly dynamic (‘fragile’) or remain stabilized, depending on the binding affinities between DNA and histones. DNA sequence as a determinant to sequence-dependent DNA–histone interactions and DNA bending mechanics, should also play a non-negligible role in regulating nucleosome stability.
Evidences for sequence-dependent nucleosome stability have been provided by previous studies. For example, free energies for nucleosome formation have been measured for many DNA sequences in competitive reconstitution assays and differences in binding affinities have been observed even between similar sequences (41). On the other hand, poly-dA/dT as a bending-resistant DNA sequence has been found frequently present in the 5′ NFRs near promoters, which demonstrates that the bending rigidity of a DNA sequence could exclude nucleosome formation (42–44). Additional experiments have shown that insertion of this sequence element into a nucleosomal DNA sequence could increase the equilibrium constant for nucleosome site exposure by 1.5- to 1.7-fold per position (30).
MNase sequence preference in DNA digestions is overwhelmed by DNA accessibility
MNase has long been known for its sequence preference when it cleaves DNA. But still, it has been extensively used in the routine to fragment chromatin for nucleosomes. The reasons can be attributed to the followings. Firstly, there is no evidence showing that MNase can cleave bound DNA under the protection of protein binding. Oppositely, existing studies and this work have demonstrated that protein binding can shield DNA from MNase digestion (19,20). Secondly, experiments have also made it clear that MNase not only cleaves its favoured DNA sequences, but also other types of sequences though at a lower rate (14–17). Based on these two aspects, it can be reasoned that MNase sequence preference in DNA digestions is overwhelmed by DNA accessibility, i.e. even for the preferred sequences, as long as in the bound state, cleavage is prohibited.
In this study, we probe the DNA accessibility in the +1 nucleosomes of yeast by using MNase. By comparing the digestions at the end and internal regions at different time scales, we identified the preferential sequences and site-exposure sequence. As the preferential sequence, TA, TC and TG are found generally well bound on histones and therefore they can only be preferentially cleaved in the exo-manner from the 5′ end of DNA (Figure 7A). Being favoured by MNase means, these dinucleotides should be able to twist and roll well enough to fit into the binding pocket of MNase for cleavage, which is consistent with earlier analyses on nucleosomal DNA (31). TA and TG ( = CA) have been recognized as the most flexible dinucleotide steps with respect to twist, roll and slide, which enables them to contribute to the smoothing bending at major grooves and the kinked bending at minor grooves in nucleosomes, respectively (31). On the other hand, AA ( = TT) dinucleotide step as the site-exposure sequence, can induce nucleosome site exposure and result the concurrent accessibility of DNA stretch for MNase cleavage in the endo-manner (Figure 7B). Since the DNA entry site of +1 nucleosomes in yeast is AA/TT-rich, it is expected that multiple sites composed of the site-exposure sequence and assembled at one end of nucleosome could effectively lower DNA–histone affinities for gene activation (Figure 7C).
Figure 7.

Sequence-dependent site exposure in nucleosome. (A) MNase digestion on preferential sequence. When the preferential sequence (e.g. TATA) is at the end region where MNase can access from the 5′ end of DNA, TATA would be favourably cleaved. However, if it is at the internal region where TATA is tightly bound on histones, cleavages are prohibited. (B) MNase digestion on site-exposure sequence. When the site-exposure sequence (e.g. AAAA) is at the end region, because MNase can access from the 5′ end of DNA and sequence-dependent site exposure occurs, cleavages on AAAA are allowed, though less favourably than TATA. When it is at the internal site, due to site exposure, cleavages will also occur. (C) When multiple sites composed of the site-exposure sequence are assembled at one end of nucleosome (i.e. DNA entry site), the overall affinities between DNA and histones will dwindle to assist nucleosome unwrapping with the presence of transcription factors or chromatin remodellers (shown in green ellipse).
Furthermore, existing studies seem to have generalized the sequence preference of MNase to A/T-rich sequences (45); however, from our analyses, we want to emphasize that MNase differentiates dinucleotides (e.g. AA and TA) even those representing the same dinucleotide step (e.g. TG and CA). In our study, we find TA is extremely favoured by MNase and exhibits a fast digestion. Accordantly, it has been reported that cleavage on poly-d(A-T) is 8 times faster than poly-d(A) and ∼120 times faster than poly-d(G-C) (17).
MNase cleavages on nucleosome core structures lead to the underestimated nucleosome positioning
Although MNase sequence preference could hardly bias the predictions for nucleosome positions because of the shielding effect of histone binding (46), it does affect the predictions for nucleosome positioning density.
Inconsistent positioning densities of +1 and −1 nucleosomes near the 5′ NFR have been observed in the yeast (21) and Drosophila (22) chromatin at very low and high MNase concentrations. Specifically, a mild digestion at low MNase strength may only fragment genes into oligo- to mono-nucleosomes. Therefore, the leading nucleosomes (+1 or −1) could be well fragmented and exhibit a distinguished positioning whereas the downstream nucleosomes (+2, +3 …) have yet been digested. On the contrary, a high MNase strength will not only result a thorough digestion up to the downstream nucleosomes, but also arouse significant cleavages on the core structures of the leading nucleosomes, which will eventually lead to an underestimated positioning.
In the study of Drosophila chromatin (22), genome-wide MNase-sensitive nucleosomes have been identified to favour A/T-rich sequences, which is consistent with our findings of the AA/TT-dependent site exposures. Similar results have also been reported recently, but concerns have been raised if the sensitivity of MNase to a nucleosome is the result of MNase sequence specificity or nucleosome stability (47,48).
In this study, we demonstrated how MNase cleaves nucleosomal DNA at a base-pair resolution, identified the sequence-driven and accessibility-driven cleavages and concluded that MNase sequence preference is restrained by DNA accessibility. Our results suggest that it is inappropriate to attribute the sensitivity of MNase to A/T-rich nucleosomes to the preference of MNase for A/T-rich sequences. The digestion especially at the inward sites of nucleosome is ultimately a reflection of DNA accessibility, regardless of MNase sequence preference. We believe that the insights provided here will help future interpretations of sequencing data generated from MNase digestion assays.
DATA AVAILABILITY
MNase-seq data generated in this study have been deposited to European Nucleotide Archive under the accession numbers: PRJEB25382, PRJEB25383 and PRJEB25388.
Supplementary Material
ACKNOWLEDGEMENTS
We thank our lab members for the valuable discussions and Dr Osakabe, A. and Dr Arimura, Y. for their helpful suggestions. We also thank the Advanced Computational Scientific Program of the Research Institute for Information Technology, Kyushu University for providing the high-performance computing resources.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Ministry of Education, Culture, Sports, Science and Technology (MEXT) Strategic Programs for Innovative Research, Computational Life Science and Application in Drug Discovery and Medical Development [hp160223, hp170255, hp18019 to H.Ko.]; Japan Society for the Promotion of Science KAKENHI [JP25116002, JP17H01408 to H.Ku., JP25116003 to H.Ko., JP251160010 to Y.O.]; Platform Project for Supporting in Drug Discovery and Life Science Research from the Japan Agency for Medical Research and Development (AMED) [JP18am0101076 to H.Ku., JP18am0101106 to H.Ko.]; Japan Science and Technology Agency CREST [JPMJCR16G1 to Y.O.]. Funding for open access charge: Ministry of Education, Culture, Sports, Science and Technology (MEXT) Strategic Programs for Innovative Research, Computational Life Science and Application in Drug Discovery and Medical Development [hp180191 to H. Ko.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Jansen A., Verstrepen K.J.. Nucleosome positioning in saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 2011; 75:301–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mavrich T.N., Ioshikhes I.P., Venters B.J., Jiang C., Tomsho L.P., Qi J., Schuster S.C., Albert I., Pugh B.F.. A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res. 2008; 18:1073–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jimeno-González S., Ceballos-Chávez M., Reyes J.C.. A positioned +1 nucleosome enhances promoter-proximal pausing. Nucleic Acids Res. 2015; 43:3068–3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jiang C., Pugh B.F.. Nucleosome positioning and gene regulation: advances through genomics. Nat. Rev. Genet. 2009; 10:161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jin C., Zang C., Wei G., Cui K., Peng W., Zhao K., Felsenfeld G.. H3.3/H2A.Z double variant-containing nucleosomes mark ‘nucleosome-free regions’ of active promoters and other regulatory regions. Nat. Genet. 2009; 41:941–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Albert I., Mavrich T.N., Tomsho L.P., Qi J., Zanton S.J., Schuster S.C., Pugh B.F.. Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature. 2007; 446:572–576. [DOI] [PubMed] [Google Scholar]
- 7. Yen K., Vinayachandran V., Pugh B.F.. SWR-C and INO80 chromatin remodelers recognize Nucleosome-free regions near +1 nucleosomes. Cell. 2013; 154:1246–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zentner G.E., Henikoff S.. Regulation of nucleosome dynamics by histone modifications. Nat. Struct. Mol. Biol. 2013; 20:259–266. [DOI] [PubMed] [Google Scholar]
- 9. Tessarz P., Kouzarides T.. Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 2014; 15:703–708. [DOI] [PubMed] [Google Scholar]
- 10. Li G., Levitus M., Bustamante C., Widom J.. Rapid spontaneous accessibility of nucleosomal DNA. Nat. Struct. Mol. Biol. 2005; 12:46–53. [DOI] [PubMed] [Google Scholar]
- 11. Li G., Widom J.. Nucleosomes facilitate their own invasion. Nat. Struct. Mol. Biol. 2004; 11:763–769. [DOI] [PubMed] [Google Scholar]
- 12. Cunningham L., Catlin B.W., de Garilhe M.P.. A deoxyribonuclease of micrococcus pyogenes1. J. Am. Chem. Soc. 1956; 78:4642–4645. [Google Scholar]
- 13. Cotton F.A., Hazen E.E., Legg M.J.. Staphylococcal nuclease: Proposed mechanism of action based on structure of enzyme—thymidine 3′,5′-bisphosphate—calcium ion complex at 1.5-Å resolution. Proc. Natl. Acad. Sci. U.S.A. 1979; 76:2551–2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hörz W., Altenburger W.. Sequence specific cleavage of DNA by micrococcal nuclease. Nucleic Acids Res. 1981; 9:2643–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sulkowski E., Laskowski M.. Hydrolysis of ribo- and deoxyribo-dinucleotides by micrococcal nuclease. Biochim. Biophys. Acta. 1970; 217:538–540. [DOI] [PubMed] [Google Scholar]
- 16. Mikulski A.J., Sulkowski E., Stasiuk L., Laskowski M.. Susceptibility of dinucleotides bearing either 3′- or 5′-Monophosphate to micrococcal nuclease. J. Biol. Chem. 1969; 244:6559–6565. [PubMed] [Google Scholar]
- 17. Dingwall C., Lomonossoff G.P., Laskey R.A.. High sequence specificity of micrococcal nuclease. Nucleic Acids Res. 1981; 9:2659–2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Axel R. Cleavage of DNA in nuclei and chromatin with staphylococcal nuclease. Biochemistry. 1975; 14:2921–2925. [DOI] [PubMed] [Google Scholar]
- 19. Sulkowski E., Laskowski M.. Degradation of thymus DNA and crab poly d(A-T) by micrococcal nuclease in the presence of actinomycin D. Biochim. Biophys. Acta. 1968; 157:207–209. [DOI] [PubMed] [Google Scholar]
- 20. Zhang L., Gralla J.D.. Micrococcal nuclease as a probe for bound and distorted DNA in lac transcription and repression complexes. Nucleic Acids Res. 1989; 17:5017–5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mieczkowski J., Cook A., Bowman S.K., Mueller B., Alver B.H., Kundu S., Deaton A.M., Urban J.A., Larschan E., Park P.J. et al. . MNase titration reveals differences between nucleosome occupancy and chromatin accessibility. Nat. Commun. 2016; 7:11485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chereji R.V., Kan T.-W., Grudniewska M.K., Romashchenko A.V., Berezikov E., Zhimulev I.F., Guryev V., Morozov A.V., Moshkin Y.M.. Genome-wide profiling of nucleosome sensitivity and chromatin accessibility in Drosophila melanogaster. Nucleic Acids Res. 2016; 44:1036–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nagalakshmi U., Wang Z., Waern K., Shou C., Raha D., Gerstein M., Snyder M.. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008; 320:1344–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brogaard K., Xi L., Wang J.-P., Widom J.. A map of nucleosome positions in yeast at base-pair resolution. Nature. 2012; 486:496–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tanaka Y., Tawaramoto-Sasanuma M., Kawaguchi S., Ohta T., Yoda K., Kurumizaka H., Yokoyama S.. Expression and purification of recombinant human histones. Methods. 2004; 33:3–11. [DOI] [PubMed] [Google Scholar]
- 26. Tachiwana H., Kagawa W., Osakabe A., Kawaguchi K., Shiga T., Hayashi-Takanaka Y., Kimura H., Kurumizaka H.. Structural basis of instability of the nucleosome containing a testis-specific histone variant, human H3T. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:10454–10459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dyer P.N., Edayathumangalam R.S., White C.L., Bao Y., Chakravarthy S., Muthurajan U.M., Luger K.. Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol. 2004; 375:23–44. [DOI] [PubMed] [Google Scholar]
- 28. Gardiner E.J., Hunter C.A., Packer M.J., Palmer D.S., Willett P.. Sequence-dependent DNA Structure: A database of octamer structural parameters. J. Mol. Biol. 2003; 332:1025–1035. [DOI] [PubMed] [Google Scholar]
- 29. Fujii S., Kono H., Takenaka S., Go N., Sarai A.. Sequence-dependent DNA deformability studied using molecular dynamics simulations. Nucleic Acids Res. 2007; 35:6063–6074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Anderson J.D., Widom J.. Poly(dA-dT) promoter elements increase the equilibrium accessibility of nucleosomal DNA target sites. Mol. Cell. Biol. 2001; 21:3830–3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Richmond T.J., Davey C.A.. The structure of DNA in the nucleosome core. Nature. 2003; 423:145–150. [DOI] [PubMed] [Google Scholar]
- 32. Hall M.A., Shundrovsky A., Bai L., Fulbright R.M., Lis J.T., Wang M.D.. High-resolution dynamic mapping of histone-DNA interactions in a nucleosome. Nat. Struct. Mol. Biol. 2009; 16:124–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xue Y., Wong J., Moreno G.T., Young M.K., Côté J., Wang W.. NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol. Cell. 1998; 2:851–861. [DOI] [PubMed] [Google Scholar]
- 34. Kingston R.E., Narlikar G.J.. ATP-dependent remodeling and acetylation as regulators of chromatin fluidity. Genes Dev. 1999; 13:2339–2352. [DOI] [PubMed] [Google Scholar]
- 35. Wysocka J., Swigut T., Xiao H., Milne T.A., Kwon S.Y., Landry J., Kauer M., Tackett A.J., Chait B.T., Badenhorst P. et al. . A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature. 2006; 442:86–90. [DOI] [PubMed] [Google Scholar]
- 36. Bannister A.J., Kouzarides T.. Regulation of chromatin by histone modifications. Cell Res. 2011; 21:381–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Strahl B.D., Allis C.D.. The language of covalent histone modifications. Nature. 2000; 403:41–45. [DOI] [PubMed] [Google Scholar]
- 38. Mattiroli F., Arcy S., Luger K.. The right place at the right time: chaperoning core histone variants. EMBO Rep. 2015; 16:1454–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Adkins M.W., Howar S.R., Tyler J.K.. Chromatin disassembly mediated by the histone chaperone asf1 is essential for transcriptional activation of the yeast PHO5 and PHO8 genes. Mol. Cell. 2004; 14:657–666. [DOI] [PubMed] [Google Scholar]
- 40. Loppin B., Bonnefoy E., Anselme C., Laurencon A., Karr T.L., Couble P.. The histone H3.3 chaperone HIRA is essential for chromatin assembly in the male pronucleus. Nature. 2005; 437:1386–1390. [DOI] [PubMed] [Google Scholar]
- 41. Thåström A., Lowary P.T., Widlund H.R., Cao H., Kubista M., Widom J.. Sequence motifs and free energies of selected natural and non-natural nucleosome positioning DNA sequences. J. Mol. Biol. 1999; 288:213–229. [DOI] [PubMed] [Google Scholar]
- 42. Behe M.J. An overabundance of long oligopurine tracts occurs in the genome of simple and complex eukaryotes. Nucleic Acids Res. 1995; 23:689–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shimizu M., Mori T., Sakurai T., Shindo H.. Destabilization of nucleosomes by an unusual DNA conformation adopted by poly(dA)⋅poly(dT) tracts in vivo. EMBO J. 2000; 19:3358–3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Iyer V., Struhl K.. Poly(dA:dT), a ubiquitous promoter element that stimulates transcription via its intrinsic DNA structure. EMBO J. 1995; 14:2570–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ishii H., Kadonaga J.T., Ren B.. MPE-seq, a new method for the genome-wide analysis of chromatin structure. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:E3457–E3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Allan J., Fraser R.M., Owen-Hughes T., Keszenman-Pereyra D.. Micrococcal nuclease does not substantially bias nucleosome mapping. J. Mol. Biol. 2012; 417:152–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chereji R.V., Ocampo J., Clark D.J.. MNase-sensitive complexes in yeast: nucleosomes and non-histone barriers. Mol. Cell. 2017; 65:565–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kubik S., Bruzzone M.J., Albert B., Shore D.. A reply to ‘MNase-Sensitive complexes in yeast: nucleosomes and non-histone barriers,’ by Chereji et al. Mol. Cell. 2017; 65:578–580. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
MNase-seq data generated in this study have been deposited to European Nucleotide Archive under the accession numbers: PRJEB25382, PRJEB25383 and PRJEB25388.