


Abstract
Intersegmental transfer (IST) is an important strategy in the target search used by sequence-specific DNA-binding proteins (DBPs), enabling DBPs to search for targets between multiple DNA strands without dissociation. We examined the IST of the tumor suppressor p53 using ensemble stopped-flow and single-molecule fluorescence measurements. The ensemble measurements demonstrated that p53 exhibits very fast IST, whose rate constant was ∼108 M−1 s−1. To determine the domains of p53 responsible for IST, two mutants with deletions of one of its two DNA binding domains were generated. The mutant lacking the disordered C-terminal (CT) domain (the CoreTet mutant) abolished IST, whereas the mutant lacking the structured core domain (the TetCT mutant) maintained IST, clearly demonstrating the importance of the CT domain. Single-molecule fluorescence measurements further demonstrated the transfer of p53 between two tethered DNA strands. The pseudo-wild-type p53 and the TetCT mutant showed significant transfer efficiencies, whereas the transfer efficiency for the CoreTet mutant was zero. These results suggest that ultrafast IST might be promoted by four copies of the CT domain, by binding to two DNA strands simultaneously. Such ultrafast IST might be important to avoid nearby-bound DBPs during the target search process of p53 in nucleus.
INTRODUCTION
Intersegmental transfer (IST) is an important strategy for facilitated target search by sequence-specific DNA-binding proteins (DBPs) in crowded environments. Sequence-specific DBPs participate in wide varieties of cellular processes by searching and binding short target DNA sequences interspersed in the vast genomic DNA (1–4). To search for the target sequence efficiently, DBPs exploit the one dimensionality of DNA and utilize both one-dimensional (1D) sliding along DNA and three-dimensional (3D) diffusion between separated DNA strands. A simple calculation based on the two-component system composed of DBP and DNA demonstrated that use of both strategies at the optimum ratio would facilitate the target search process significantly (5). However, the actual cellular environment around DBPs and DNA is far from ideal for the simple two-component system. The target search of DBPs in cells needs to be efficient in the presence of crowding macromolecules as well as obstacles bound to DNA. IST is the transfer of DBPs from one site of DNA to a distal site through the formation of a three-body complex composed of the DBP and two sites of DNA (Figure 1A). If the three-body complex is a transient and low-populated state, the IST can be described as the second-order process and depends linearly on the concentration of the second DNA (6) (Figure 1B). IST is expected to be an important strategy for DBPs to avoid obstacles in 1D sliding without utilizing 3D diffusion in cells. However, the precise molecular mechanism of IST based on the structural properties of DBPs is unclear.
Figure 1.
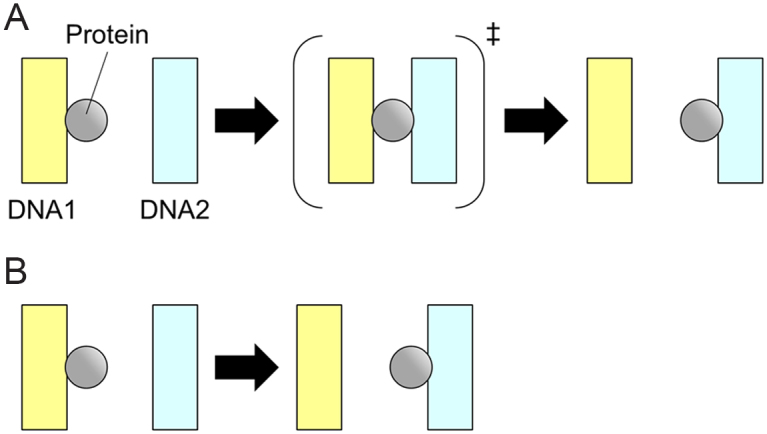
A schematic drawing of the elementary steps of IST reactions. A DBP transfers from DNA1 to DNA2 through the formation of the three-body complex with two DNA strands in panel (A). If the three-body complex is a transient and low-populated state, the reaction can be described as the second-order process in panel (B). Gray circles and rectangles represent proteins and DNA strands, respectively.
We and other researchers have studied the facilitated diffusion of p53, an essential tumor suppressor that prevents tumorigenesis in eukaryotic cells (4). p53 binds to its target sequence and regulates the expression of downstream genes involved in DNA repair, cell cycle arrest or apoptosis. p53 forms a homotetramer whose monomeric unit consists of the N-terminal (NT) (residues 1–93), core (residues 94–292), linker (residues 293–324), tetramerization (Tet) (residues 325–355) and C-terminal (CT) (residues 356–393) domains (7). The core domain is structured and is used for the specific recognition of the target sequence. The CT domain is intrinsically disordered with five net positive charges and is used for non-specific binding to DNA. The 1D sliding of p53 along DNA has been characterized by single-molecule fluorescence observations (8–14) and by molecular dynamics (MD) simulations (15–17). Two sliding modes were identified: the fast mode in which the CT domain mainly interacts with DNA and the slow mode in which the core and CT domains interact with DNA (13). The core domain in the slow mode is recruited to DNA by the disordered linker (14). Moreover, the sliding p53 passes over the target sequence frequently, leading to the proposal that target recognition probability regulates the target binding of p53 (12). The kinetic association of p53 to DNA shows an extremely fast rate constant, ∼ 109 M−1 s−1, for short 30 bp DNA (12), which may be achieved by strong electrostatic interaction between p53 and DNA (18). Accordingly, various aspects of facilitated diffusion in p53 have been investigated comprehensively except for IST.
However, several lines of evidence strongly suggest that IST of p53 may be important for facilitated diffusion in the crowded environment. First, the unique tetrameric structure of p53, possessing eight DNA-binding domains, might enable the protein to bind to two DNA strands simultaneously. In particular, the disordered CT domain, possessing a strong affinity toward non-specific DNA, might also be used for IST, as suggested for the disordered N-terminal domain of homeodomain proteins by Levy et al. (19–21). Second, the coarse-grained MD simulations predicted IST for p53 (15,22). Khazanov and Levy demonstrated that p53 can bind to two DNA strands simultaneously with the disordered CT domain (15). Takada et al. further confirmed that IST of p53 occurs predominantly by attaching the CT domain to DNA and sub-dominantly by attaching the core domain (22). Third, p53 is known to cause DNA looping by binding to more than two sites of DNA, suggesting the possibility of IST of p53 (23).
Here, we examined the IST of p53 using ensemble stopped-flow and single-molecule fluorescence observations. The kinetic ensemble measurements revealed that the IST of p53 occurs efficiently with a rate constant of ∼108 M−1 s−1. Moreover, to elucidate the role of the CT domain on the IST, we constructed the TetCT and CoreTet mutant that maintains and lacks the CT domain, respectively. The ensemble measurements with those two mutants demonstrated that the CT domain but not the core domain is responsible for IST. Single-molecule observation of IST determined the transfer efficiency of p53 on crisscrossed DNA. Our results suggest that the molecular architecture of p53 might be optimized for IST to facilitate target search in the crowded nuclear environment.
MATERIALS AND METHODS
Preparation of p53 mutants
Expression systems for the pseudo-wild-type (WT), inactive (R248Q), CoreTet and TetCT mutants were constructed and the mutants were expressed and purified following our previous methods (11–13). In brief, Escherichia coli (BL21(DE3)pLysS) containing the plasmid for GST-tagged p53 mutant was cultured at 37°C. ZnCl2 and IPTG were added at a final concentration of 100 μM when the culture medium showed an OD600 of ∼0.5. For the TetCT mutant, we did not add ZnCl2 because it does not have the Zn2+-binding site. After induction, E. coli was further cultured at 20°C for 16 h. Escherichia coli was collected and disrupted by sonication. p53 was attached to the GST column (GSTrap FF; GE Healthcare) via the GST tag and eluted after digestion of the GST tag with PreScission protease (GE Healthcare). p53 was further purified using a heparin column (HiTrap Heparin HP; GE Healthcare).
For single-molecule measurements, the pseudo-WT p53, CoreTet mutant, and TetCT mutant were labeled with ATTO532 (ATTO-TEC, Siegen, Germany) using the cysteine-maleimide reaction. The free dye was removed using a cation exchange column (HiTrap SP HP; GE Healthcare). The labeling efficiencies for the pseudo-WT and CoreTet mutants were estimated by using optical absorbance at 280 nm and at 532 nm, and were 1.1 and 1.5 dye per monomer, respectively. The labeling efficiency of the TetCT mutant containing no tryptophan was estimated to be 0.97 dye per monomer by using Bradford assay and optical absorbance at 532 nm.
DNA used for the stopped-flow measurements
DNA fragments were purchased from Sigma-Aldrich and used without further purification. The sequences of the target DNA are 5′-6-FAM-ATCAGGAACATGTCCCAACATGTTGAGCTC-3′ and 3′-TAGTCCTTGTACAGGGTTGTACAACTCGAG-5′, which are derived from the p21 5′ promoter site. The consensus sequences are underlined. The sequence of the non-target DNA was 5′-6-FAM-AATATGGTTTGAATAAAGAGTAAAGATTTG-3′ and 3′-TTATACCAAACTTATTTCTCATTTCTAAAC-5′. The non-labeled target DNA was used as the competitor DNA.
Stopped-flow measurements
To examine the IST of p53, we used a stopped-flow apparatus (Unisoku, Osaka, Japan) equipped with a home-made detection unit of fluorescence polarization as explained previously (11,12). Briefly, a vertically-polarized light from a 473-nm laser (Laser Quantum, Cheshire, UK) was irradiated to the flow cell. A polarizer (Edmund Optics, Japan) and a long pass filter (FF01-520/35; Sermock, New York, USA) were set in front of the photo detector (H10722; Hamamatsu Photonics, Hamamatsu, Japan). The mixing dead time of the mixer was about 1 ms. The time resolution of fluorescence detection was 1 ms. We calculated the anisotropy value using the following equation:
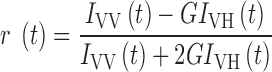 |
where r(t), IVH(t), IVV(t) and G represent time course of anisotropy, fluorescence intensity excited at vertical polarized light and detected as horizontal polarized light, fluorescence intensity excited at vertical polarized light and detected as vertical polarized light, and the G factor that was set to 1, respectively. To obtain the kinetic changes of r(t), four kinetic traces for each of IVH(t) and IVV(t) were obtained independently and cumulated. For IST experiments, 40 nM of 6-FAM labeled DNA and 60 nM per tetramer of p53 mutant were premixed. The complex was mixed with the solution containing 100−400 nM of non-labeled p21 target DNA at a mixing ratio of 1:1. We used a buffer containing 20 mM HEPES, 0.5 mM EDTA, 0−100 mM KCl, 0−3 mM MgCl2, 0.2 mg/ml BSA, 1 mM DTT and 2 mM Trolox at pH 7.9. All the measurements were performed at 25°C. To prevent the dissociation of p53 tetramers to dimers or monomers, we finished all the measurements within 50 min after the preparation of the p53 solutions from the concentrated stock solution, because the half-life of the dissociation from the tetramer into the two dimers was 50 min (24). The obtained anisotropy decay curves were fitted with a single-exponential function. The apparent dissociation rate constants (kobs) obtained by the fitting were plotted against the concentration of competitor DNA and fitted with a linear function.
Labeling of λDNA with biotin or digoxigenin for single-molecule experiments
We constructed two types of λDNA. One was λDNA which possessed two biotins at both ends and the other possessed one biotin and one digoxigenin at each end. To label λDNA, we followed a reported method with some modifications (25). λDNA (New England Biolabs, Massachusetts, USA) was mixed with an oligo DNA (5′-pAGGTCGCCGCCC-biotin-3′; oligo-1) at a molar ratio of 1:10 (λDNA:oligo-1). The mixture was heated at 65°C for 15 min and cooled to room temperature for annealing of the DNA. Next, T4 ligase (New England Biolabs) was added to the solution and incubated for 30 min at room temperature. After ligation, an oligo DNA (5′-pGGGCGGCGACCT-biotin-3′; oligo-2) was added at a molar ratio of 1:50 (λDNA:oligo-2). The mixture was heated at 65°C for 15 min and cooled to room temperature for annealing of the DNA. In this step, we added an oligo DNA labeled with digoxigenin instead of biotin for dig-labeled λDNA. Again, T4 ligase was added to the solution and incubated for 30 min at room temperature. The solution was then heated at 65°C for 10 min to inactivate the T4 ligase. The excess oligos were removed by using MicroSpin S-400 HR Columns (GE Healthcare). The purified λDNA was stored at 4°C.
Polydimethylsiloxane (PDMS) stamp
For single-molecule observations, we designed a PDMS stamp possessing lattice-shaped patterns. The custom-made PDMS stamp was purchased from Fluidware Technologies (Saitama, Japan). The patterns of the stamp are depicted in Supplementary Figure S1A and B. The line widths of the lattices were 2 and 3 μm. The distances between two parallel lines of the lattices were 10, 11, 12 and 13 μm. In total, eight types of lattices were designed in one stamp. The stamped area of the lattices was a square whose size was 0.5 mm by 0.5 mm. The number of lattices in one preparation of the flow cell was ∼10 000 in total. The height of the lattice patterns in the stamp was 1 μm.
Modification of the DNA garden method for preparation of crisscross DNA strands
To construct crisscrossed DNA strands, we first stamped neutravidin onto a washed coverslip coated with the MPC polymer following the protocol developed for the DNA garden method (23). A coverslip (Matsunami Glass Ind. Ltd.) was immersed in the solution containing 10% NH3 and 10% H2O2 for a 20 min wash. After the wash, 100 μl of 0.5% MPC polymer (Lipidure-CM5206; NOF Corp., Tokyo, Japan) in 99.5% ethanol was dropped onto the coverslip and dried to coat the surface with the polymer to prevent the adsorption of DNA and p53. Next, 20 μl solution containing 1 mg/ml neutravidin and 10 mM phosphate at pH 7.4 was dropped onto the PDMS stamp and incubated for 45 min. The stamp was washed thrice with 1 ml of ultrapure water. To transfer neutravidin from the stamp to the coverslip, the coated coverslip was set on the stamp on which 300 g of weight was applied for 5 min. After the stamping, the flow cell with a cross-shaped flow path was constructed using the stamped coverslip, a slide glass with four holes, and a double-sided tape whose thickness was 100 μm. The cross-shaped flow path having a width of 4 mm and a length of 1.4 or 3 cm was made by cutting the double-sided tape. The stamped area was placed at the center of the cross-shaped flow path. A solution of 5% polyvinylpyrrolidone K 15 (Tokyo Chemical Industry, Tokyo, Japan) in 10 mM phosphate buffer at pH 7.4 was introduced into the flow cell and incubated for 20 min. Furthermore, 1 mg/mL of bovine serum albumin (Sigma-Aldrich Japan) (BSA) in 10 mM phosphate buffer at pH 7.4 was introduced and incubated for 20 min.
λDNA was attached to the stamped neutravidin at one end by introducing 150–500 pM of double-biotin λDNA or biotin-λDNA-digoxigenin in 10 mM phosphate buffer at pH 7.4 into one flow path at a flow rate of 40 μl/min and by incubating for 30 min. To attach both ends of λDNA to two neutravidins stamped at separate positions, we used two methods described below. In case of the double-biotin λDNA, we introduced a buffer containing 20 mM HEPES at pH 7.9, 0.5 mM EDTA, 25 mM KCl, 0−2 mM MgCl2, 0.2 mg/ml BSA, 1 mM DTT and 2 mM Trolox, in one flow path at a rate of 200 μl/min for 5 min, followed by the flow of the same solution in the other flow path at the same rate for 5 min. In case of biotin–λDNA–digoxigenin, we introduced a buffer containing 20 mM HEPES at pH 7.9, 0.5 mM EDTA, 25 mM KCl, 0–2 mM MgCl2, 0.2 mg/ml BSA, 1 mM DTT, 2 mM Trolox and 1000-fold diluted anti-digoxin-biotin (Sigma-Aldrich Japan) in one flow path at a flow rate of 500 μl/min for 4 min, followed by the flow of the same solution in the other flow path at the same rate for 2 min.
Single-molecule observation of IST
To observe IST at the single-molecule level, we used the home-built single-molecule fluorescence microscope described previously with some modifications (11–14,23). In brief, 532-nm laser light (Compact-TS-20G-532; OTPO-LINE, Tokyo, Japan or CL-532-050-L; CrystaLaser, Nevada, USA) reflected with a dichroic mirror (Di01-R405/488/532/635-25 × 36; Semrock, NY, USA) was collimated on the back focal plane of an objective lens with N.A. = 1.4 (Plan Apo VC 100×; Nikon, Tokyo, Japan) to illuminate the molecules sliding on tethered DNA selectively based on the principle of total internal reflection fluorescence (TIRF) microscopy. The flow cell with the cross-shaped flow path was placed above the objective lens and fluorescence was detected with a TDI-EM CCD camera (Hamamatsu Photonics; Hamamatsu, Japan) after passing through an optical filter (BLP01-532R-24; Semrock). For the single-molecule experiments, we introduced 250−500 pM per tetramer of ATTO532-p53 in a buffer containing 20 mM HEPES, 0.5 mM EDTA, 25 mM KCl, 0 or 2 mM MgCl2, 0.2 mg/ml BSA, 1 mM DTT and 2 mM Trolox at pH 7.9 into the flow cell. All the measurements were performed at room temperature, 22°C. Sequential images of p53 were taken at 33 ms intervals under a flow at a rate of 500 μl/min. To prevent the dissociation of p53 tetramers to dimers or monomers, we finished all the observations within 50 min after the preparation of the p53 solutions from the concentrated stock solution (24).
For the analysis, movement of the p53 molecule was tracked using ImageJ and its plugin, Particle Track and Analysis, developed by Dr Yoshiyuki Arai (Osaka University, Japan). The position of the p53 molecule was determined by fitting with the 2D Gaussian function. To analyze the IST of p53, the method developed by Greene's group was referred (26). First, to determine the position of DNA strands and the intersection, we tracked p53 molecules in a series of frames and plotted the tracked coordinates of all molecules in one image (Supplementary Figure S2A). The plots, corresponding to the tethered DNA strands, were fitted with a linear function when there was no flow. If the horizontally tethered DNA strands were curved by a flow pressure in a vertical direction, the plots were fitted with a quadratic function. The coordinates of the intersections were calculated from the fitted equations of the two crisscrossed DNA strands. We defined the intersection as a circle with a radius of 4 pixels (320 nm) considering the fluctuation of DNA. Two crisscrossed DNA strands were denoted as DNA1 and DNA2, and divided into five areas, the intersection, one side of DNA1, the other side of DNA1 across the intersection, one side of DNA2 and the other side of DNA2 across the intersection. The movement of p53 after entering the intersection was classified into four cases. First, when p53 entered the intersection from one side of DNA1 by the 1D sliding, resided in the intersection for a while, and went out of the intersection to either side of DNA2, the movement was categorized as ‘transfer’, that is, IST occurred. Second, when p53 entered the intersection from one side of DNA1 and went out of the intersection to the opposite side of DNA1, the movement was categorized as a ‘pass’, that is, p53 passed over the intersection without IST. Third, when p53 entered the intersection from one side but returned to the same side, the movement was categorized as ‘return’. Fourth, when p53 disappeared after entering the intersection, the movement was categorized as ‘dissociation’ of p53 from DNA or photobleaching/photoblinking of the fluorescent dye, ATTO532. The number of each of the four events was counted and the transfer efficiency, which is the ratio of the number of the transfer events to that of the whole events, was estimated.
RESULTS
IST of p53 is extremely fast
To examine whether p53 undergoes IST, we conducted ensemble stopped-flow measurements detected by fluorescence anisotropy (11). Since WT p53 aggregates easily, we constructed pseudo-WT p53 that retains the functions of WT p53 (11). A 30 bp fragment of double-stranded DNA was labeled with the fluorescent dye 6-FAM, and was initially complexed with p53. The solution was mixed with various concentrations of target DNA without a fluorescent dye, used as a competitor, in the presence of 50 mM K+ and 2 mM Mg2+ (Figure 2A). The kinetic dissociation of p53 from the 6-FAM labeled DNA can be detected as decay of the fluorescence anisotropy of 6-FAM (Figure 2B). While the absolute anisotropies of some traces were shifted due to the experimental uncertainty caused by independent detections of the fluorescence data at different polarization angles (12), we confirmed the reproducibility of the kinetic anisotropy changes. If IST occurs, the apparent dissociation rate constant (kobs) should increase as the competitor concentration increases. To obtain the kobs values, we fitted the decay curves with a single-exponential function based on the assumption of a pseudo-first-order process (Figure 2B). The rate constant from non-target DNA linearly depended on the competitor concentration, and the rate constant of IST (kIST) was estimated by fitting the plot of the kobs values in the presence of 100–200 nM of the competitor DNA with a linear function (Figure 2C). The kIST value for pseudo-WT p53 corresponding to the slope was (3.6 ± 0.3) × 108 M−1 s−1. The kIST value of p53 from non-target DNA is extremely large. The validity of the above-mentioned analyses was discussed in Supplementary Data and Supplementary Figure S2.
Figure 2.

(A) A schematic drawing of the experimental set up for stopped-flow fluorescence anisotropy measurements. Kinetic dissociation of p53 from the 6-FAM labeled DNA was monitored at different concentrations of competitor DNA by following fluorescence anisotropy. (B) Time course of fluorescence anisotropy of the 6-FAM labeled non-target DNA premixed with p53 after mixing with the competitor DNA. The black, red, blue, and orange lines correspond to the traces obtained in the presence of 50, 100, 150, and 200 nM of the competitor DNA. The curve-fitting was done according to the single-exponential function. (C) The apparent dissociation rate constant of pseudo-WT p53 (kobs) from the target (circle) and the non-target (triangle) 30 bp DNA strands against the competitor DNA concentration. The data in the presence of 200 nM of competitor DNA were adapted from our previous work (12). The data in the presence of 50 nM of competitor DNA were obtained in the condition that did not satisfy the pseudo first order approximation, and were presented for comparison (see discussion in Supplementary Data). (D) The apparent dissociation rate constant of the inactive mutant of p53 (kobs) from the target (circle) and the non-target (triangle) 30 bp DNA strands against the competitor DNA concentration. The lines in panels (C) and (D) show a linear-fitting of the results obtained in the competitor concentrations between 100 and 200 nM, where slope corresponds to kIST. Panels (B)–(D) are the data obtained in the presence of 50 mM K+ and 2 mM Mg2+. The error bars represent fitting errors of the kinetic data.
We observed that IST of p53 occurs only from non-target DNA. First, the apparent dissociation rate constant of pseudo-WT p53 from target DNA did not depend on the competitor concentration (Figure 2c). Second, the inactive mutant (R248Q) of p53 that lost binding specificity for target DNA (12) showed competitor-dependent dissociation both from the target and non-target DNAs, corresponding to a kIST of (2.1 ± 0.5) × 108 M−1 s−1 and (2.0 ± 0.3) × 108 M−1 s−1, respectively (Figure 2D). The difference in IST from the target and non-target sequences might be attributed to the different quaternary conformations of the p53–DNA complexes.
Roles of the two DNA-binding domains in IST
To clarify the roles of the two DNA-binding domains in IST, we prepared two mutants of p53, CoreTet and TetCT, that lack the disordered CT domain and the structured core domain, respectively (13), and determined their IST rate constants from non-target DNA in the presence of 50 mM K+ and 1 mM Mg2+ (Figure 3A). The apparent dissociation rate constant for TetCT increased linearly with the increase in competitor concentration, corresponding to kIST ((4.7 ± 0.8) × 108 M−1 s−1), close to that of pseudo-WT ((2.5 ± 0.2) × 108 M−1 s−1) (Figure 3B, red and black). In contrast, the apparent dissociation rate constant for CoreTet was not dependent on competitor concentration (Figure 3B, blue). Thus, the CT domain is critically important for IST, which might be consistent with suggestions from theoretical investigations (22). In contrast, the current results clearly demonstrate that the core domain is not directly involved in IST.
Figure 3.

Ensemble kinetic observation of IST for the p53 mutants. (A) Domain organization of the p53 mutants. The pseudo-WT p53 is composed of the NT, core, linker, Tet and CT domains. Thick and thin rectangles represent the structured domain and disordered domain, respectively. (B) The apparent dissociation rate constants (kobs) of the pseudo-WT (black), TetCT (red), and CoreTet (blue) mutants of p53 from the non-target 30 bp DNA in the presence of 50 mM K+ and 1 mM Mg2+ plotted against the competitor DNA concentration. The error bars represent fitting errors of kinetic anisotropy changes using an exponential function. The data in the presence of 50 nM of competitor DNA were obtained in the condition that did not satisfy the pseudo first order approximation, and were presented for comparison. The lines show a linear-fitting of the results obtained in the competitor concentrations between 100 and 200 nM, whose slope corresponds to kIST.
To further understand the roles of the CT domain, we next examined how salt concentrations affect the kinetics of IST. If the CT domain is used to bind to two DNA segments simultaneously in IST by electrostatic interaction between the positive charges of the CT domain and the negative charges of DNA, kIST might be modulated by ionic strength. Accordingly, we estimated the rate constant of IST at different concentrations of K+, which screens the long-range electrostatic interactions, and at different concentrations of Mg2+, which binds to DNA and neutralizes the negative charges. In the presence of 2 mM Mg2+ (Figure 4A), kIST increased by about six fold with the increase in K+ from 0 to 100 mM (Figure 4B). In addition, in the presence of 50 mM K+ (Figure 4C), kIST also increased by about nine fold with the increase in Mg2+ from 0 to 3 mM (Figure 4D). The estimated kIST values are summarized in Supplementary Table S1. The results demonstrate the strong dependency of IST on electrostatic interactions, and are consistent with the involvement of the positively charged CT domain in IST.
Figure 4.

Salt concentration dependency of the IST rate constant for pseudo-WT p53. (A) The apparent dissociation rate constant of pseudo-WT p53 (kobs) from the non-target 30 bp DNA against the competitor DNA concentration in the presence of 0 (red), 50 (black), and 100 mM (blue) of K+ and 2 mM Mg2+. (B) kIST values obtained in the presence of 2 mM Mg2+ at different concentrations of K+. (C) The dissociation rate constant of pseudo-WT p53 (kobs) from the non-target 30 bp DNA against the competitor DNA concentration in the presence of 50 mM of K+ and 0 (red), 1 (orange), 2 (black), and 3 (blue) mM of Mg2+. (D) kIST values obtained in the presence of 50 mM K+ at different concentrations of Mg2+. The data in the presence of 50 nM of competitor DNA in panels (A) and (C) were presented for comparison. The lines in panels (A) and (C) show linear-fitting of the results obtained in the competitor concentrations between 100 and 200 nM, where slope corresponds to kIST. In panels (A) and (C), the data with 50 mM K+ and 2 mM Mg2+ (black) in the presence of 200 nM of competitor DNA were adapted from our previous work (12). The error bars show fitting errors.
Single-molecule observation of IST
We next characterized IST of p53 by single-molecule fluorescence microscopy. To this end, we prepared DNA strands aligned in crisscrossed patterns by modifying the DNA garden method (23) (Figure 5A), and observed the dynamics of fluorescent p53 on the crisscrossed DNA strands using a TIRF microscope. With this experimental setup, the IST events of fluorescent p53 can be identified as 90-degree changes in the sliding direction.
Figure 5.

Single-molecule observation of IST of p53. (A) Schematic drawing of the crisscrossed DNA garden. (Left) The crisscrossed flow cell. Orange cylinders, blue and gray denote connectors to the sample supply and disposal lines, the crisscrossed flow channel, and the coverslip, respectively. (Right) The enlarged view of the coverslip surface at the center of the flow channel. Yellow circles are neutravidin stamped on the coverslip, black curves are DNA, and black circles are biotin. The dynamics of ATTO532-p53 (red circle) near the intersection were observed by TIRF microscopy. (B) Representative fluorescence image of ATTO532-p53 observed on the crisscrossed DNA. Black dots represent single p53. Red curves are DNA strands obtained by fitting all the p53 coordinates sliding along DNA strands with a linear (vertical) and quadratic (horizontal) function. Scale bar in panel (B): 2 μm. (C)–(E) Representative trajectories of the pseudo-WT p53 near the intersection. Panels (C), (D) and (E) represent the images in which pseudo-WT p53 performed IST, passed over the intersection and dissociated from DNA at the intersection, respectively. Panels (F) and (G) represent the trajectories of the TetCT mutant performing IST and the CoreTet mutant which passed over the intersection, respectively. Black solid curves and rainbow curves in panels (C)–(G) denote DNA and the time course of trajectories in which red is the starting point, 0 s, respectively. Scale bars in panels (C)–(G): 0.5 μm. Panels (B)–(G) are the data obtained in the presence of 25 mM K+ and in the absence of Mg2+. (H) The observed numbers of events for p53 mutants entering the intersection. Errors were calculated by the bootstrap method with 1000 replicates (43).
The crisscrossed DNA strands were prepared as follows. First, neutravidins were stamped on a coverslip in lattice-shaped patterns using a PDMS stamp (Supplementary Figure S1). The crisscrossed flow channels were constructed so that the stamped area could be placed at the center of the channels (Figure 5A). Second, a solution containing λDNA conjugated with biotin at both ends was introduced into the flow channel by applying a flow pressure in the horizontal direction. In some experiments, λDNAs conjugated with biotin at one end and digoxigenin at the other end were tethered to the stamped neutravidin by flowing the anti-digoxin conjugate of biotin simultaneously. In this step, most of the DNA strands attached to the stamped biotins were tethered only by one end and were collapsed in the absence of flow. However, a small number of DNA strands, less than one per lattice, attaches the two ends to biotins across the lattice in the extended conformation. Third, we applied a flow pressure in the vertical direction to extend the single tethered DNA strands and promoted the attachment of other ends across the lattice. We could prepare several crisscrossed DNA strands in one preparation of a flow cell consisting of ∼10 000 lattices. In some cases, we continuously applied a flow pressure to stretch single-tethered DNA strands in the vertical direction in order to increase the number of crisscrossed DNA strands.
To observe IST at the single molecule level, we labeled p53 with a fluorescent dye, ATTO532 (13), and introduced ATTO532-pseudo-WT p53 into the flow cell. To increase the residence time of p53 on DNA (13), we conducted observations with lower concentrations of cations: 25 mM K+ and in the absence of Mg2+. Figure 5B shows a representative image of ATTO532-pseudo-WT p53 on the crisscrossed DNA. All the black spots in the image correspond to p53, which is the sole fluorescent species in this experiment. The lattice-shaped patterns correspond to immobile p53s attached to the collapsed DNA adsorbed on the stamped neutravidins (23). However, some p53 molecules showed sliding dynamics along the tethered DNA strands in the region surrounded by the lattices. Accordingly, the location of the tethered DNA strands could be identified by plotting all coordinates of the fluorescent spots in one measurement in one image (Supplementary Figure S3A). The images of tethered DNA strands were fitted with a linear or quadratic function (lines of Figure 5B and Supplementary Figure S3A). We found that many p53s showed 1D sliding and sometimes entered the intersection of two DNA strands. For the quantitative analysis, the radius of the intersection was set to 4 pixels (320 nm) by considering the DNA fluctuation. Figure 5C shows a typical trajectory where pseudo-WT p53 entered the intersection by 1D sliding from the bottom and moved to the right. The turn of p53 at ∼90° in the intersection implies its transfer from one DNA to the other. Among the 59 events, we identified 10 events of IST for pseudo-WT p53.
Efficiency of IST of p53
We next quantitated the differences among the mutants of p53 by comparing the efficiency of IST. As shown in Figure 5C, we identified pseudo-WT p53 performing IST. In addition, larger numbers of pseudo-WT p53 passed over or returned from the intersection without IST (Figure 5D and E). Furthermore, others disappeared in the intersection, which represents dissociation of p53 from DNA (Figure 5E). Accordingly, the movement of p53 after its entrance into the intersection can be classified into four cases: transfer, pass, return and dissociation. The numbers of the four events were compared in Figure 5H and were listed in Supplementary Table S2. We defined transfer efficiency (TE) as the ratio of the number of transfer events to that of the overall events.
The TE value of pseudo-WT was estimated to be 17 ± 5% (n = 59). The IST of the TetCT mutant was observed similarly (Figure 5F), whose TE was 15 ± 3% (n = 162). We also detected IST in the presence of 25 mM K+ and 2 mM Mg2+ (Supplementary Figure S3B and C), and estimated that the TE values for pseudo-WT and TetCT were 9 ± 4% (n = 44) and 26 ± 4% (n = 120), respectively. In contrast, IST of the CoreTet mutant was not observed, corresponding to a TE of 0% (n = 20) (Figure 5G). Notably, the obtained TE values were minimal estimates because the intersection with 320 nm of the radius used for the analysis is much larger than the actual size of the intersection (2 nm, diameter of DNA helix). Accordingly, some molecules might return to the original direction before reaching the actual intersection as shown in the large fraction of return events (Figure 5H). In addition, the variation of the distance between the two DNA strands, estimated within ∼200 nm, might also cause the uncertainty of the obtained TE values (Supplementary Data). However, the single-molecule results were consistent with those obtained in the ensemble measurements demonstrating the importance of the CT domain for IST.
DISCUSSION
In this investigation, we demonstrate that p53 displays extremely fast IST from non-target DNA to other DNA, with a rate constant of ∼108 M−1 s−1. The ultrafast IST of p53 may be attributed to its unique tetrameric structure having a positively-charged and disordered CT domain. In cells, p53 sliding along DNA would be frequently blocked by other DBPs bound to DNA; however, IST can rescue p53 from trapped sites and resume target search immediately. We will discuss the formation of the ternary complex in IST, the molecular architecture that enables facile IST, and the physiological importance of IST for the target search in nuclei.
We propose that IST of p53 involves the formation of a ternary complex comprising two DNA strands and p53. We detected an increased dissociation rate of p53 from non-specific DNA upon an increase in the competitor concentration; however, Sidorova et al. suggested that a similar observation for EcoRI does not imply the formation of a ternary complex and explained that rebinding of the protein after hopping might be inhibited by competitor DNA (27). We suggest that transfer via hopping is less likely to explain the current results for p53. Tafvizi et al. demonstrated that full-length p53 slides along DNA but that the mutant lacking the CT domain hops along DNA (9). If p53 transfers via hopping, the CoreTet mutant should display a faster dissociation upon increase in the competitor compared to pseudo-WT. The absence of IST for the CoreTet mutant contradicts this explanation, and demonstrates that IST for p53 proceeds through an intermediate formed by binding to two DNA strands simultaneously.
The molecular architecture of p53 possessing four copies of the disordered and positively charged CT domain should enable p53 to bind to two DNA strands simultaneously and facilitate IST. We observed that the pseudo-WT p53 and the TetCT mutant exhibited IST, but the CoreTet mutant did not (Figure 3). The transfer efficiency of the pseudo-WT and the TetCT mutant was ∼ 15%, but that for the CoreTet mutant was zero (Figure 5). These results were consistent with the coarse-grained MD simulations demonstrating IST triggered by positively-charged tails (20,22). The increase of kIST with increased concentrations of K+ and Mg2+ can also be understood by considering the roles of the CT domain. Terakawa et al. revealed that the number of the CT domains contacting DNA during the 1D sliding decreases with the increase in the concentration of monovalent ion in their MD simulation (16). The larger number of unbound CT domains at higher salt concentrations might allow p53 to bind to another DNA more frequently and facilitate IST. The positive charge of the CT domain might also be important to screen the negatively charged DNA to which p53 is bound and to enable collision with the second DNA strand. Accordingly, the positively-charged and disordered CT domain is indispensable for fast IST.
The kIST value of p53 determined in this investigation is one of the fastest among the reported values of kIST for DBPs (Table 1) (28–33). The structures of DBPs listed in Table 1 are as follows: dimeric folded protein (Fis), a domain composed of three α-helices (Sox2), one structured domain and disordered N-terminal tail (HoxD9), and three folded zinc finger domains connected by linkers (Egr-1). The rate constant kIST-non-target for Egr-1 was estimated for the single binding site spanning 9 bp (microscopic rate constant). We can convert the value to the IST rate for the 30-bp DNA by multiplying 22 (macroscopic rate constant), since the 30 bp DNA possesses 22 possible binding sites for Egr-1 (6). The macroscopic kIST-non-target value of Egr-1 becomes comparable to that for p53. Interestingly, all the DBPs in the list possess multiple binding sites to DNA, likely enabling them to capture two DNA strands simultaneously. Schmidt et al. reported that the ratio of DBPs forming oligomers is higher than that of total proteins (3). In addition, some of the DBPs in the list possess the disordered region, which might also be important for IST (19–21). We suggest that the architecture of p53, possessing multiple copies of the disordered binding sites, is optimized for efficient IST and distinguishes p53 among the DBPs.
Table 1.
k IST values of a variety of DBPs
| Protein | Fisa | Sox2b | HoxD9b | Egr-1c | Pseudo-WT p53b | TetCT mutant p53b |
|---|---|---|---|---|---|---|
| k IST-target / M−1 s−1 | N. D. | No IST | ∼6 × 104 | 0.84 ± 0.16 | No IST | N. D. |
| k IST-non-target / M−1 s−1 | ∼1 × 104 | (1.3 ± 0.2) × 106 | ∼1.4 × 106 | 105–107 | ∼108 | ∼108 |
| Oligomerization state | 2 | 1 | 1 | 1 | 4 | 4 |
| # of basic tails | 0 | 0 | 1 | 0 | 4 | 4 |
| References | (33) | (32) | (30,31) | (28,29) | This study | This study |
aThe kIST value of Fis was estimated by examining the competitor-dependent dissociation of Fis from a long DNA strand. The observed kIST value was converted from the weight concentration unit [(ng/μl)−1 s−1] into the unit of the Fis binding-site concentration (M−1 s−1), assuming the length of the Fis binding site of 21 bp.
bThe kIST values for Sox2, HoxD9 and p53s were obtained based on the competitor dependent dissociation experiments and were presented in the unit of the concentration of the DNA strand used. The lengths of the competitor DNAs used were 29, 24 and 30 bp for Sox2, HoxD9 and p53, respectively.
cThe kIST value of Egr-1 for the non-target DNA was obtained by mixing the solution of Egr-1 with that containing the FAM-labeled target DNA and the competitor DNA. The time courses of the association of Egr-1 to the target site were fitted with the equations assuming the IST, giving the microscopic kIST value based on the Egr-1 binding-site concentration, whose length was assumed as 9 bp.
N.D.: not determined.
The ultrafast kIST is physiologically significant in the target search of p53 in nuclei. The concentration of DNA in nuclei is ∼150 mM by base pairs (31) and ∼5 mM in the unit of 30 bp. The accessible DNA in vivo is assumed to be ∼1%, because the other DNA regions are wrapped around histone proteins and form nucleosomes and chromatin structures (34). Thus, the concentration of accessible DNA in nuclei is 50 μM in the unit of 30 bp. The diffusion coefficient of the p53-DNA complex in cells was estimated as 0.014 μm2/s (35), which was ∼3400-fold smaller than that for p53 in solution (48 μm2/s) (9). Since the diffusion coefficient of the complex between p53 and 30 bp-DNA in solution should be comparable to that of free p53 (24), the kIST in nuclei can be estimated as 2.9 × 104 M−1 s−1. Thus, the apparent rate constant of IST in nuclei can be estimated by multiplying 2.9 × 104 M−1 s−1 and 50 μM giving 1.5 s−1. This is 2.5-fold faster than the dissociation rate constant of p53 from nonspecific DNA (0.6 s−1) in nuclei (35), and demonstrates that the IST event should occur at least at a rate comparable to that of the dissociation of p53 from DNA. We suggest that p53 requires ultrafast kIST to conduct IST efficiently even in the crowded nuclear environment.
IST can be categorized as one mechanism of the facilitated dissociation, in which the third biomolecules such as proteins and DNA increase the apparent dissociation rate of DBPs bound to DNA in a concentration-dependent manner (36–42). In contrast to the simple dissociation, in which the dissociation is independent from the concentration of other molecules, the facilitated dissociation including IST should be important in cells crowded with biomolecules. Further investigations on the other mechanisms of the facilitated dissociation for p53 are warranted.
In summary, we demonstrate that IST of p53 occurs extremely fast with a bimolecular rate constant of ∼108 M−1 s−1 using ensemble stopped-flow measurements. In addition, single-molecule fluorescence methods also showed the transfer of p53 between two tethered DNA strands in a crisscrossed pattern. A simple consideration suggested that the IST of p53 should contribute to a facilitated target search in the crowded nuclear environment. The presence of four copies of the positively charged and disordered C-terminal domain enables p53 to conduct ultrafast IST.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Saori Kanbayashi for construction of the p53 mutants.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Ministry of Education, Culture, Sports, Science and Technology/Japan Society for the Promotion of Science [KAKENHI 16K07313 and 16KK0157 to K.K. and JP25104007 to S.T.]. Funding for open access charge: Ministry of Education, Culture, Sports, Science and Technology/Japan Society for the Promotion of Science [KAKENHI 16K07313].
Conflict of interest statement. None declared.
REFERENCES
- 1. Halford S.E., Marko J.F.. How do site-specific DNA-binding proteins find their targets. Nucleic Acids Res. 2004; 32:3040–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sheinman M., Kafri Y.. The effects of intersegmental transfers on target location by proteins. Phys. Biol. 2009; 6:016003. [DOI] [PubMed] [Google Scholar]
- 3. Schmidt H.G., Sewitz S., Andrews S.S., Lipkow K.. An integrated model of transcription factor diffusion shows the importance of intersegmental transfer and quaternary protein structure for target site finding. PLoS One. 2014; 9:e108575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kamagata K., Murata A., Itoh Y., Takahashi S.. Characterization of facilitated diffusion of tumor suppressor p53 along DNA using single-molecule fluorescence imaging. J. Photochem. Photobiol. C Photochem. Rev. 2017; 30:36–50. [Google Scholar]
- 5. Slutsky M., Mirny L.A.. Kinetics of protein-DNA interaction: facilitated target location in sequence-dependent potential. Biophys. J. 2004; 87:4021–4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Esadze A., Iwahara J.. Stopped-flow fluorescence kinetic study of protein sliding and intersegment transfer in the target DNA search process. J. Mol. Biol. 2014; 426:230–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Joerger A.C., Fersht A.R.. The tumor suppressor p53: from structures to drug discovery. Cold Spring Harb. Perspect. Biol. 2010; 2:a000919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tafvizi A., Huang F., Leith J.S., Fersht A.R., Mirny L.A., van Oijen A.M.. Tumor suppressor p53 slides on DNA with low friction and high stability. Biophys. J. 2008; 95:L01–L03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tafvizi A., Huang F., Fersht A.R., Mirny L.A., van Oijen A.M.. A single-molecule characterization of p53 search on DNA. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:563–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Leith J.S., Tafvizi A., Huang F., Uspal W.E., Doyle P.S., Fersht A.R., Mirny L.A., van Oijen A.M.. Sequence-dependent sliding kinetics of p53. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:16552–16557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Murata A., Ito Y., Kashima R., Kanbayashi S., Nanatani K., Igarashi C., Okumura M., Inaba K., Tokino T., Takahashi S. et al. . One-dimensional sliding of p53 along DNA is accelerated in the presence of Ca(2+) or Mg(2+) at millimolar concentrations. J. Mol. Biol. 2015; 427:2663–2678. [DOI] [PubMed] [Google Scholar]
- 12. Itoh Y., Murata A., Sakamoto S., Nanatani K., Wada T., Takahashi S., Kamagata K.. Activation of p53 facilitates the target search in DNA by enhancing the target recognition probability. J. Mol. Biol. 2016; 428:2916–2930. [DOI] [PubMed] [Google Scholar]
- 13. Murata A., Itoh Y., Mano E., Kanbayashi S., Igarashi C., Takahashi H., Takahashi S., Kamagata K.. One-dimensional search dynamics of tumor suppressor p53 regulated by a disordered C-terminal domain. Biophys. J. 2017; 112:2301–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Subekti D.R.G., Murata A., Itoh Y., Fukuchi S., Takahashi H., Kanbayashi S., Takahashi S., Kamagata K.. The disordered linker in p53 participates in nonspecific binding to and one-dimensional sliding along DNA revealed by single-molecule fluorescence measurements. Biochemistry. 2017; 56:4134–4144. [DOI] [PubMed] [Google Scholar]
- 15. Khazanov N., Levy Y.. Sliding of p53 along DNA can be modulated by its oligomeric state and by cross-talks between its constituent domains. J. Mol. Biol. 2011; 408:335–355. [DOI] [PubMed] [Google Scholar]
- 16. Terakawa T., Kenzaki H., Takada S.. p53 searches on DNA by rotation-uncoupled sliding at C-terminal tails and restricted hopping of core domains. J. Am. Chem. Soc. 2012; 134:14555–14562. [DOI] [PubMed] [Google Scholar]
- 17. Terakawa T., Takada S.. p53 dynamics upon response element recognition explored by molecular simulations. Sci. Rep. 2015; 5:17107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Halford S.E. An end to 40 years of mistakes in DNA-protein association kinetics. Biochem. Soc. Trans. 2009; 37:343–348. [DOI] [PubMed] [Google Scholar]
- 19. Vuzman D., Levy Y.. DNA search efficiency is modulated by charge composition and distribution in the intrinsically disordered tail. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:21004–21009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vuzman D., Azia A., Levy Y.. Searching DNA via a “Monkey Bar” mechanism: the significance of disordered tails. J. Mol. Biol. 2010; 396:674–684. [DOI] [PubMed] [Google Scholar]
- 21. Vuzman D., Levy Y.. Intrinsically disordered regions as affinity tuners in protein-DNA interactions. Mol. Biosyst. 2012; 8:47–57. [DOI] [PubMed] [Google Scholar]
- 22. Takada S., Kanada R., Tan C., Terakawa T., Li W., Kenzaki H.. Modeling structural dynamics of biomolecular complexes by coarse-grained molecular simulations. Acc. Chem. Res. 2015; 48:3026–3035. [DOI] [PubMed] [Google Scholar]
- 23. Igarashi C., Murata A., Itoh Y., Subekti D.R.G., Takahashi S., Kamagata K.. DNA garden: a simple method for producing arrays of stretchable DNA for single-molecule fluorescence imaging of DNA binding proteins. Bull. Chem. Soc. Jpn. 2017; 90:34–43. [Google Scholar]
- 24. Rajagopalan S., Huang F., Fersht A.R.. Single-Molecule characterization of oligomerization kinetics and equilibria of the tumor suppressor p53. Nucleic Acids Res. 2011; 39:2294–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Graneli A., Yeykal C.C., Prasad T.K., Greene E.C.. Organized arrays of individual DNA molecules tethered to supported lipid bilayers. Langmuir. 2006; 22:292–299. [DOI] [PubMed] [Google Scholar]
- 26. Gorman J., Wang F., Redding S., Plys A.J., Fazio T., Wind S., Alani E.E., Greene E.C.. Single-molecule imaging reveals target-search mechanisms during DNA mismatch repair. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:E3074–E3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sidorova N.Y., Scott T., Rau D.C.. DNA concentration-dependent dissociation of EcoRI: direct transfer or reaction during hopping. Biophys. J. 2013; 104:1296–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Takayama Y., Sahu D., Iwahara J.. NMR studies of translocation of the Zif268 protein between its target DNA Sites. Biochemistry. 2010; 49:7998–8005. [DOI] [PubMed] [Google Scholar]
- 29. Esadze A., Kemme C.A., Kolomeisky A.B., Iwahara J.. Positive and negative impacts of nonspecific sites during target location by a sequence-specific DNA-binding protein: origin of the optimal search at physiological ionic strength. Nucleic Acids Res. 2014; 42:7039–7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Iwahara J., Zweckstetter M., Clore G.M.. NMR structural and kinetic characterization of a homeodomain diffusing and hopping on nonspecific DNA. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:15062–15067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Iwahara J., Clore G.M.. Direct observation of enhanced translocation of a homeodomain between DNA cognate sites by NMR exchange spectroscopy. J. Am. Chem. Soc. 2006; 128:404–405. [DOI] [PubMed] [Google Scholar]
- 32. Takayama Y., Clore G.M.. Interplay between minor and major groove-binding transcription factors Sox2 and Oct1 in translocation on DNA studied by paramagnetic and diamagnetic NMR. J. Biol. Chem. 2012; 287:14349–14363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Giuntoli R.D., Linzer N.B., Banigan E.J., Sing C.E., de la Cruz M.O., Graham J.S., Johnson R.C., Marko J.F.. DNA-segment-facilitated Dissociation of Fis and NHP6A from DNA detected via single-molecule mechanical response. J. Mol. Biol. 2015; 427:3123–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hesselberth J.R., Chen X., Zhang Z., Sabo P.J., Sandstrom R., Reynolds A.P., Thurman R.E., Neph S., Kuehn M.S., Noble W.S. et al. . Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat. Methods. 2009; 6:283–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Loffreda A., Jacchetti E., Antunes S., Rainone P., Daniele T., Morisaki T., Bianchi M.E., Tacchetti C., Mazza D.. Live-cell p53 single-molecule binding is modulated by C-terminal acetylation and correlates with transcriptional activity. Nat. Commun. 2017; 8:313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Graham J.S., Johnson R.C., Marko J.F.. Concentration-dependent exchange accelerates turnover of proteins bound to double-stranded DNA. Nucleic Acids Res. 2011; 39:2249–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Loparo J.J., Kulczyk A.W., Richardson C.C., van Oijen A.M.. Simultaneous single-molecule measurements of phage T7 replisome composition and function reveal the mechanism of polymerase exchange. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:3584–3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Joshi C.P., Panda D., Martell D.J., Andoy N.M., Chen T.Y., Gaballa A., Helmann J.D., Chen P.. Direct substitution and assisted dissociation pathways for turning off transcription by a MerR-family metalloregulator. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:15121–15126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gibb B., Ye L.F., Gergoudis S.C., Kwon Y., Niu H., Sung P., Greene E.C.. Concentration-dependent exchange of replication protein A on single-stranded DNA revealed by single-molecule imaging. PLoS One. 2014; 9:e87922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen T.Y., Santiago A.G., Jung W., Krzeminski L., Yang F., Martell D.J., Helmann J.D., Chen P.. Concentration- and chromosome-organization-dependent regulator unbinding from DNA for transcription regulation in living cells. Nat. Commun. 2015; 6:7445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kamar R.I., Banigan E.J., Erbas A., Giuntoli R.D., Olvera de la Cruz M., Johnson R.C., Marko J.F.. Facilitated dissociation of transcription factors from single DNA binding sites. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E3251–E3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lewis J.S., Spenkelink L.M., Jergic S., Wood E.A., Monachino E., Horan N.P., Duderstadt K.E., Cox M.M., Robinson A., Dixon N.E. et al. . Single-molecule visualization of fast polymerase turnover in the bacterial replisome. eLife. 2017; 6:e23932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Efron B., Tibshirani R.J.. An Introduction to the Bootstrap. 1993; Chapman and Hall/CRC. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.