


Abstract
The ubiquitous RarA/Mgs1/WRNIP protein plays a crucial, but poorly understood role in genome maintenance. We show that Bacillus subtilis RarA, in the apo form, preferentially binds single-stranded (ss) over double-stranded (ds) DNA. SsbA bound to ssDNA loads RarA, and for such recruitment the amphipathic C-terminal domain of SsbA is required. RarA is a DNA-dependent ATPase strongly stimulated by ssDNA–dsDNA junctions and SsbA, or by dsDNA ends. RarA, which may interact with PriA, does not stimulate PriA DNA unwinding. In a reconstituted PriA-dependent DNA replication system, RarA inhibited initiation, but not chain elongation. The RarA effect was not observed in the absence of SsbA, or when the host-encoded preprimosome and the DNA helicase are replaced by proteins from the SPP1 phage with similar function. We propose that RarA assembles at blocked forks to maintain genome integrity. Through its interaction with SsbA and with a preprimosomal component, RarA might impede the assembly of the replicative helicase, to prevent that recombination intermediates contribute to pathological DNA replication restart.
INTRODUCTION
The bacterial replication associated recombination protein A, RarA, belongs to a highly-conserved family of AAA+ ATPases, where also the yeast Mgs1 (Maintenance of genome stability 1) and mammal WRNIP1 (Werner [WRN] Interacting Protein 1) proteins are included (1). The Mgs1/WRNIP1/RarA enzymes play a crucial, although poorly characterised role in replication restart (2–6). Interacting partners support their role in DNA replication and recombination. The eukaryotic homologs, Mgs1 and WRNIP1, interact with WRN helicase, Rad18, RAD51, ubiquitinated PCNA sliding clamp and polymerase δ (7–13). Bacterial RarA is also found to be associated with the replisome (2), and to interact with the essential SSB/SsbA protein (6,14).
Lack of Escherichia coli RarA (RarAEco) improves the growth defect of a thermosensitive mutation in the core polymerase subunit α (dnaEtsEco) and also partially suppresses a mutation in the non-essential Ψ subunit (holDEco) of the replicase (4,9,15). In contrast, inactivation of Bacillus subtilis rarA (formerly termed yrvN) affects growth at semipermissive temperature of mutants in the preprimosomal DnaB protein (no counterpart in E. coli) and the replicative DNA helicase DnaC (homolog of DnaBEco) (H. Romero personal communication). Recently two mutually exclusive models of RarAEco action were proposed. In model 1, RarAEco may contribute to the rescue of an inactivated replication fork by creating a flap at broken replication forks, a suitable substrate for the replicative helicase DnaBEco to continue the unwinding of parental dsDNA. This will allow DNA replication to continue without replisome disassembly (16). In model 2, RarAEco could be required at stalled forks for the replacement of the replicative DNA polymerase by a translesion synthesis DNA polymerase as PolIVEco (5,15). Similarly, it has been proposed that eukaryotic Mgs1/WRNIP1 may stimulate detachment of the replicative Polδ from ubiquitylated PCNA and facilitate the recruitment of the translesion synthesis polymerase Polη to sites of DNA damage (1). All this suggests a role for RarA in DNA replication restart. Bacterial RarA also shows 21% sequence identity with discrete segments of the clamp loader subunit DnaX (also termed τ subunit) and the branch migration translocase RuvB (2). The crystal structure of RarAEco resembles the structure of the τ subunit (DnaXEco) of the clamp loader complex (6).
Nothing is known about RarA from other bacterial phyla. A recent estimate of the frequency of stalling of replication forks assembled at oriC suggested that it can be as frequent as five times per generation in B. subtilis cells, as determined by single-molecule microscopy (17). The specific factors that contribute to the rescue of stalled or collapsed replication forks differ among distantly related bacteria, as those from Proteobacteria (e.g. Escherichia coli) from those of the Firmicutes phylum (e.g. B. subtilis) (18–22). Bacillus subtilis cells lack counterparts for the E. coli preprimosomal PriB, PriC and DnaT proteins, and in this bacterium, the loading of the replicative helicase DnaC relies on two discrete mechanisms: DnaA at oriC, in concert with preprimosomal DnaB–DnaD (no counterpart in E. coli) and with helicase loader DnaI (homolog of DnaCEco), or PriA (in concert with DnaB–DnaD–DnaI) at regions outside the replication origin (23–25). In other words, the essential preprimosome proteins DnaD and DnaB and the helicase loader DnaI are utilized once at oriC and in a DnaA-dependent manner, and also at blocked forks in a PriA-dependent manner. The replisomes of the two bacteria also show differences. Bacillus subtilis lacks counterparts for the E. coli DnaQ, HolC, HolD, HolE and the γ subunits of the replicase, and it has two C-type DNA polymerases, PolC and DnaE3 (for simplicity termed DnaE) (21,23–33).
Bacillus subtilis PriA binds displaced-loops (D-loop) or forked DNA and recruits the tetrameric DnaD and DnaB and hexameric DnaI proteins (28–33). Assembly of the preprimosome (PriA–DnaD–DnaB) complex and DnaI on a template created by homologous recombination proteins commits to the assembly of the replicative hexameric DNA helicase DnaC (30,34–37). DnaG primase and DnaE polymerase via interaction with DnaC and the tetrameric single stranded binding protein, SsbA, bind to the pre-initiation complex, forming the complete primosome (14,25,38,39). The association of DnaG with DnaC triggers the release of the preprimosome proteins and activates the helicase, which unwinds DNA progressively in the 5′→3′ direction along the lagging strand template (30,39). Activated DnaC helicase also interacts with the C-terminal domain of DnaX of the DnaX–HolA–HolB (also termed τδδ′) clamp loader complex (40–42). The clamp loader plays a central role in anchoring the PolC replicase and recruiting the sliding clamp (DnaN or β-clamp) at leading and lagging strands. The B. subtilis DnaE polymerase, which lacks a proofreading activity and it has been demonstrated to be error-prone (43,44), synthesises, in concert with DnaG, the RNA-DNA hybrid primers used by PolC on the lagging strand, initiating each Okazaki fragment (23,45). DnaE and DnaG, as the eukaryotic Polα enzyme, also prime leading strand synthesis at a replication origin and when there is a blocked replication fork (25). PolC, which has polymerase and proof reading exonuclease activities, elongates these hybrid RNA–DNA primers both at leading and lagging strands (23,45).
To gain insight into the role of B. subtilis RarA in replication restart we have used biochemical techniques. Our work reveals that RarA binds with 3-fold higher affinity single-stranded (ss) than double-strand (ds) DNA. RarA is a DNA-dependent ATPase strongly stimulated by hairpin structures or DNA ends. SsbA stimulates the ssDNA dependent ATPase activity of RarA. RarA may interact with SsbA and PriA. The interaction of RarA with the amphipathic C-terminal domain of SsbA-bound to ssDNA significantly stimulates the RarA ATPase and its ssDNA binding activities. In the presence of SsbA and the preprimosomal (PriA–DnaD–DnaB in concert with DnaI) proteins, initiation of DNA replication is inhibited by the addition of RarA, but DNA chain elongation is insensitive to RarA action. We propose that an intricate web of molecular interactions affect the fate of damaged DNA at the blocked replication fork; RarA might regulate such decision by preventing the re-assembly of the replicative helicase to circumvent that potentially dangerous recombination might be used for pathologic re-initiation of DNA replication.
MATERIALS AND METHODS
Bacterial strains and plasmids
Escherichia coli BL21(DE3) [pLysS] cells bearing pCB906-borne rarA gene and its variant with a mutation in codon 51 (pCB1056-borne rarAK51A) under the control of a phage T7 promoter, were used to overexpress RarA and its mutant variant RarAK51A in E. coli BL21(DE3) [pLysS] cells. Note that, unless stated otherwise, the indicated genes and products are of B. subtilis origin. The nomenclature used to denote the origin of proteins from other bacteria is based on the bacterial genus and species (e.g. E. coli RarA is referred as RarAEco).
Enzymes, reagents, DNA and proteins
All chemicals used in this study were of analytical grade. Isopropyl β-d-1-thiogalactopyrano-side (IPTG) was from Calbiochem. Disuccinimidyl suberate (DSS), polyethyleneimine, DTT, ATP, dATP and ATPγS were from Sigma. DNA restriction enzymes, T4 polynucleotide kinase and DNA ligase were supplied by New England Biolabs. DEAE, Q- and SP-sepharose were from GE healthcare, hydroxyapatite from BioRad and phosphocellulose was from Whatman. The radioactive nucleotides, [γ-32P]-ATP, [α-32P]-dCTP and [α-32P]-dGTP, were from Perkin Elmer.
All proteins were expressed in E. coli cells, from either pT712-, pET-, or pA1-based vectors (23,45–47). The B. subtilis SsbA, SsbB and the SsbBA chimera (previously termed SsbB*) were purified as described (47). The protocols for purification of B. subtilis replication proteins (DnaE, PolC, DnaG, δ, δ′, PriA, β, τ, DnaD, DnaB and DnaC and DnaI) and for SPP1-encoded replication proteins (G36P, G38P G39P and G40P) were reported (23,45). All protein concentrations were calculated using their estimated extinction coefficient and are expressed taking into account its native state: monomers (DnaE, PolC, DnaG, δ, δ′, PriA, G38P and G39P), dimers (β), trimers (τ), tetramers (SsbA, SsbB, SsbBA, DnaD, DnaB and G36P) and hexamers (DnaC, DnaI and G40P).
The wt RarA and the RarAK51A variant were purified as follows: E. coli BL21 (DE3) (pLysS) cells bearing the pCB906-borne rarA or pCB1056-borne rarAK51A gene were grown at 30°C to mid-exponential phase and the expression of the 46.3-kDa RarA protein was induced by addition of 1 mM IPTG (30 min). Then, rifampicin was added to inhibit bacterial RNA synthesis and the cells were collected 90 min later. The cell mass was resuspended in buffer A (50 mM Tris–HCl, pH 7.5, 1 mM DTT, 15% glycerol) containing 250 mM NaCl and lysed with a French press. After removal of insoluble proteins and cell debris by centrifugation at 18 000 rpm for 45 min, polyethylenimine was added to a final concentration of 0.25% (A260 ∼120) for precipitation of the RarA protein and chromosomal DNA. RarA was solubilised from the polyethylenimine pellet with buffer A containing 400 mM NaCl and 150 mM amonium sulfate, and the pellet containing chromosomal DNA and other proteins was discarded. RarA or RarAK51A was precipitated by addition of solid ammonium sulfate (80% saturation). The ammonium sulfate pellet was resuspended in buffer A, and loaded onto a phosphocellulose column equilibrated with buffer A containing 80 mM NaCl. RarA or RarAK51A was eluted with a linear gradient from 100 to 400 mM NaCl. Fractions containing the protein were recovered and loaded onto a hydroxyapatite column equilibrated with the buffer A containing 400 mM NaCl. The column was washed with buffer A containing 10 mM K+ phosphate and eluted with a 10–200 mM K+ phosphate gradient. A nuclease contaminant co-purified with RarA or RarAK51A protein. The peak fractions containing RarA without nuclease contaminant were pooled, concentrated, and stored in buffer A containing 300 mM NaCl and 50% glycerol. The concentrated proteins were aliquoted, frozen in liquid nitrogen, and stored at −80°C. The concentration of homotetrameric RarA or RarAK51A was calculated using its estimated extinction coefficient and is expressed as moles of protein tetramers.
The synthetic mini-circular DNA template used for in vitro replication assays is a 409-nt circle containing a 396-nt tail described in (23), and prepared as described (48). This synthetic DNA template, which has a strong (50:1) dG + dC strand bias, was designed in such a way that radiolabeled dCTP is incorporated, nearly exclusively, into the leading strand product, and radiolabeled dGTP into the lagging strand product (23). The list of oligonucleotides used to construct the different DNA substrates for electrophoretic mobility shift assays (EMSAs) is shown in Supplementary Table S1. The source of circular ssDNA or dsDNA used in the ATPase assays was derived from 3199-bp pGEM-3zf(+) phagemid. In some ATPase assays an 80-nt long polydT substrate was used. Unless stated, DNA concentrations are expressed as moles of nucleotides.
ATPase assays
The ATPase activity of RarA was assayed using an NAD/NADH-linked assay as previously described (47). Reactions (50 μl), containing the indicated concentration of RarA, DNA effector, 5 mM ATP and the NADH enzyme mix (620 μM NADH, 100 U/ml of lactic dehydrogenase, 500 U/ml pyruvate kinase, and 2.5 mM phosphoenolpyruvate), were performed during 25 min at 37°C and in buffer B (50 mM Tris–HCl pH 7.5, 50 mM NaCl, 10 mM magnesium acetate [MgOAc], 1 mM DTT, 5% glycerol, 0.1 mg/ml BSA). The steady-state ATPase activity was determined by measuring the rate of NADH absorbance decrease at 340 nm due to NAD production, using a Shimadzu CPS-20A dual-beam spectrophotometer. A standard curve with known amounts of NADH was obtained and used to calculate the rate of ADP production from absorbance/time. In the text the protein concentrations are expressed in their stoichiometric ratios with DNA whereas in the Figure legends the molar concentrations of proteins and DNA are presented.
Protein–DNA and protein–protein complexes
For EMSAs, the different DNA substrates were first assembled by annealing different oligonucleotides as represented in Supplementary Table S1, having one of the oligonucleotides radiolabeled with [γ32P]-ATP and T4 polynucleotide kinase. DNA structures (e.g. forked-DNA) were gel-purified as described (49). Typically, radiolabeled DNA (0.4 nM in molecules) was incubated with different amounts of proteins (15 min, 30°C) in buffer B or buffer C (50 mM Tris–HCl pH 7.5, 50 mM NaCl, 1 mM EDTA, 1 mM DTT, 5% glycerol, 0.1 mg/ml BSA) in a 20 μl final volume as stated in the figures legends. The reactions were stopped by addition of loading buffer (1 mM EDTA, 0.1% bromophenol blue, and 0.1% xylene cyanol) and the samples were subjected to 4% or 6% polyacrylamide gel electrophoresis (PAGE). Gel electrophoresis was conducted using 0.25× TBE as running buffer, at 200 V, 4°C, and the gels were dried prior to autoradiography.
To obtain apparent binding constant (KDapp) values, the concentration of free DNA and protein–DNA complexes was determined using a Personal Molecular Imager and the Image Lab software (Bio-Rad). Since the nucleoprotein complexes formed by RarA do not always migrate as well-defined DNA species, protein–DNA complexes were estimated quantifying the disappearance of the free DNA substrate, using a Personal Molecular Imager and the Image Lab software (Bio-Rad). The protein concentrations that transfer 50% of the labeled DNA into complexes are approximately equal to the KDapp under conditions where the DNA concentration is much lower than the KDapp.
The cross-linking agent DSS was used to study protein-protein interactions as described previously (50). Cross-linking was performed by incubating RarA with PriA or SsbA (1 μg each) in buffer D (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.05 mg/ml BSA, 4 mM DTT, 5% glycerol) for 15 min at 37°C. Then DSS was added to a final concentration of 50 μM (10 min, 37°C) and then stopped by addition of 10 μl stop buffer (50 mM Tris–HCl pH 7.5, 400 mM glycine, 3% β-mercaptoethanol, 2% SDS, 10% glycerol), and subjected to 7% or 12% SDS-PAGE. Polyclonal antibodies raised against RarA and PriA were used to confirm the proteins present in the cross-linked complex.
Helicase assays
Helicase assay was performed under the conditions used for PriA (29). Briefly, they contained 0.4 nM radiolabeled forked DNA, and the indicated concentrations of PriA and RarA proteins in buffer E (20 mM Tris–HCl pH 7.5, 50 mM NaCl, 0.05 mg/ml BSA, 4 mM DTT, 3 mM MgCl2, 5% glycerol). After 30 min incubation at 30°C, reactions were stopped, deproteinized and run in a 12% PAGE in 1× TBE. Gels were dried and revealed in a Personal Molecular Imager apparatus. Products and substrates were quantified with the Image Lab software (Bio-Rad).
DNA replication assays
Reactions conditions for reconstituted PriA-dependent rolling circle DNA replication were performed as described (23,45). They consisted of 15 nM DnaE, 20 nM PolC, 8 nM DnaG, 25 nM HolA (δ), 25 nM HolB (δ′), 15 nM PriA, 24 nM DnaN (β), 90 nM SsbA, 25 nM DnaX (τ), 50 nM DnaD, 100 nM DnaB, 30 nM DnaC, 40 nM DnaI, 4 μM mini-circular DNA template in nucleotides (5 nM as molecules), 350 μM ATP, 100 μM CTP, GTP and UTP and 48 μM dNTPs (except 18 μM dCTP or dGTP for the leading and lagging strand DNA synthesis, respectively) and 0.2 μCi/reaction [α-32P]-dCTP and/or [α-32P]-dGTP, all in buffer BsRC (40 mM Tris-acetate (pH 7.8), 12 mM MgOAc, 200 mM potassium glutamate, 3 μM ZnSO4, 2% (w/v) polyethylene glycol (MW ∼8000), 0.02% Pluronic F68 and 1 mM DTT).
In experiments shown in Figures 3, 4 and Supplementary Figure S4 an enzyme mix consisting of all proteins except SsbA was generated, and added to a substrate mix composed of template DNA, rNTPs, dNTPs and SsbA. In the assays where RarA was added to ongoing DNA replication, the B. subtilis replisome was first preassembled incubating proteins with the DNA substrate (5 min, 37°C) in presence of 5 μM ATPγS and in absence of dNTPs and ATP. The replication reactions were then initiated by the addition of dNTPs and ATP, and after 20 s, RarA protein was added. Aliquots were removed at the indicated times. An equal volume of a stop mix consisting of 40 mM Tris–HCl (pH 8.0), 0.4% (w/v) SDS, 100 mM EDTA and 50 μg/ml proteinase K was added, and after 15 min the samples were applied onto Sephadex G-50 columns to eliminate non-incorporated dNTPs. Quantification was then performed by scintillation counting. For visualization of products, an aliquot was brought to 50 mM NaOH, 5% (v/v) glycerol and 0.05% bromphenol blue and fractionated in alkaline 0.8% agarose gels for ∼5 h at 60 V. Gels were fixed in 7% (w/v) trichloroacetic acid, dried and analysed with the Personal Molecular Imager and the Image Lab (Bio-Rad) software.
Figure 3.

SsbA-dependent RarA-mediated inhibition of B. subtilis PriA-dependent DNA replication. (A) Total DNA synthesis obtained in the presence of increasing RarA concentrations (15 min, 37°C). Reaction mixes contained all replisome components (preprimosomal proteins [PriA, DnaB, DnaD, DnaI), DnaC, DnaG, SsbA, τ-complex, β, PolC, DnaE), the indicated RarA concentration, template DNA, rNTPs, dNTPs and [α-32P]-dCTP and [α-32P]-dGTP. An enzyme mix consisting of all proteins except SsbA was generated and added to a substrate mix composed of template DNA, rNTPs, dNTPs, and SsbA. Then, samples were placed at 37°C. (B) Visualization of products obtained in the presence of 100 nM RarA or RarAK51A (15 min, 37°C). In the presence of [α-32P]-dCTP very large DNA fragments derived from rolling circle leading strand DNA synthesis is observed. A parallel reaction in the presence of [α-32P]-dGTP renders visible the small Okazaki fragments due to lagging strand DNA synthesis. Quantification of leading (C) and lagging strand (D) synthesis in the absence/presence of 100nM RarA and the indicated SsbA concentrations (15 min, 37°C). The quantification of the results is expressed as the mean ± SEM of six independent experiments. On the right part, a representative alkaline gel visualized by autoradiography showing the products of the DNA synthesis obtained in the presence or absence of RarA and SsbA.
Figure 4.
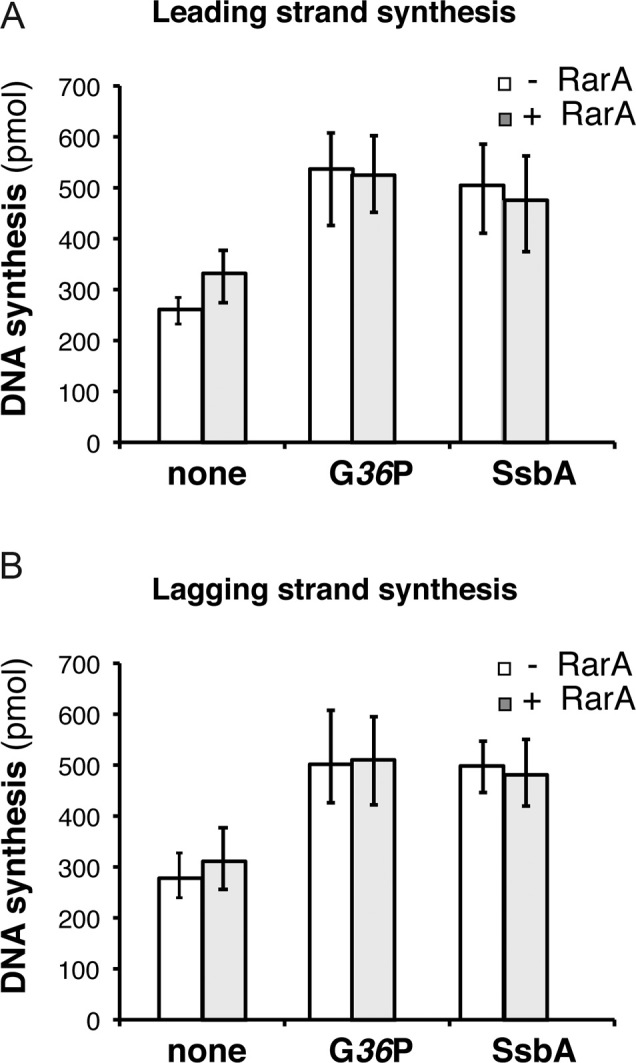
RarA does not inhibit SPP1 DNA replication. Quantification of leading (A) and lagging (B) strand synthesis obtained in standard SPP1 rolling circle DNA replication assays in the absence or in the presence of 100 nM RarA. Reaction mixes contained the SPP1 replisome, which is composed by SPP1 preprimosomal proteins (G38P and G39P) and DNA helicase G40P, and host proteins (DnaG, τ-complex, β, PolC and DnaE). The SPP1 replisome works with both SSB proteins (SsbA or G36P) and the effect of RarA on reactions having either viral G36P or host SbsA was tested. An enzyme mix consisting of all proteins except the SSB was generated, and added to a substrate mix composed of template DNA, rNTPs, dNTPs, and the indicated SSB (none, 30 nM G36P, or, 90 nM SsbA). Then reactions were placed at 37°C and incubated for 10 min. Leading strand synthesis was quantified by [α-32P]-dCTP incorporation and lagging strand synthesis by [α-32P]-dGTP incorporation. The results are expressed as the mean ± SEM of >3 independent experiments.
SPP1 rolling circle DNA replication reactions were similar than described above but with a different enzyme mix: 30 nM G40P, 300 nM G39P, 300 nM G38P, 8 nM DnaG, 15 nM DnaE, 20 nM PolC, 25 nM τ, 25 nM δ, 25 nM δ′, 24 nM β and 30 nM G36P or 90 nM SsbA. Reactions were incubated for 10 min at 37°C. The SSBs concentrations used (30 nM G36P and, 90 nM SsbA) are the optimal concentrations in the SPP1 replisome (25).
RESULTS
RarA ATPase is stimulated by ssDNA–dsDNA junctions or dsDNA ends
All RarA/Mgs1/WRNIP proteins studied so far show an ATPase activity stimulated upon binding to ssDNA and/or dsDNA (6,12,16,51). Bacillus subtilis RarA was purified under native conditions and its purity was estimated to be 95% (Supplementary Figure S1). First, RarA-mediated hydrolysis of ATP was analysed. In the absence of DNA, RarA hydrolysed ATP at very low efficiency, and with a catalytic rate constant (Kcat) of 6 ± 0.7 min−1 (Figure 1A, Supplementary Figure S2 and Supplementary Table S2). The potential stimulatory effect of ssDNA on the ATPase activity of RarA was tested using a fixed amount of circular 3199-nt ssDNA (20 μM in nt) and increasing RarA concentrations. The steady state rate of RarA-mediated ATP hydrolysis was observed without a significant lag time. ATP hydrolysis increased by increasing RarA concentrations from 12.5 to 50 nM (Kcat from ∼170 to ∼213 min−1), but at the highest RarA concentrations used (100 and 200 nM) the Kcat obtained was lower (∼172 and ∼102 min−1) (Supplementary Figure S2A and Supplementary Table S2), suggesting that the ssDNA added in the reaction is limiting. Stimulation of the RarA ATPase by ssDNA containing secondary structures has been observed also for yeast Mgs1 and RarAEco (6,7,16,51).
Figure 1.

RarA ATPase activity is stimulated by dsDNA-ssDNA junctions and dsDNA ends. (A) Stimulation by ssDNA containing secondary structures. RarA (25 nM) was incubated with increasing concentrations of circular 3199-nt ssDNA (7.5 to 20 μM) or linear 80-nt polydT ssDNA (15 and 30 μM) in buffer B containing 5 mM ATP, and the ATPase activity was measured (25 min, 37°C). The ATPase activity of RarA was also measured in the absence of ssDNA (no ssDNA, light blue) and in the presence of polydT and SsbA (gray). (B) Stimulation by dsDNA ends. RarA (25 nM) was incubated with two concentrations (7.5 and 15 μM) of various duplex DNA substrates: supercoiled 3199-bp dsDNA [cdsDNA], or dsDNA linearized with EcoRI [5′-ldsDNA], KpnI [3′-ldsDNA], SmaI [bl-dsDNA] or AluI [dsDNA-ends]. (C) Stimulation by SsbA bound to ssDNA. RarA (25 nM) was incubated with circular 3199-nt ssDNA (15 μM) and increasing concentrations of SsbA (9–75 nM). No ATPase activity is detected in the absence of ssDNA but presence of SsbA (18 nM and 37 nM, orange and blue) or when RarA was replaced by RarAK51A (ssDNA+K51A, dark brown). (D) ATPAse is stimulated by the C-terminal end of SsbA. RarA (25 nM) was incubated with circular 3199-nt ssDNA (15 μM) and increasing SsbB or SsbBA concentrations (18–75 nM). As a control, ATPase activity of RarA in the absence of ssDNA (no ssDNA, blue), in the presence of only ssDNA (magenta), or with ssDNA and SsbA (red) is shown. The amount of ATP hydrolysed was calculated as described (see Materials and Methods). Representative graphics are shown and quantification of the results are expressed as the mean ± SEM of >3 independent experiments (see Supplementary Table S2).
To test this hypothesis, we performed the assays varying the concentration of circular ssDNA in the presence of a fixed RarA concentration (25 nM). Increasing concentrations of ssDNA (7.5, 10, 15 and 20 μM) stimulated 11- to 30-fold the RarA ATPase activity (Figure 1A, Supplementary Table S2). These results suggest that RarA may bind to a component that is rate-limiting in the reaction rather than to the ssDNA regions. Native circular ssDNA can form secondary structures, where the bases hybridise to form a duplex with mismatches, surrounded by ssDNA–dsDNA junctions. Thus, the circular ssDNA was replaced by linear 80-nt polydT ssDNA, which cannot form secondary structures. No stimulation of the RarA ATPase was observed, even at high polydT concentrations, with a Kcat of ∼6.8 min−1 (Figure 1A, Supplementary Table S2). It is likely that RarA hydrolyses ATP upon binding to the DNA secondary structure (duplex DNA), to the ssDNA–dsDNA junctions present on the circular ssDNA or to both.
To analyze the former hypothesis, we used the 3199-bp dsDNA, either supercoiled, KpnI-, EcoRI- or SmaI-linearized. When increasing concentrations of circular duplex or KpnI-linearized dsDNA (3′-ldsDNA in Supplementary Table S2) were incubated with a fixed amount of RarA (25 nM), the RarA ATPase activity was stimulated up to 6-fold (Figure 1B, Supplementary Table S2). The RarA ATPase activity, however, was further increased up to 14-fold when EcoRI-linearized DNA (5′-ldsDNA) or up to 19-fold when the SmaI-linearized (bl-dsDNA substrate) was used as effector (Figure 1B, Supplementary Table S2). Similar results were observed when the RarAEco ATPase activity was measured in the presence of dsDNA with different DNA ends (16).
Then, it was tested whether the concentration of blunted DNA ends directly contributes to such stimulation. The total concentration of DNA ends was increased in the reaction by digesting the 3199-bp dsDNA with the AluI restriction enzyme that gives 19 different blunt-ended fragments. The AluI-cleaved duplex DNA (7.5 or 15 μM, dsDNA-ends in Supplementary Table S2) stimulated the rate of ATP hydrolysis of RarA 50-fold, with Kcat of ∼257 and ∼308 min−1, respectively (Figure 1B, Supplementary Table S2). Under this condition, however, the concentration of DNA ends was increased 19-fold, but the RarA ATPase activity only increased 2.6-fold when compared to the activity observed in the presence of SmaI-linearized duplex DNA (Figure 1B, Supplementary Table S2).
These data altogether suggest that: (i) RarA is a DNA-dependent ATPase; (ii) unstructured ssDNA does not stimulate and 3′-protruding ends or supercoiled circular duplex DNA poorly stimulate the RarA ATPase activity; (iii) RarA efficiently hydrolyses ATP upon binding to 5′-ssDNA–dsDNA junctions or to blunted dsDNA ends. The ATPase activity of RarAEco was also maximally stimulated by the presence of increasing concentrations of blunted DNA ends (16).
ssDNA-dependent ATPase of RarA is stimulated by SsbA bound to ssDNA
Bacillus subtilis encodes two tetrameric single stranded binding (SSB) proteins (SsbA and competence specific SsbB), and SsbA interacts with RarA (14). To study whether SsbA bound to ssDNA stimulates the ATPase activity of RarA, the circular ssDNA (15 μM in nt) was pre-incubated with increasing but limiting SsbA concentrations (9–75 nM) for 5 min. RarA (∼5 RarA tetramers/ssDNA molecule or 1 RarA/600-nt) was then added and the rate of ATP hydrolysis was measured. As revealed in Figure 1C, ∼2 SsbA tetramers/ssDNA molecule (9 nM SsbA) slightly stimulated the ATPase activity of RarA (Kcat of ∼174 min−1) when compared to absence of SsbA (Kcat 169.2 min−1) (see Supplementary Table S2). In the presence of higher SsbA concentrations (∼4 and ∼8 SsbA/ssDNA molecule) the rate of RarA-mediated ATP hydrolysis was significantly increased (Kcat of ∼200 and ∼267 min−1, respectively). SsbA bound to the ssDNA may recruit RarA by protein-protein interaction to the ssDNA–dsDNA junction, increasing its ATPase activity. However, further increase in the amounts of SsbA (75 nM SsbA, which corresponds to 16 SsbA/ssDNA molecule, or 1 SsbA/200-nt) reduced the RarA ATPase activity (Kcat of ∼246 min−1) when compared to 37 nM SsbA (Kcat of ∼267 min−1) (Figure 1C, Supplementary Table S2). In the presence of sub-saturating to saturating SsbA concentrations (1 SsbA/100-, 50- or 25-nt) the total rate of ATP hydrolysis was further reduced (data not shown). An interpretation of these data is that a fraction of DNA secondary structures is removed by SsbA, and thus the rate of RarA-mediated ATP hydrolysis is reduced. Low concentrations of SSBEco had little effect on ATP hydrolysis, and stoichiometric to saturating SSBEco concentrations inhibited RarAEco-mediated ATP hydrolysis (6), probably because saturating concentrations of SSBEco also disrupt DNA secondary structures and ssDNA substrates with no secondary structures fail to stimulate the ATPase activity of RarAEco (16).
To analyze whether the RarA interaction with SsbA, or with SsbA-bound to ssDNA, is responsible for the stimulation of the RarA ATPase activity, RarA (25 nM) was incubated with SsbA (18 or 37 nM) in the absence of ssDNA. Under these conditions, SsbA did not stimulate the ATPase activity of RarA (Kcat ∼6.4 min−1) that was indistinguishable from RarA alone (no ssDNA) (Figure 1C, Supplementary Table S2).
The previous results suggest that SsbA stimulates the ATPase activity of RarA bound at ssDNA–dsDNA junctions. Alternatively, SsbA bound to ssDNA could recruit and stimulate the ATPase activity of RarA bound to ssDNA without secondary structures. To test this, ATP hydrolysis was measured in the presence of SsbA-polydT ssDNA complexes, because polydT is an homopolymer that lacks secondary structures (i.e, no ss-ds junctions). Two SsbA concentrations (18 and 37 nM, i.e. ∼4 and ∼8 SsbA/ssDNA molecule) were preincubated with linear polydT ssDNA (15 μM), then RarA was added and the rate of ATP hydrolysis measured. The ATPase activity in the presence of the SsbA-polydT ssDNA complexes or with only polydT ssDNA was similar (Kcat ∼8.4 and ∼6.6 min−1) (Figure 1A, Supplementary Table S2). It is likely that limiting concentrations of SsbA bound to ssDNA at, or near, the dsDNA–ssDNA junctions are necessary and sufficient to stimulate the rate of ATP hydrolysis by RarA.
To further confirm that the measured ssDNA–dsDNA junction-dependent ATPase activity was related to the RarA protein, and not to a minor contaminant, the conserved Lys 51 of the Walker A motif was replaced by Ala, and the RarAK51A mutant protein was purified using a similar protocol. We did not detect ATPase activity with the mutant variant in the presence of ssDNA (Figure 1C, Supplementary Table S2).
The C-terminal domain of SsbA-bound to ssDNA plays a crucial role in the stimulation of the ATPase activity of RarA
The interaction of RarA with SsbA-bound to ssDNA is responsible for the stimulation of the RarA ATPase activity (Figure 1C, Supplementary Table S2). To evaluate whether the C-terminal amphipathic tail of SsbA plays a role in such protein-protein interaction, the 172-residue long SsbA was replaced in the ATPase assays by the 113-residue long SsbB. SsbB shares 63% identity with the N-terminal DNA binding domain of SsbA (residues 1–106). SsbB has a similar structure to SsbA (47) but it lacks the amphipathic C-terminal domain of SsbA, which is crucial for interaction with many recombination and replication proteins (52). In the presence of ∼4 to ∼8 SsbB tetramer/ssDNA molecule (18 and 37 nM SsbB) the maximal rate of RarA-mediated ATP hydrolysis was not significantly increased when compared to the RarA–ssDNA condition (Kcat ∼134 and ∼144 min−1). The presence of sub-saturating SsbB concentrations (75 nM, ∼12 SsbB/ssDNA molecule) significantly reduced the total rate of ATP hydrolysis (Kcat ∼110 min−1) (Figure 1D, Supplementary Table S2). These results suggest that SsbB does not stimulate RarA-mediated ATP hydrolysis, and that SsbB-mediated removal of a fraction of the secondary structures reduces the ATPase activity of RarA.
To confirm that the amphipathic C-terminus of SsbA interacts with RarA, and that a protein-protein interaction was required for ATPase stimulation, the SsbBA (formerly termed SsbB*) protein was used. The SsbBA chimera contains the last 9 terminal acidic residues of SsbA fused to SsbB, resulting in a 122-long protein (53). The SsbBA variant stimulated the RarA ATPase activity. The maximal rate of ATP hydrolysis was observed at ∼8 and ∼16 SsbBA/ssDNA molecule (Kcat ∼214 and ∼226 min−1) (Figure 1D, Supplementary Table S2). It is likely that the C-terminal nine residues of SsbA are necessary and sufficient to recruit RarA to the ssDNA–dsDNA substrate.
SsbA does not stimulate the ATPase of RarA bound to dsDNA ends
Previously it has been shown that RarAEco bound to a duplex end may partially unwind it, to create a flap (16). To test whether RarA bound to dsDNA ends can separate the strands, we took advantage of the fact that SsbA, which efficiently binds to ≥30-nt ssDNA (52), stimulates the RarA ATPase activity. SsbA, however, forms unstable complexes with a 20-nt long ssDNA segment and does not interact with duplex or duplex DNA with short tails (up to 10-nt long) (47). Therefore, if RarA unwinds duplex DNA to give ssDNA tails ≥30 nt we should expect an increase in the ATPase activity when duplex DNA and SsbA are both present in the reaction. The ATPase activity of RarA significantly increased when incubated with EcoRI-linearized 3199-bp dsDNA (4-nt 5′-overhang substrate) as observed previously, but this activity was not further stimulated by the addition of SsbA (Supplementary Figure S2B). Similar results were obtained when KpnI-linealized DNA (i.e. 3′-overhang) and SsbA were used (Supplementary Figure S2B). These data suggest that if RarA promotes strand separation, the length of the unwound strands was not sufficient for stable SsbA binding and subsequent loading of RarA onto duplex DNA.
RarA preferentially binds ssDNA and replication fork structures
To gain insight into the type of substrate that RarA binds, the interaction of RarA with structures that accumulate at replication forks (fork DNA), at stalled forks (ssDNA regions and replicated forked structures) or at collapsed forks (dsDNA ends) was assayed by EMSA.
In the absence of a nucleotide cofactor, RarA bound [γ32P]-91-nt ssDNA or [γ32P]-fork DNA in a concentration-dependent manner with apparent dissociation constant (KDapp) of ∼12 nM, followed by the replicated fork (KDapp of ∼19 nM) (Figure 2A and B) At low protein:DNA ratios one complex (CRarA) was observed, and at higher protein concentrations more complexes that migrated close to the well were observed with the two substrates (Figure 2A). RarA bound [γ32P]-dsDNA (KDapp of ∼37 nM) with ∼3-fold lower affinity than ssDNA (Figure 2B). RarA binding affinity to the different substrates tested was not significantly increased by the presence of ATP (data not shown). In contrast, RarAEco preferentially binds dsDNA in the presence of ATPγS (KDapp of ∼58 nM), and ssDNA in the apo form (KDapp of ∼890 nM) (16). The yeast Mgs1 protein binds DNA with flaps in an ATP independent manner, and does not bind dsDNA (51), and human WRNIP binds forked DNA in an ATP dependent manner (12).
Figure 2.

Binding affinity of RarA to different DNA substrates. (A) Binding of RarA to forked DNA and to a replicated fork (a replication fork with a fully synthesized leading-strand end and a gap in the lagging strand) determined by EMSA. [γ32P]-radiolabelled DNA (0.4 nM) was incubated with increasing concentrations of RarA (from 1.5 to 200 nM) in buffer B. Protein-DNA complexes were separated as described in methods. (B) RarA binding affinity for [γ32P]-dsDNA, [γ32P]-ssDNA, [γ32P]-fork or a [γ32P]-replicated fork was quantified from EMSA analysis. The results are expressed as the mean ± SEM of >3 independent experiments. (C–E) Cooperative binding of RarA and SsbA to forked DNA, replicated fork or ssDNA. The indicated combinations of SsbA (0.05–0.4 nM) and RarA (3–12 nM) were incubated with [γ-32P]-fork (C), [γ-32P]-replicated fork (D) or [γ-32P]-ssDNA (E) in buffer B. Protein–DNA complexes were analysed by native PAGE and autoradiography. Abbreviations: FD, free DNA, CSsb1 and CSsb2, SsbA-DNA complexes; CRarA, RarA–DNA complex and CSR, SsbA–DNA–RarA ternary complexes.
SsbA stimulates RarA binding to ssDNA
To analyse whether SsbA recruits RarA onto the different DNA substrates, EMSAs were performed with the two proteins and the fork DNA, the replicated fork, and ssDNA. SsbA bound with high affinity to the ssDNA region present in all the substrates. Two types of complexes (CSsb1 and CSsb2) were observed with 91-nt ssDNA, which has the potential of forming secondary structures, or with fork DNA (Figure 2C and E, lanes 2–5). As expected, SsbA bound the single tailed-ssDNA of replicated fork formed only one type of complex (CSsb1) (Figure 2D, lanes 2–5). This is consistent with binding of the protein in the 35-nt mode to the ssDNA regions available in the different substrates (53,54).
Under the conditions used, apo RarA bound fork DNA, the replicated fork, and the ssDNA forming one complex (CRarA) (Figure 2C–E, lanes 6–8). Similar results were observed when RarA was replaced by RarAK51A, a variant which neither binds nor hydrolyses ATP due to a mutation in the Walker A motif, but here the binding affinity was reduced (Supplementary Figure S3A, lanes 6–8). In the presence of limiting concentrations of both, SsbA (0.05 nM, ∼2-fold below KDapp) and RarA (3 nM, ∼4-fold below KDapp), the formation of CSsb1 and CRarA complexes and new slow and diffuse moving complexes (CSR) were observed (Figure 2C–E, lanes 9–11). The formation of CSR complexes increased with increasing RarA concentrations (Figure 2C–E, lanes 9–11). The ‘cooperative’ binding leading to CSR complexes cannot be attributed to molecular crowding, because addition of higher amounts of BSA did not function as a substitute for SsbA or RarA (data not shown). In the presence of KDapp or an excess of SsbA, both, CSsb and CSR, complexes were observed, but not the CRarA complex (Figure 2C–E, lanes 15–19). The same cooperativity in the appearance of CSR complex was observed when the RarAK51A variant was used, but here the CRarA complexes disappeared even at low RarAK51A:SsbA ratios (Supplementary Figure S3A).
These data all together suggest that SsbA, via a protein-protein interaction, led to a cooperative increase in the formation of CSR complexes even in the absence of a nucleotide cofactor, and that SsbA might recruit RarA onto ssDNA or the duplex region. If the former hypothesis is correct, the formation of a CSR complex should not be observed if SsbA is replaced by SsbB. This was indeed the case (Supplementary Figure S3B). When SsbA was replaced by SsbB, SsbB bound the 91-nt [γ32P]-ssDNA with KDapp of ∼6 nM and formed several complexes (Supplementary Figure S3B). In these assays, the CSR complex formation was not observed, and increasing SsbB concentrations inhibited RarA binding to the ssDNA (Supplementary Figure S3B, lanes 9–19). When SsbB was replaced by the SsbBA chimera, the CSR complexes were again formed (Supplementary Figure S3C, lanes 9–17), suggesting that SsbA or SsbBA interaction with RarA is necessary for the formation of the CSR complexes. In contrast, it has been suggested that SSBEco competes with RarAEco for binding to the ssDNA substrate (6).
SsbA modulates RarA-mediated inhibition of DNA replication
To test whether RarA contributes to DNA replication re-start we have used a reconstituted in vitro replication system with a substrate that mimics a paused replication fork with a gap in the lagging strand (see Supplementary Figure S4A). This is a synthetic circular 409-bp DNA template that contains a 3′-end for leading strand and a 5′-unpaired flap for lagging strand synthesis and it has a strong (50:1) dG:dC strand bias for separate analysis of leading and lagging strand synthesis (Supplementary Figure S4A). For leading strand synthesis with this 3′-OH primed substrate, the preprimosomal components (PriA–DnaD–DnaB complex and DnaI), DnaC helicase, PolC polymerase, the clamp loader (τδδ′), and the β-sliding clamp processivity factor are required. Lagging strand synthesis additionally requires DnaG primase and DnaE polymerase, which synthesise the hybrid RNA-DNA hybrids used by PolC (Supplementary Figure S4B) (23,25,45). First, we conducted in vitro replication assays substituting τ (DnaX) or the τ-complex (τδδ′) by RarA to check if RarA could act as an alternative clamp loader, as suggested (6). No DNA synthesis was obtained (Supplementary Figure S4C). Second, we analysed if RarA could have any effect on PriA-dependent DNA replication. Reactions were performed with all the replisome components and in addition RarA. PriA-dependent initiation of DNA replication was inhibited by increasing the RarA concentration (Figure 3A). In the presence of RarA, leading and lagging strand synthesis were inhibited ∼5-fold and ∼3-fold, respectively, (Figure 3B). When RarA was replaced by the RarAK51A variant, in vitro replication was also inhibited (Figure 3B), suggesting that ATP hydrolysis is not required for RarA-mediated inhibition of DNA synthesis. Alternatively, traces of a contaminant inhibit DNA synthesis.
To address the latter hypothesis, in vitro replication in the presence of RarA and various SsbA concentrations was analysed. As previously reported with this substrate (23,45), leading strand synthesis is strongly reduced, and lagging strand DNA synthesis is only partially affected when the replication reactions do not have a SSB protein (Figure 3C and D, lane 1). This occurs because in the absence of SsbA, the DnaC helicase binds to the DNA template by threading onto the free 5′-end available on the lagging strand, and some DNA synthesis is obtained (55). Interestingly, under this condition (absence of SsbA), addition of RarA neither affected leading nor lagging strand DNA synthesis (Figure 3C and D, lane 2). In the presence of optimal SsbA concentrations DNA synthesis was sensitive to the addition of RarA (Figure 3C, lane 4), and when an excess of SsbA was added, leading and lagging strand DNA synthesis took place albeit with lower efficiency, and both were still sensitive to RarA action (Figure 3C, lane 6). These results rule out that traces of a contaminant could inhibit DNA synthesis.
RarA inhibits PriA-dependent initiation of DNA replication
RarA may inhibit initiation of DNA replication by two different mechanisms: either affecting replisome assembly or by directly inhibiting DNA synthesis. To address if the presence of RarA may affect DNA synthesis, we analysed its effect on the replication of the B. subtilis SPP1 bacteriophage. The differences between both in vitro replication systems are just the helicase loaders (preprimosome PriA-DnaB-DnaD and DnaI) and the DNA helicase (DnaC) in B. subtilis versus helicase loader (G38P–G39P) and the replicative DNA helicase (G40P) in SPP1. Both replication systems share the polymerases, the primase, the processivity factor and the clamp loader (45). In addition, the SPP1 replisome can use both SSB proteins (viral G36P, or the host SsbA protein) (45). In other words, SPP1 DNA replication has the same components as B. subtilis replication, except the helicase and its loading system. We found that addition of RarA neither affected leading nor lagging strand DNA synthesis by the SPP1 replisome, independently of which was the SSB used (G36P or SsbA) (Figure 4A and B), suggesting that SsbA is necessary but not sufficient for RarA-mediated inhibition of initiation of DNA replication.
These results, apart from discarding any negative effect due to a contamination in our RarA preparation, suggest that the negative effect exerted by RarA must be at the stage of preprimosomal assembly, or inhibiting DnaC-mediated DNA unwinding during chain elongation. The helicase loaders (preprimosome components plus DnaI) and the DNA helicase are essential for PriA-dependent initiation of DNA replication, but once the replicative helicase is loaded, the preprimosome components are not required for the chain elongation reaction, whereas the helicase is required at all stages of DNA replication (23,45). To analyze if RarA inhibits DnaC-mediated DNA unwinding, we allowed the assembly of the full B. subtilis replisome, incubating all protein components except RarA with the DNA in the presence of limiting ATPγS for 5 min. Then ATP and the dNTPs were added to initiate the replication reaction and RarA was added 20 s after replication start, and aliquots were taken at several times and processed. Addition of RarA to an active replication elongation reaction did not affect DNA synthesis (Figure 5). In a control reaction set up in the same way, but adding RarA at the time of replisome assembly, RarA inhibited, even at early times of the replication reaction (Supplementary Figure S5). Here, we observed less DNA synthesis in the presence of RarA, but the DNA products obtained had a similar length, again suggesting that RarA does not affect the rate of DNA synthesis and that on ongoing DNA replication RarA has no effect.
Figure 5.

RarA has no effect on ongoing DNA replication. (A) Scheme of the experimental design. The B. subtilis replisome was assembled on the DNA in the absence of RarA and in the presence of limiting ATPγS and then DNA replication was started by dNTP (including [α-32P]-dCTP) and ATP addition. After 20 s of initiating the reaction, 100 nM RarA was added or not, and reactions were continued for the indicated times. (B) Quantification of leading strand synthesis (mean ± SEM of >3 independent experiments). (C) The leading strand DNA products obtained in one of these assays are visualized by denaturing gel electrophoresis and autoradiography.
RarA may interact with the PriA protein in vitro
The previous results show that RarA does not inhibit the DnaC helicase during chain elongation and may act during helicase assembly. This suggests that the negative effect exerted by RarA could be at the stage of preprimosomal assembly. The first step in replication restart is preprimosome assembly by the binding of PriA to a paused replication fork. EMSA experiments were performed with purified RarA and PriA proteins to detect if RarA has any effect on PriA binding to the paused fork. The buffer conditions optimal for PriA-DNA interaction (buffer C, 1 mM EDTA (see 29)) are different than for RarA-DNA interaction (buffer B, 10 mM MgOAc), and here PriA conditions were used. In buffer C conditions, RarA bound DNA with ∼4 times less affinity as in buffer B conditions (see Figure 2).
PriA preferentially recognizes a replicated fork (fully synthesized leading-strand and a gap in the lagging strand) (reviewed in 21). PriA bound this replicated fork DNA forming one discrete protein–DNA complex (CPriA) with high affinity (KDapp of ∼3 nM) (Figure 6A and B, lane 2). In the presence of PriA and RarA a new protein–DNA complex (CPR) that migrated more slowly than the CRarA complex could be observed even in the presence of free DNA, suggesting an interaction between both proteins (Figure 6A, lanes 6–8). At saturating PriA concentrations CPR complexes were also observed (Figure 6B, lanes 6–8) suggesting the binding of both proteins to different arms of the replicated fork DNA. To determine whether RarA interacts with the PriA protein, we performed protein-protein crosslinking assays with DSS in the absence of replicated fork DNA. As control the RarA interaction with SsbA was also analysed by crosslinking with DSS (Supplementary Figure S6A). A small fraction of the RarA and PriA proteins could be cross-linked by DSS and this novel protein band cross reacted against the specific polyclonal antibodies raised against PriA (Supplementary Figure S6C).
Figure 6.

RarA may interact with PriA but does not stimulate its helicase activity. (A and B) Simultaneous binding of RarA and PriA to a replicated fork. PriA (2.5 nM [A], and 5 nM [B]) and RarA (25–100 nM) were incubated with [γ-32P]-replicated fork (0.4 nM in molecules) in buffer C. Protein-DNA complexes were analysed by native PAGE and autoradiography. Abbreviations: FD, free DNA, CPriA, PriA-DNA complex; CRarA, RarA–DNA complex and CPR, PriA–DNA–RarA ternary complex. (C) RarA does not stimulate the helicase activity of PriA. The indicated combinations of PriA (5 nM) and RarA (25, 50, 100 nM) were incubated with the helicase substrate ([γ-32P]-fork) in buffer E (30 min, 30°C). Products were separated after deproteinization by PAGE and visualized by autoradiography. RarA was unable to unwind this substrate under these experimental conditions (lanes 2–4).
PriA is a 3′→5′ DNA helicase (reviewed in 21). To determine whether RarA has any effect on the helicase activity of PriA we employed a DNA unwinding assay described previously which uses a fork DNA substrate (29). PriA efficiently unwound a fork DNA substrate, and the presence of increasing RarA concentrations did not affect PriA-dependent DNA unwinding (Figure 6C, lanes 6–8). Under these conditions, RarA could not unwind the fork substrate (Figure 6C, lanes 2–4). Similarly, no DNA helicase activity has been associated with RarAEco on a fork substrate (16).
DISCUSSION
In this report, we have made seven principal observations pertaining the mode of action of the ubiquitous RarA protein. First, RarA is a DNA-dependent ATPase specifically stimulated by hairpin structures present on circular ssDNA or ssDNA–dsDNA junctions, and by the interaction with the SsbA protein bound to this ssDNA, as shown in Figure 1. Bacterial RarA proteins interact with the SSB protein through the C-terminal domain (this work) (6,14), whereas an interaction of the eukaryotic homologs with RPA has not been described. Second, blunted and 5′-tailed duplex DNA stimulate the ATPase activity of RarA, as reported for mammalian WRNIP and RarAEco (13,16). Third, in vitro the DNA binding capability of the ubiquitous RarA/Mgs1/WRNIP proteins, and the effect of ATP on this activity, varies. RarA in the apo form bound forks, replicated forks or ssDNA with a similar affinity, and it bound duplex DNA with ∼3-fold lower affinity. No significant difference was observed when ATP was added to the reaction mixture or when the mutant lacking ATPase activity was assayed (Supplementary Figure S3). In contrast, RarAEco preferentially binds dsDNA in the presence of ATPγS (16), the yeast Mgs1 protein binds DNA with flaps in an ATP independent manner, and does not bind dsDNA (51), and human WRNIP binds forked DNA in an ATP dependent manner (12). The nature of these discrepancies remains unknown, but they may indicate the different DNA substrates recognized by the protein in vivo. We found that SsbA, bound to one of the arms or to a ssDNA region contributes to recruit RarA trough a protein-protein interaction to the other DNA arm or to the ssDNA–dsDNA junction (Figure 2 and Supplementary Figure S3). Due to the high abundance of SsbA in the cell and its high affinity to ssDNA it is tempting to speculate that in the cell RarA will be bound preferentially at ssDNA or ss-dsDNA junctions through its interaction with SsbA.
Fourth, RarA does not act as an alternative clamp loader system in our in vitro replication assay (Supplementary Figure S4), as suggested from the X-ray structure of RarAEco, and the sequence similarity with DnaXEco (6). Fifth, RarA, upon interaction with SsbA, inhibits PriA-dependent initiation of DNA replication, suggesting that RarA regulates PriA-dependent replication restart, perhaps by inhibiting primosome assembly. Indeed, elongation of DNA replication was refractory to RarA action (Figures 3 to 5). Sixth, RarA may interact with PriA, but neither inhibited its binding activity nor stimulated its 3′→5′ unwinding activity (Figure 6). Seventh, RarA lacks the ability to unwind fork DNA (Figure 6). It was proposed that RarAEco, in the ATPγS bound form, separates the strands of a duplex to create flaps (16). Those flaps could contribute to rescue a stalled replication fork by allowing DnaBEco helicase and the associated replisome to continue DNA synthesis without disassembly (model 1 for RarAEco action, see Introduction). We cannot discard this activity for B. subtilis RarA, but our ATPase experiments did not show any further stimulation by dsDNA ends when SsbA was also present. If RarA promotes strand separation, the length of the unwound strands is not sufficient for SsbA binding (>25-nt), and subsequent loading of RarA.
Previous results have found that the Mgs1/WRNIP1/RarA protein is associated with the replication fork and involved in replication restart. It was proposed that Mgs1/WRNIP1 might stimulate detachment of the replicative Polδ from ubiquitylated PCNA, and facilitate the recruitment of the translesion synthesis polymerase Polη to sites of DNA damage (1). Similarly, model 2 for RarAEco action (see Introduction) proposed that this protein facilitates the replacement of Pol IIIEco by a bypass DNA polymerase (5,15). The authors proposed that when DNA replication is arrested, due to Pol III holoenzyme instability, the complex may fall off DNA. RarAEco may then bind to the available 3′-end of the leading strand and unwind it. When RarAEco reaches a βEco-clamp it may stop unwinding and place the DNA 3′-end at the βEco-clamp position thereby facilitating DinBEco loading (15). This model suggests an interaction of RarA with the clamp loader, and we did not observe this in our in vitro replication assays. Although we did not find a helicase activity for the isolated protein, a helicase activity could explain the observation that SOS induction, which requires a RecA nucleoprotein filament onto ssDNA, is reduced in the ΔrarA context (P. Cardenas personal communication). Further work is required to explain the differences we found between the two bacteria.
The biochemical activities associated with B. subtilis RarA suggest that this family of proteins contribute to solve problems at blocked forks. Such RarA activity might be necessary for transient response to replicative stress. A stalled fork can be repaired by different DNA damage tolerance mechanisms, and failure in recovery of the stalled fork may result in a blocked fork (reviewed in 22,56,57–59). After replisome disassembly, RarA might control the early stage of preprimosomal reassembly, which is crucial for the reassembly of the replicative DnaC helicase, as suggested in the model depicted in Figure 7. We propose that when a replication fork encounters a lesion in the leading or lagging strand template, replisome disassembly can occur. SsbA, which rapidly protects the resulting ssDNA region, might recruit recombination proteins onto ssDNA, and RarA toward the ssDNA–dsDNA junction. Our model proposes that RarA, by interacting with SsbA and PriA, might prevent PriA-dependent premature re-initiation of DNA replication. Once alternative repair pathways remove and/or circumvent the DNA lesion a PriA-dependent DNA replication might re-initiate. Indeed, the absence of RarA renders cells very sensitive to H2O2, which introduces nicks in the template DNA, and the absence of both RarA and RecA compromise viability in E. coli or B. subtilis cells (4, our unpublished results).
Figure 7.

Model of RarA action on blocked forks. (i) When a lesion (filled yellow square) blocks DNA replication, DNA synthesis is stopped and the replisome might disassemble. (ii) SsbA binds to the lagging strand of a stalled replication fork. (iii) By protein interaction PriA is recruited. (iv) PriA bound to the lagging strand recruits, by protein-protein interaction, DnaB and DnaD and the DnaC-DnaI helicase–loader complex to the stalled fork. Then the damage is repaired. The helicase is then activated by DnaI release and subsequent preprimosome disassembly. DnaC recruits DnaG primase and the replisome and DNA replication can restart (not depicted). (v and vi) If the DNA damage is not removed, SsbA loads RarA at the stalled fork. The loading of PriA is not avoided, but the SsbA-RarA-PriA-DNA complex impedes the recruitment of the replisome and initiation of DNA synthesis is inhibited.
Investigation of events that occur during replication restart is essential for understanding the complex mechanisms cells have evolved to deal with a replicative stress. The inhibition caused by RarA in initiation of DNA replication is complex and probably reflects its multiple interactions with replication and recombination proteins as detected earlier (reviewed in 1). Inhibition of initiation of DNA replication was observed even in the presence of an excess of SsbA, which suggests that RarA not only interacts with SsbA, but also with another component of the host-encoded preprimosome assembly machinery. We found here that RarA may directly interact with PriA, however it did not inhibit its helicase or DNA binding activity. Both, RarA and PriA directly interact with the C-terminal region of SsbA (14, this work,60). How PriA-directed preprimosome assembly is modulated by RarA and SsbA requires further analysis.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Charles S. McHenry for providing us with B. subtilis replication proteins, and Alan D. Grossman for polyclonal antibodies against PriA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Ministerio de Economía, Industria y Competitividad (MINECO/FEDER) [BFU2015-67065-P to J.C.A., S.A.]. Funding for open access charge: Ministerio de Economía y Competitividad (MINECO/FEDER).
Conflict of interest statement. None declared.
REFERENCES
- 1. Yoshimura A., Seki M., Enomoto T.. The role of WRNIP1 in genome maintenance. Cell Cycle. 2017; 16:515–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barre F.X., Soballe B., Michel B., Aroyo M., Robertson M., Sherratt D.. Circles: the replication-recombination-chromosome segregation connection. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:8189–8195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lau I.F., Filipe S.R., Soballe B., Okstad O.A., Barre F.X., Sherratt D.J.. Spatial and temporal organization of replicating Escherichia coli chromosomes. Mol. Microbiol. 2003; 49:731–743. [DOI] [PubMed] [Google Scholar]
- 4. Shibata T., Hishida T., Kubota Y., Han Y.W., Iwasaki H., Shinagawa H.. Functional overlap between RecA and MgsA (RarA) in the rescue of stalled replication forks in Escherichia coli. Genes Cells. 2005; 10:181–191. [DOI] [PubMed] [Google Scholar]
- 5. Lestini R., Michel B.. UvrD controls the access of recombination proteins to blocked replication forks. EMBO J. 2007; 26:3804–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Page A.N., George N.P., Marceau A.H., Cox M.M., Keck J.L.. Structure and biochemical activities of Escherichia coli MgsA. J. Biol. Chem. 2011; 286:12075–12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hishida T., Iwasaki H., Ohno T., Morishita T., Shinagawa H.. A yeast gene, MGS1, encoding a DNA-dependent AAA(+) ATPase is required to maintain genome stability. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:8283–8289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hishida T., Ohno T., Iwasaki H., Shinagawa H.. Saccharomyces cerevisiae MGS1 is essential in strains deficient in the RAD6-dependent DNA damage tolerance pathway. EMBO J. 2002; 21:2019–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hishida T., Ohya T., Kubota Y., Kamada Y., Shinagawa H.. Functional and physical interaction of yeast Mgs1 with PCNA: impact on RAD6-dependent DNA damage tolerance. Mol. Cell. Biol. 2006; 26:5509–5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Saugar I., Parker J.L., Zhao S., Ulrich H.D.. The genome maintenance factor Mgs1 is targeted to sites of replication stress by ubiquitylated PCNA. Nucleic Acids Res. 2012; 40:245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vijeh Motlagh N.D., Seki M., Branzei D., Enomoto T.. Mgs1 and Rad18/Rad5/Mms2 are required for survival of Saccharomyces cerevisiae mutants with novel temperature/cold sensitive alleles of the DNA polymerase delta subunit, Pol31. DNA Repair (Amst.). 2006; 5:1459–1474. [DOI] [PubMed] [Google Scholar]
- 12. Yoshimura A., Seki M., Kanamori M., Tateishi S., Tsurimoto T., Tada S., Enomoto T.. Physical and functional interaction between WRNIP1 and RAD18. Genes Genet. Syst. 2009; 84:171–178. [DOI] [PubMed] [Google Scholar]
- 13. Tsurimoto T., Shinozaki A., Yano M., Seki M., Enomoto T.. Human Werner helicase interacting protein 1 (WRNIP1) functions as a novel modulator for DNA polymerase delta. Genes Cells. 2005; 10:13–22. [DOI] [PubMed] [Google Scholar]
- 14. Costes A., Lecointe F., McGovern S., Quevillon-Cheruel S., Polard P.. The C-terminal domain of the bacterial SSB protein acts as a DNA maintenance hub at active chromosome replication forks. PLoS Genet. 2010; 6:e1001238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Michel B., Sinha A.K.. The inactivation of rfaP, rarA or sspA gene improves the viability of the Escherichia coli DNA polymerase III holD mutant. Mol Microbiol. 2017; 104:1008–1026. [DOI] [PubMed] [Google Scholar]
- 16. Stanage T.H., Page A.N., Cox M.M.. DNA flap creation by the RarA/MgsA protein of Escherichia coli. Nucleic Acids Res. 2017; 45:2724–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mangiameli S.M., Merrikh C.N., Wiggins P.A., Merrikh H.. Transcription leads to pervasive replisome instability in bacteria. eLife. 2017; 6:e19848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kuzminov A. Recombinational repair of DNA damage in Escherichia coli and bacteriophage lambda. Microbiol. Mol. Biol. Rev. 1999; 63:751–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Michel B., Flores M.J., Viguera E., Grompone G., Seigneur M., Bidnenko V.. Rescue of arrested replication forks by homologous recombination. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:8181–8188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Atkinson J., McGlynn P.. Replication fork reversal and the maintenance of genome stability. Nucleic Acids Res. 2009; 37:3475–3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gabbai C.B., Marians K.J.. Recruitment to stalled replication forks of the PriA DNA helicase and replisome-loading activities is essential for survival. DNA Repair (Amst.). 2010; 9:202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yeeles J.T., Poli J., Marians K.J., Pasero P.. Rescuing stalled or damaged replication forks. Cold Spring Harb. Perspect. Biol. 2013; 5:a012815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sanders G.M., Dallmann H.G., McHenry C.S.. Reconstitution of the B. subtilis replisome with 13 proteins including two distinct replicases. Mol. Cell. 2010; 37:273–281. [DOI] [PubMed] [Google Scholar]
- 24. McHenry C.S. DNA replicases from a bacterial perspective. Annu. Rev. Biochem. 2011; 80:403–436. [DOI] [PubMed] [Google Scholar]
- 25. Seco E.M., Ayora S.. Bacillus subtilis DNA polymerases, PolC and DnaE, are required for both leading and lagging strand synthesis in SPP1 origin-dependent DNA replication. Nucleic Acids Res. 2017; 45:8302–8313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dervyn E., Suski C., Daniel R., Bruand C., Chapuis J., Errington J., Janniere L., Ehrlich S.D.. Two essential DNA polymerases at the bacterial replication fork. Science. 2001; 294:1716–1719. [DOI] [PubMed] [Google Scholar]
- 27. Bruck I., O’Donnell M.. The DNA replication machine of a Gram-positive organism. J. Biol. Chem. 2000; 275:28971–28983. [DOI] [PubMed] [Google Scholar]
- 28. Marsin S., McGovern S., Ehrlich S.D., Bruand C., Polard P.. Early steps of Bacillus subtilis primosome assembly. J. Biol. Chem. 2001; 276:45818–45825. [DOI] [PubMed] [Google Scholar]
- 29. Polard P., Marsin S., McGovern S., Velten M., Wigley D.B., Ehrlich S.D., Bruand C.. Restart of DNA replication in Gram-positive bacteria: functional characterisation of the Bacillus subtilis PriA initiator. Nucleic Acids Res. 2002; 30:1593–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Velten M., McGovern S., Marsin S., Ehrlich S.D., Noirot P., Polard P.. A two-protein strategy for the functional loading of a cellular replicative DNA helicase. Mol. Cell. 2003; 11:1009–1020. [DOI] [PubMed] [Google Scholar]
- 31. Bruand C., Velten M., McGovern S., Marsin S., Serena C., Ehrlich S.D., Polard P.. Functional interplay between the Bacillus subtilis DnaD and DnaB proteins essential for initiation and re-initiation of DNA replication. Mol. Microbiol. 2005; 55:1138–1150. [DOI] [PubMed] [Google Scholar]
- 32. Zhang W., Carneiro M.J., Turner I.J., Allen S., Roberts C.J., Soultanas P.. The Bacillus subtilis DnaD and DnaB proteins exhibit different DNA remodelling activities. J. Mol. Biol. 2005; 351:66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li Y.C., Naveen V., Lin M.G., Hsiao C.D.. Structural analyses of the bacterial primosomal protein DnaB reveal that it is a tetramer and forms a complex with a primosomal re-initiation protein. J. Biol. Chem. 2017; 292:15744–15757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Soultanas P. A functional interaction between the putative primosomal protein DnaI and the main replicative DNA helicase DnaB in Bacillus. Nucleic Acids Res. 2002; 30:966–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ioannou C., Schaeffer P.M., Dixon N.E., Soultanas P.. Helicase binding to DnaI exposes a cryptic DNA-binding site during helicase loading in Bacillus subtilis. Nucleic Acids Res. 2006; 34:5247–5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Smits W.K., Goranov A.I., Grossman A.D.. Ordered association of helicase loader proteins with the Bacillus subtilis origin of replication in vivo. Mol. Microbiol. 2010; 75:452–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Smits W.K., Merrikh H., Bonilla C.Y., Grossman A.D.. Primosomal proteins DnaD and DnaB are recruited to chromosomal regions bound by DnaA in Bacillus subtilis. J. Bacteriol. 2011; 193:640–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bird L.E., Pan H., Soultanas P., Wigley D.B.. Mapping protein-protein interactions within a stable complex of DNA primase and DnaB helicase from Bacillus stearothermophilus. Biochemistry. 2000; 39:171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rannou O., Le Chatelier E., Larson M.A., Nouri H., Dalmais B., Laughton C., Janniere L., Soultanas P.. Functional interplay of DnaE polymerase, DnaG primase and DnaC helicase within a ternary complex, and primase to polymerase hand-off during lagging strand DNA replication in Bacillus subtilis. Nucleic Acids Res. 2013; 41:5303–5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Martinez-Jimenez M.I., Mesa P., Alonso J.C.. Bacillus subtilis τ subunit of DNA polymerase III interacts with bacteriophage SPP1 replicative DNA helicase G40P. Nucleic Acids Res. 2002; 30:5056–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Haroniti A., Anderson C., Doddridge Z., Gardiner L., Roberts C.J., Allen S., Soultanas P.. The clamp-loader-helicase interaction in Bacillus. Atomic force microscopy reveals the structural organisation of the DnaB-tau complex in Bacillus. J. Mol. Biol. 2004; 336:381–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Afonso J.P., Chintakayala K., Suwannachart C., Sedelnikova S., Giles K., Hoyes J.B., Soultanas P., Rafferty J.B., Oldham N.J.. Insights into the structure and assembly of the Bacillus subtilis clamp-loader complex and its interaction with the replicative helicase. Nucleic Acids Res. 2013; 41:5115–5126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bruck I., Goodman M.F., O’Donnell M.. The essential C family DnaE polymerase is error-prone and efficient at lesion bypass. J. Biol. Chem. 2003; 278:44361–44368. [DOI] [PubMed] [Google Scholar]
- 44. Paschalis V., Le Chatelier E., Green M., Kepes F., Soultanas P., Janniere L.. Interactions of the Bacillus subtilis DnaE polymerase with replisomal proteins modulate its activity and fidelity. Open Biol. 2017; 7:170146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Seco E.M., Zinder J.C., Manhart C.M., Lo Piano A., McHenry C.S., Ayora S.. Bacteriophage SPP1 DNA replication strategies promote viral and disable host replication in vitro. Nucleic Acids Res. 2013; 41:1711–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Carrasco B., Manfredi C., Ayora S., Alonso J.C.. Bacillus subtilis SsbA and dATP regulate RecA nucleation onto single-stranded DNA. DNA Repair (Amst.). 2008; 7:990–996. [DOI] [PubMed] [Google Scholar]
- 47. Yadav T., Carrasco B., Myers A.R., George N.P., Keck J.L., Alonso J.C.. Genetic recombination in Bacillus subtilis: a division of labor between two single-strand DNA-binding proteins. Nucleic Acids Res. 2012; 40:5546–5559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yuan Q., McHenry C.S.. Cycling of the E. coli lagging strand polymerase is triggered exclusively by the availability of a new primer at the replication fork. Nucleic Acids Res. 2014; 42:1747–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zecchi L., Lo Piano A., Suzuki Y., Cañas C., Takeyasu K., Ayora S.. Characterization of the Holliday junction resolving enzyme encoded by the Bacillus subtilis bacteriophage SPP1. PLoS One. 2012; 7:e48440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cañas C., Carrasco B., Ayora S., Alonso J.C.. The RecU Holliday junction resolvase acts at early stages of homologous recombination. Nucleic Acids Res. 2008; 36:5242–5249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kim J.H., Kang Y.H., Kang H.J., Kim D.H., Ryu G.H., Kang M.J., Seo Y.S.. In vivo and in vitro studies of Mgs1 suggest a link between genome instability and Okazaki fragment processing. Nucleic Acids Res. 2005; 33:6137–6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shereda R.D., Kozlov A.G., Lohman T.M., Cox M.M., Keck J.L.. SSB as an organizer/mobilizer of genome maintenance complexes. Crit. Rev. Biochem. Mol. Biol. 2008; 43:289–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yadav T., Carrasco B., Hejna J., Suzuki Y., Takeyasu K., Alonso J.C.. Bacillus subtilis DprA recruits RecA onto single-stranded DNA and mediates annealing of complementary strands coated by SsbB and SsbA. J. Biol. Chem. 2013; 288:22437–22450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yadav T., Carrasco B., Serrano E., Alonso J.C.. Roles of Bacillus subtilis DprA and SsbA in RecA-mediated genetic recombination. J. Biol. Chem. 2014; 289:27640–27652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Manhart C.M., McHenry C.S.. The PriA replication restart protein blocks replicase access prior to helicase assembly and directs template specificity through its ATPase activity. J. Biol. Chem. 2013; 288:3989–3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kuzminov A. Collapse and repair of replication forks in Escherichia coli. Mol. Microbiol. 1995; 16:373–384. [DOI] [PubMed] [Google Scholar]
- 57. Mirkin E.V., Mirkin S.M.. Replication fork stalling at natural impediments. Microbiol. Mol. Biol. Rev. 2007; 71:13–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Branzei D., Foiani M.. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell. Biol. 2008; 9:297–308. [DOI] [PubMed] [Google Scholar]
- 59. Gaillard H., Aguilera A.. Transcription as a threat to genome integrity. Annu. Rev. Biochem. 2016; 85:291–317. [DOI] [PubMed] [Google Scholar]
- 60. Lecointe F., Serena C., Velten M., Costes A., McGovern S., Meile J.C., Errington J., Ehrlich S.D., Noirot P., Polard P.. Anticipating chromosomal replication fork arrest: SSB targets repair DNA helicases to active forks. EMBO J. 2007; 26:4239–4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.