


Abstract
DNA methylation is an epigenetic mechanism known to affect gene expression and aberrant DNA methylation patterns have been described in cancer. However, only a small fraction of differential methylation events target genes with a defined role in cancer, raising the question of how aberrant DNA methylation contributes to carcinogenesis. As recently a link has been suggested between methylation patterns arising in ageing and those arising in cancer, we asked which aberrations are unique to cancer and which are the product of normal ageing processes. We therefore compared the methylation patterns between ageing and cancer in multiple tissues. We observed that hypermethylation preferentially occurs in regulatory elements, while hypomethylation is associated with structural features of the chromatin. Specifically, we observed consistent hypomethylation of late-replicating, lamina-associated domains. The extent of hypomethylation was stronger in cancer, but in both ageing and cancer it was proportional to the replication timing of the region and the cell division rate of the tissue. Moreover, cancer patients who displayed more hypomethylation in late-replicating, lamina-associated domains had higher expression of cell division genes. These findings suggest that different cell division rates contribute to tissue- and cancer type-specific DNA methylation profiles.
INTRODUCTION
Genes encoding epigenetic modifiers are frequently mutated in cancer patients (1–4) and the resulting epigenetic aberrations complement genetic aberrations during carcinogenesis. Genome-wide epigenetic aberrations displayed by malignant cells include differential DNA methylation, changes in histone post-translational modifications, and abnormalities in chromatin structure (1,5). Modern techniques allow studying these epigenetic aberrations on a genome-wide scale. Specifically, differential DNA methylation in cancer has been studied extensively and is the epigenetic mechanism for which the most data are available. The extent and consistency of DNA methylation patterns in certain cancer types (such as prostate cancer) suggest that differential DNA methylation could be as important for carcinogenesis as genetic aberrations (6,7).
Systematic evaluation of the DNA methylation landscape in different cancer types has revealed several common features. In tumour samples, methylation is generally lost (hypomethylation) over wide stretches of the genome and selectively gained (hypermethylation) in promoter regions of specific genes (8). This has been thought to have functional implications. Specifically, transcriptional silencing of tumour suppressor genes by hypermethylation of their promoters has been observed in cancer and initially proposed as one of the main epigenetic contributors to carcinogenesis (9). Complementarily, promoters of genes beneficial for cancer development have been shown to be hypomethylated (10,11), although the link between hypomethylation and restored gene expression is generally debated (12,13). Cancer-associated DNA hypomethylation has been linked to late-replicating structural elements of the chromatin such as lamina-associated domains (LADs) and long stretches of histone H3 lysine 9 dimethylation (LOCKs), suggesting it is rather influenced by chromatin organization (14–16). However, the mechanisms behind hypomethylation of these regions are poorly understood (17). In addition to these common methylation patterns observed in multiple types of cancer, tissue-specific patterns of methylation exist as well (18). In conclusion, defining causes and consequences of aberrant methylation in cancer development is not straightforward, complicating interpretation of available data.
The aberrant methylation landscape in cancer shares common features with that in ageing (19,20). Ageing is a complex process that involves the decline of multiple physiological functions over time and is often accompanied by changes in DNA methylation (21). The revolutionary work of Horvath has shown that it is possible to predict chronological age based on methylation values measured at 353 sites, suggesting a certain directionality to the process (22). An overlap has been found between sites that become gradually hypermethylated during ageing and sites that are hypermethylated in colon cancer (23). Recently, Cedar and colleagues have proposed a model in which the accumulation of methylation events that occur during ageing makes cells more vulnerable to tumour-initiating mutations (20). A direct comparison between ageing-associated methylation events and cancer-associated methylation events may help elucidate the mechanisms behind these two processes.
In our study, we wanted to better describe the patterns of differential DNA methylation that are established in cancer and determine how much these patterns differed between cancer types. We first determined which genes were affected by differential methylation in cancer, but only a small number of these genes had been previously linked to carcinogenesis. We then wondered what could drive methylation in cancer rather than selection for changing the activity of cancer genes. To address this question, we compared regions that became differentially methylated in cancer to regions that became differentially methylated during ageing in the corresponding normal tissue (Figure 1). In addition, we compared tissues between each other with respect to their changes in cancer and in ageing. To our surprise, most changes in methylation that we observed in ageing did not overlap with those in cancer for most of the tissues we tested. Nevertheless, by accounting for ageing-related methylation changes, we were able to find several structural features of the chromatin that were either specifically or more strongly associated with cancer-related methylation changes. Specifically, the loss of methylation in late-replicating, lamina-associated domains was more prominent in cancer and was associated with cell division rates. We ruled out the possibility of our observations being the result of a population-level effect only. However, how exactly replication timing and genome structural rearrangements in cancer each contribute to the observed hypomethylation remains to be determined. Our findings provide insights into tissue-specific methylation in cancer and provide another link between the intrinsic nature of different cell types and their propensity for carcinogenesis.
Figure 1.

Generation of linear regression models of ageing and cancer. Publicly available data generated on methylation arrays is used to create two types of linear regression models. In the ageing models, the change of methylation levels with respect to age is studied. In the cancer models, the change of methylation levels in tumours versus patient-matched normal tissue is studied. Models are generated for every probe on the array. The results from the models are then compared between different tissues and between ageing and cancer within the same tissue.
MATERIALS AND METHODS
Methylation data collection
We downloaded TCGA level 3 data generated using the Illumina Infinium HumanMethylation450 BeadChip array from the FireBrowse portal (March 2017). The cohort list consisted of bladder urothelial carcinoma (BLCA) (24), breast invasive carcinoma (BRCA) (25), colon adenocarcinoma (COAD) (26), esophageal carcinoma (ESCA) (27), glioma (GBMLGG) (28,29), head and neck squamous cell carcinoma (HNSC) (30), the pan-kidney cohort (KIPAN) (31–33) liver hepatocellular carcinoma (LIHC) (34), lung adenocarcinoma (LUAD) (35), lung squamous cell carcinoma (LUSC) (36), pancreatic adenocarcinoma (PAAD) (37), prostate adenocarcinoma (PRAD) (38), skin cutaneous melanoma (SKCM) (39), thyroid carcinoma (THCA) (40), and uterine corpus endometrial carcinoma (UCEC) (41).
To increase the number of samples from normal tissue, we downloaded additional datasets from the Gene Expression Omnibus. These were GSE32146 (colon) (42), GSE40360 (brain, frontal lobe) (43), GSE49149 (pancreas) (44), GSE51954 (skin) (45), GSE61107 (brain, frontal cortex) (46), GSE77954 (colon) (47), GSE88890 (brain, cortex) (48), GSE89702 (brain, cerebellum) (49), GSE89703 (brain, hippocampus) (49), GSE89705 (brain, striatum) (49), GSE90124 (skin) (50).
Finally, we considered other phenotypes that share similar features with cancer. To look at phenotypes which display abnormalities in nuclear envelope structure, we downloaded data from senescent cells (GSE56719, GSE69046 (51)) and patients with progeria (GSE42865 (52)). To study fast-dividing cells during normal human development, we downloaded data from foetal tissue (GSE31848 (53)).
Methylation data preparation
From all downloaded data, we removed probes that were defined by TCGA as aligning to multiple sites in the genome, overlapping with common SNPs within 10 bp from the CpG site, or being located within 15 bp from a repetitive element. In addition, we removed probes that were shown to cross-hybridize to sex chromosomes (54). If required, methylation values were converted to beta-values using the lumi package (55). If applicable, beta-values were corrected for potential batch effects using the sva package (56). We did not observe major differences in the final results for the gene-level linear models that depended on whether the batch effect correction was performed before or after calculating the methylation per gene.
Before proceeding with further analyses, we excluded the possibility of copy number aberrations confounding the beta-values obtained from tumour samples (57). We downloaded TCGA level 3 data generated on the Genome-Wide Human SNP Array 6.0 from the FireBrowse portal (March 2017). In patients who had both copy number aberration and methylation data available, we calculated the correlations between the copy number aberration signal and the difference in methylation between cancer and adjacent normal tissue. We were unable to find a consistent pattern across patients and tissues. Thus, we believe that our beta-values were not influenced by copy number aberrations that occur during cancer.
Linear regression model creation
Probes were annotated using the IlluminaHumanMethylation450kanno.ilmn12.hg19 package (58). Four different types of the gene level linear regression models were generated using probes located in different regions of the genome, namely in the first exon, in the 5′ UTR, within 200 base pairs from the gene's transcription start site (TSS200), and within 1500 bp from the gene's transcription start site (TSS1500). If multiple probes mapped to a single gene, the mean beta-value of these probes was used as the beta-value of the gene. Gene level linear regression models were generated for 14 588 genes (first exon), 13 463 genes (5′ UTR), 16 545 genes (TSS200) or 19 715 genes (TSS1500). Probe level linear regression models were generated for 378 471 probes.
For the cancer linear regression models, we determined the change in beta-value in the tumour when compared to normal tissue. In cohorts where at least 15 samples from patient-matched adjacent normal tissue were available, the patient identification number was included as a variable in the models. For the ageing linear regression models, we determined the change in beta-value as a function of age. Only samples from normal tissue were used. To ensure linearity of age-related methylation changes, ages were transformed as defined previously (22):
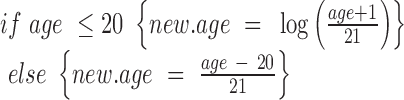 |
The total number of samples used in the cancer and ageing linear regression models can be found in Table 1. The complete list of samples used can be found in Supplementary Table S1.
Table 1.
Number of samples used in generated linear models for extended set of tissues (asterisk indicates tissues with <25 samples from normal tissue available on the TCGA).
| Cohort | Ageing | Cancer |
|---|---|---|
| BLCA* | 21 | 434 |
| BRCA | 89 | 162 |
| COAD | 58 | 96 |
| ESCA* | 16 | 202 |
| GBMLGG* | 122 | 685 |
| HNSC | 50 | 96 |
| KIPAN | 205 | 400 |
| LIHC | 48 | 92 |
| LUAD | 26 | 58 |
| LUSC | 39 | 78 |
| PAAD* | 39 | 388 |
| PRAD | 46 | 94 |
| SKCM* | 357 | 475 |
| THCA | 56 | 110 |
| UCEC | 41 | 64 |
For selecting differentially methylated genes in ageing, we applied a cut-off of 0.05 on the P-value adjusted for false discovery rate. For selecting differentially methylated genes in cancer, we used a cut-off of 0.05 on the P-value adjusted for false discovery rate and an additional cut-off of 0.1 on the absolute slope. The additional slope cut-off was applied as otherwise more than half of the genes in several cancer types were designated as differentially methylated, making functional interpretation of the results difficult.
For the senescence linear regression model, methylation in cells defined to be in deep senescence was compared to methylation in cells defined to be from early passages. The original data were generated in two separate experiments. We included the experiment number as an additional variable in the linear model. For the progeria linear regression model, methylation in patients with premature ageing syndromes (Hutchinson-Gilford Progeria or Werner syndrome) was compared to methylation in lymphoblastoid cell lines. For the normal human development linear regression model, methylation in foetal tissues was compared to methylation in the corresponding adult tissues. Due to the small number of samples, linear regression analysis was performed using the package limma (59).
Correlation with expression data
We downloaded TCGA level 3 normalized RNA-Seq data for 10 of the previously mentioned cohorts (BRCA, COAD, HNSC, KIPAN, LIHC, LUAD, LUSC, PRAD, THCA, UCEC) from the FireBrowse portal (May 2017). RSEM values calculated per gene were logarithmically transformed. For every patient, the difference between expression values in cancer samples and their matched normal samples was calculated. Similarly, the difference between beta-values in cancer samples and their matched normal samples was calculated. For the correlations, we considered only genes that were significantly affected by methylation changes in cancer.
Analysis of paralogs
The table containing human paralogs was obtained from Ensembl via BioMart (May 2017) (60). For every gene, we counted the number of paralog pairs it occurred in. A Mann–Whitney U test was used to compare average methylation values.
Analysis of tumour suppressors and oncogenes
The list of tumour suppressor genes and oncogenes was obtained from the Cancer Gene Census in the COSMIC database (July 2017) (61). We only considered genes labelled as tsg or tsg/fusion for tumour suppressors and oncogene or oncogene/fusion for oncogenes. Genes labelled as both tumour suppressors and oncogenes were not considered.
Genomic region categories
We defined genomic region categories as the position of a probe with respect to annotated genes. Probes were assigned the following categories: within 1500 base pairs from the transcription start site (TSS1500), within 200 base pairs from the transcription start site (TSS200), 5′ untranslated region (5′ UTR), first exon (1stExon), gene body (Body), 3′ untranslated region (3′ UTR), enhancer and intergenic. We considered separately the 6605 probes located in regions defined as Human Enhancers by the FANTOM5 Enhancer Atlas (62) because enhancers have previously been shown to be targeted by aberrant methylation in cancer in a manner different from other intergenic regions (63,64).
Chromatin feature mapping
Data on histone marks and chromatin states were downloaded from the NIH Roadmap Epigenomics Mapping Consortium (April 2017) (65). The mapping between TCGA cohorts and Roadmap Epigenomics tissues is provided in Table 2. To assign chromatin states to probes, we downloaded the expanded 18-state chromHMM model and mapped probes based on their locations in the genome (66). To assign histone marks to probes, we downloaded the genome-wide signal coverage tracks for all marks that were available for each tissue. We calculated the average –log10(P-value) of the signal over a window of 300 bp surrounding each probe. As advised by the data source, a threshold of 2 was used to assign a histone mark as present for a specific probe. For kidney, two annotation files were available for each histone mark. Therefore, we assigned histone marks as present when at least one of the signals was higher than the threshold.
Table 2.
Mapping between cohorts in TCGA, tissues in the NIH Roadmap Epigenomics Mapping Consortium, and tissues in GTEx
| TCGA | Roadmap Epigenomics Consortium | GTEx |
|---|---|---|
| BLCA | Bladder | |
| BRCA | E028 – breast variant human mammary epithelial cells (vHMEC) | Breast – Mammary Tissue |
| COAD | E075 – colonic mucosa | Colon – Transverse |
| ESCA | Esophagus – Mucosa | |
| GBMLGG | Brain – Cortex | |
| HNSC | Minor Salivary Gland | |
| KIPAN | Adult kidney (unconsolidated epigenome) | Kidney – Cortex |
| LIHC | E066 – liver | Liver |
| LUAD | E096 – lung | Lung |
| LUSC | Lung | |
| PAAD | Pancreas | |
| PRAD | Prostate | |
| SKCM | Skin – Sun Exposed (Lower Leg) | |
| THCA | Thyroid | |
| UCEC | Uterus |
Definition of lamina-associated domains
For lamina-associated domains, we used the genomic coordinates defined for the Tig-3, human embryonic stem cell (HESC), and HT1080 cell lines (67,68). In HT1080, two experiments were performed using Lamin A and Lamin B1 for mapping lamina-associated domains. These are further referred to as HT1080A and HT1080B. Coordinates were translated to the hg19 genome build using liftOver (69). Regions that overlapped between all four definitions were labelled as constitutive LADs.
Replication timing probe assignment
Percentage-normalized signal RepliSeq data from the dermal fibroblast cell line BJ (originally generated in (70)) were downloaded from the UCSC Genome Browser. For every stage of the cell cycle, we determined the average signal over a window of 100 bp surrounding the probes. Probes were assigned to a specific stage of the cell cycle either when the determined signal exceeded 30 or when the determined signal was higher than that from the other stages of the cell cycle. When using RepliSeq data from BG02es, IMR90, MCF7 and HepG2, we obtained a similar assignment, with over 90% of probes being assigned to the same stage of the cell cycle. As a result, we proceeded with only the BJ cell line for the enrichment analysis.
Enrichment analysis
For the enrichment analysis, probes were sorted by slope and the 75700 most hypermethylated or most hypomethylated probes (∼20% of all probes) were selected. The cut-off of 20% was chosen as it resulted in moderate effect sizes that were more consistent across different cohorts than those obtained using lower thresholds. Probes were not selected based on a P-value cut-off because this occasionally resulted in small numbers of probes overlapping with certain chromatin features, inflating the resulting odds ratios. Fisher's exact test was performed to determine whether these probes were enriched in a specific category when compared to the rest of the probes on the array. To gain better insight into non-physiological patterns of methylation, the probes affected in cancer were directly compared to probes affected in ageing.
Mitotic index
The mitotic index reflects the fraction of dividing cells in a population. Previously, it was shown that the mitotic index is proportional to the average mRNA expression over a set of 9 genes: CDKN3, ILF2, KDELR2, RFC4, TOP2A, MCM3, KPNA2, CKS2 and CDC2 (71). For calculating the mitotic index in cancer, we used RSEM expression values from cancer samples in the TCGA. For calculating the mitotic index in normal tissue, we used RPKM expression values from GTEx (72). The matching between TCGA cohorts and tissues is provided in Table 2. To make the resulting mitotic indexes comparable, we created a linear regression model that described the association between GTEx and TCGA expression values using all expression data from the two databases. We then transformed the normal tissue mitotic indexes using the slope and intercept obtained from the linear regression model:
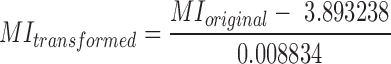 |
To calculate the correlations with the results from the LAD enrichment analysis, we took the log10 of the mitotic indexes.
DNA methylation and hypoxia
Because changes in DNA methylation in cancer have previously been linked to hypoxia (73), we wanted to exclude that our observations could be confounded by the degree of hypoxia in the tumour. Therefore, we calculated a hypoxia score for each tumour type by averaging over the TCGA expression values of 26 hypoxia marker genes: ALDOA, ANGPTL4, ANLN, BNC1, C20orf20, CA9, CDKN3, COL4A6, DCBLD1, ENO1, FAM83B, FOSL1, GNAI1, HIG2, KCTD11, KRT17, LDHA, MPRS17, P4HA1, PGAM1, PGK1, SDC1, SLC16A1, SLC2A1, TPI1 and VEGFA (74). We next tested whether the hypoxia score would correlate with the enrichment of hypomethylation in LADs and did not detect a significant correlation (r = 0.33, r2 = 0.11, P-value = 0.22; Pearson correlation).
Validation with bisulphite sequencing
To validate the results obtained using array data, we downloaded bisulphite sequencing data for three patients from whom an adjacent normal tissue sample was available—TCGA-A7-A0CE (BRCA), TCGA-AA-3518 (COAD), TCGA-AX-A1CI (UCEC). As there was only one sample per cohort, the difference in methylation between the tumour and the normal sample for each measured CpG site was used instead of the output of the linear regression model. For the enrichment analysis, 20% of the measured CpG sites were used.
Model of methylation transfer during cell division
The observed hypomethylation could be a result of a population level effect in which dividing cells contribute to low methylation values because of a delay in maintenance methylation on their newly-synthesized DNA strands. We therefore created a model of methylation information transfer based on the most conservative assumptions we could make in favour of this explanation. Our simple model did not account for possible technical variation in the signal measured for different probes, however, it would lead to the highest possible expected values of hypomethylation enrichment. As a proof by contradiction, obtaining a higher value of hypomethylation enrichment from experimental data would indicate that the hypomethylation observed in this specific case is unlikely to be the result of a population level effect only. We defined the following rules for our model:
In non-dividing cells, levels of methylation are constant.
In dividing cells, methylation in regions that are methylated early is transferred to the new strand immediately upon division. Therefore, these regions retain 100% of their initial methylation.
In dividing cells, methylation in regions that are methylated late is transferred to the new strand in the last possible moment of division. Therefore, these regions retain 50% of their initial methylation.
Let L be the fraction of the probes located in late methylated regions. Let U be the fraction of unmethylated probes. For a non-dividing cell, the contingency table would therefore be:
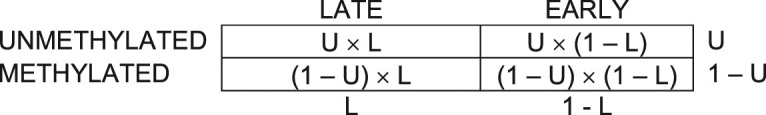
For a dividing cell, the contingency table would therefore be:
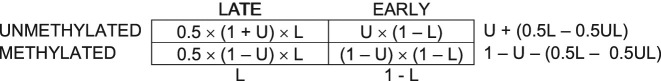
Let D be the fraction of cells in the population that are currently undergoing division. Both dividing and non-dividing cells contribute to the final contingency table proportionally to their fraction:
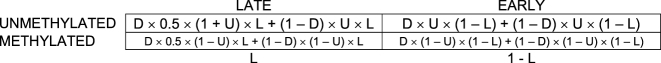
Which simplifies to:
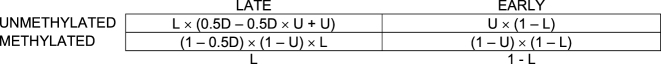
The resulting expected odds ratio would therefore be:
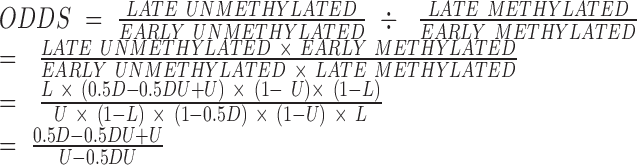 |
RESULTS
Differential methylation in cancer correlates weakly with expression and affects few known cancer genes
DNA methylation has been initially believed to contribute to cancer development through modifying the expression of specific cancer driver genes (9,75). To identify differentially methylated genes, we generated a linear regression model for each gene using the average level of methylation from all probes located either in its first exon, or its 5′ UTR, or its TSS200 region, or its TSS1500 region. Linear regression models were generated for a collection of 10 tissues that had at least 25 samples from normal tissue in their TCGA cohorts, namely breast (BRCA), colon (COAD), head and neck squamous cells (HNSC), kidney (KIPAN), liver (LIHC), lung (LUAD), lung squamous cells (LUSC), prostate (PRAD), thyroid (THCA) and uterus (UCEC). We noticed that the number of genes significantly affected by hypermethylation or hypomethylation (P-value adjusted for false discovery rate < 0.05, absolute slope > 0.1) differed between tissues (Figure 2A, Supplementary Table S2).
Figure 2.

Differential methylation of gene TSS1500 regions in cancer. (A) Total number of differentially methylated genes, number of differentially methylated tumour suppressors (TSGs), and number of differentially methylated oncogenes in every tissue. (B) Comparison of all linear regression model slopes between genes without paralogs and genes with one to five paralogs. Asterisks indicate significance (Mann–Whitney U test): *P-value < 0.05, **P-value < 0.01, ***P-value < 0.001. (C) Percentage of previously annotated 139 tumour suppressors (TSGs) and 175 oncogenes affected by differential methylation.
We then asked whether the differential methylation of these genes was complemented by changes in their expression levels. We calculated differential expression between cancer and normal tissues based on RNA-Seq data from the corresponding patients. Differentially hypermethylated genes showed a tendency for lower expression (Supplementary Figure S1A). The negative correlation between differential hypermethylation and differential expression was significant in all tissues (P-values < 10−9; Pearson correlation). By contrast, differentially hypomethylated genes showed no consistent pattern of expression across tissues. The correlations between differential hypomethylation and differential expression were significant in all tissues (P-values < 0.05) excluding head and neck squamous cells in the TSS1500 region (Supplementary Figure S1A). All correlations between differential expression and differential hypo and hypermethylation were weak, most absolute correlation coefficients not exceeding 0.20 and only three of the absolute correlation coefficients exceeding 0.30.
Having observed that hypermethylation is associated with lower gene expression in a weak but consistent manner across tissues, we asked whether we can identify traces of selection acting on methylation during cancer development. To answer this question, we compared TSS1500 region methylation levels in genes with and without paralogs. Paralogs provide functional redundancy, and less genes with paralogs were shown to be essential than genes without paralogs (76,77). We thus reasoned that genes with paralogs would be under weaker selection pressure against high methylation levels because genes from the same family could perform their function if expression was lost. Indeed, we observed a lower extent of hypermethylation in genes without paralogs than in genes with up to five paralogs. This difference was significant in all tissues (P-values < 0.016; Mann–Whitney U test) excluding uterus (Figure 2B). This finding suggested that gain of methylation is constrained in the absence of functional redundancy. This was in agreement with our previous observation that genes without paralogs are more strongly protected from damaging point mutations during cancer progression (78). By contrast, genes without paralogs were generally more hypomethylated than genes with a few paralogs (Figure 2B). This association was statistically significant for all tissues aside from breast, kidney and thyroid (P-values < 0.022; Mann–Whitney U test), although the interpretation of this association with respect to functional selection is difficult.
Finally, we determined to what extent do these methylation events target cancer genes—tumour suppressors and oncogenes that were previously defined based on their point mutation profile. Differentially methylated cancer genes accounted for a minority of methylation events that occur during cancer development (Figure 2A, Supplementary Table S2). The majority of tumour suppressors and oncogenes were not differentially methylated (Figure 2C, Supplementary Figure S1B). Affected tumour suppressors did not display a preference for hypermethylation or hypomethylation (P-value ≥ 0.08; t-test), but affected oncogenes were preferentially hypermethylated (P-values < 0.0006; t-test). We concluded that differential methylation can target cancer genes, but these gene methylation events account for a small fraction of differential methylation that occurs during cancer development. We therefore aimed to better understand which other genes and genomic regions become differentially methylated during cancer development and what could be their contribution to carcinogenesis.
Tissues differ in their overlap of methylation events that occur in cancer and ageing
DNA methylation events in ageing have previously been suggested to precede cancer development (20,23). We therefore asked whether the methylation patterns we observed in cancer overlapped with methylation patterns established during ageing. To answer this question, we generated linear models of ageing using the average level of TSS1500 region methylation in normal tissue. We then compared the overlap between differentially methylated genes in ageing (P-value adjusted for false discovery rate < 0.05) and cancer (P-value adjusted for false discovery rate < 0.05, absolute slope > 0.1) in each tissue (Figure 3A). In four of the tissues (colon, kidney, liver and thyroid), the overlap between the two gene sets was significantly higher than expected by chance.
Figure 3.

Differences and similarities of differential methylation across the whole genome in ageing and cancer in different tissues. (A) Overlap of genes that are differentially methylated in their TSS1500 region in ageing (green) and cancer (purple). P-value indicates significance of the overlap when compared to the overlap between 1000 random gene selections of the same size. (B) Within tissue correlations of slopes output from the probe-level linear regression models of ageing and cancer. All correlations are significant excluding LIHC (Pearson correlation; P-value: 0.34). (C) Inter-tissue correlations of slopes output by the linear regression model of ageing. All correlations are significant (P-values < 10−5; Pearson correlation). (D) Inter-tissue correlations of slopes output by the linear regression model of cancer. All correlations are significant (P-values = 0; Pearson correlation).
We next expanded our focus from methylation in the TSS1500 region of a gene to methylation in the whole genome by generating new linear regression models for each probe on the array. We then looked at correlations between slopes from all ageing and all cancer linear regression models to quantify the overall similarity of DNA methylation trajectories in these two processes for each tissue (Figure 3B). All correlations were significant (Pearson correlation; P-values < 10−226) excluding liver (Pearson correlation; P-value = 0.34). Only colon displayed a high correlation between the cancer and ageing models (0.57). Several tissues showed intermediate correlations between 0.30 and 0.40 (head and neck squamous cells, kidney, and lung squamous cells). Other tissues either displayed much lower or no correlation between the two groups of events. In thyroid and liver, the discrepancy between gene-level and probe-level overlap between cancer and ageing could have been the result of not accounting for direction of differential methylation in the gene-level test. Our findings suggest that in most tissues methylation changes that occur during ageing do not strongly overlap with those that occur during cancer development.
Having observed that some tissues had more similar DNA methylation changes in ageing and cancer than other tissues, we next wanted to separately compare these changes in ageing and in cancer between tissues. We determined the overlap in all ageing-associated methylation events across all tissues and all cancer-associated methylation events across all tissues in our collection. No consistent pattern appeared when comparing ageing-associated events in different tissues, although all correlations were significant (P-values < 10−5; Pearson correlation) (Figure 3C). All cancer-associated events, however, significantly positively correlated across tissues (P-values = 0; Pearson correlation) (Figure 3D). Cancer-associated events also generally correlated between patients within the same cohort (Supplementary Figure S2). Patients from the THCA cohort displayed less concordance than those from other cohorts, which could explain the low numbers of differentially methylated genes observed in this cohort. Taken together, the results suggested a cancer-specific methylation pattern that was common for multiple tissues.
Hypermethylation is associated with regulatory regions and hypomethylation is associated with structural features of the chromatin
We next asked if there are any particular genomic and epigenomic features associated with differential methylation in cancer versus ageing to better understand the surprising differences that we observed between cancer and ageing. First, we annotated probes with respect to their location relative to genes. Probes were divided into the following categories: within 1500 base pairs from the transcription start site (TSS1500), within 200 base pairs from the transcription start site (TSS200), 5′ untranslated region (5′ UTR), first exon (1stExon), gene body (Body), 3′ untranslated region (3′ UTR), intergenic, and enhancers, which we separated from other intergenic regions due to their potential effect on gene expression. We then compared the enrichment of hypermethylated or hypomethylated probes in each of these categories between cancer and ageing. Although there was no consistent pattern across all cohorts, hypermethylation in cancer was significantly enriched in enhancer regions for all cohorts excluding COAD and THCA, in the 5′ untranslated region in five cohorts, and in the first exon in five cohorts (Figure 4A, for all P-values see Supplementary Table S3). Hypomethylation in cancer was predominantly enriched in intergenic regions (Figure 4B, for all P-values see Supplementary Table S3).
Figure 4.

Association of differential methylation to genomic functional categories and chromatin features. In all graphs, the y-axis depicts the log2 of the enrichment of differentially methylated probes in cancer versus that in ageing. (A) Association between differential hypermethylation and genomic functional categories. (B) Association between differential hypomethylation and genomic functional categories. (C) Association between differential hypermethylation and chromatin features. RoadMap Epigenomics data were not available for H3K9ac in BRCA and LUAD, H3K27ac in BRCA, and H3K27me3 in KIPAN. (D) Association between differential hypomethylation and chromatin features. RoadMap Epigenomics data were not available for H3K9ac in BRCA and LUAD, H3K27ac in BRCA, and H3K27me3 in KIPAN. Chromatin states: 1_TssA – active transcription start site, 2_TssFlnk – region flanking transcription start site, 3_TssFlnkU – region flanking transcription start site upstream, 4_TssFlnkD – region flanking transcription start site downstream, 5_Tx – strong transcription, 6_TxWk – weak transcription, 7_EnhG1 – genic enhancer type 1, 8_EnhG2 – genic enhancer type 2, 9_EnhA1 – active enhancer type 1, 10_EnhA2 – active enhancer type 2, 11_EnhWk – weak enhancer, 12_ZNF/Rpts – zinc finger genes and repeats, 13_Het – heterochromatin, 14_TssBiv – bivalent/poised transcription start site, 15_EnhBiv – bivalent enhancer, 16_ReprPC – repressed Polycomb, 17_ReprPCWk – weak repressed PolyComb, 18_Quies – quiescent chromatin (no histone marks are present).
Because only a small fraction of methylation events in cancer were associated with differential gene expression, we asked whether the observed methylation patterns could be influenced by factors other than functional selection on the gene level. To answer our question, we looked for associations between hypermethylation, hypomethylation, and various features of the structural and spatial organization of chromatin. We obtained data on chromatin state annotations for three cohorts (COAD, LIHC, and LUAD) and histone mark locations for five of the cohorts (BRCA, COAD, KIPAN, LIHC and LUAD). In addition, we considered lamina-associated domains—large stretches of transcriptionally repressed and generally hypermethylated chromatin that are located close to the inner face of the nuclear envelope—the nuclear lamina (67).
Cancer-associated hypermethylation predominantly occurred in regulatory regions that could potentially affect gene expression (Figure 4C, for all P-values see Supplementary Table S4). In line with our previous observations, regions flanking the transcription start site were more hypermethylated in cancer than in ageing. Multiple types of enhancers were strongly hypermethylated in cancer. To determine whether we would also observe differential expression of genes regulated by differentially methylated enhancers, we mapped probes indicated to be in enhancers to transcription starts sites using the FANTOM5 definition. We could not find a correlation between the methylation of these probes and expression of the corresponding genes. Finally, we observed an association between cancer-associated hypermethylation, H3K27me3 and repressed Polycomb.
By contrast, cancer-associated hypomethylation predominantly occurred in transcriptionally inactive stretches of the chromatin (Figure 4D, for all P-values see Supplementary Table S4). Specifically, we observed stronger hypomethylation in cancer for repetitive regions, heterochromatin and its associated histone mark H3K9me3, and LADs, suggesting hypomethylation is at least partially associated with chromatin or nuclear structure. In conclusion, hypermethylation and hypomethylation were associated with different features of chromatin organization and might therefore be linked to cancer development in different ways.
Increase of hypomethylation in LADs is linked to the late replication of these regions and faster cell division rates
We further investigated the possible contribution of chromatin structure to hypomethylation in cancer by focusing on LADs. We compared the level of hypomethylation in LADs determined for multiple cell lines and constitutive LADs, which we defined as genomic regions that overlapped between LADs in all the cell lines. The enrichment of hypomethylation in constitutive LADs was higher in cancer when compared to that in ageing (Figure 5A). LADs determined for individual cell lines displayed the same pattern (Supplementary Figure S3A). In three cancer patients (one from the BRCA, one from the COAD, and one from the UCEC cohort) from whom bisulphite sequencing data were available, we also confirmed enrichment of hypomethylation in LADs (Supplementary Figure S3B).
Figure 5.

Differential methylation in lamina-associated domains. (A) Enrichment of hypomethylated probes in constitutive lamina-associated domains. The log2 of the odds ratio from Fisher's exact test is plotted for various conditions. Purple bars correspond to output from the linear regression models of cancer. Green corresponds to output from the linear regression models of ageing. Grey corresponds to probes output from the linear regression model of senescence and progeria. (B) Enrichment of hypomethylated probes depending on the replication timing of genomic regions in cancer (top) and ageing (bottom). The log2 of the odds ratio from Fisher's exact test is plotted per cell cycle stage. (C) Enrichment of hypomethylation in constitutive LADs (log2 of the odds ratio) in cancer versus estimated mitotic index in tumour samples (r = 0.66, P-value = 0.007; Pearson correlation). (D) Enrichment of hypomethylation in constitutive LADs (log2 of the odds ratio) in ageing versus estimated mitotic index in normal tissue (r = 0.76, P-value = 0.001; Pearson correlation). Correspondence between normal tissue labels and cohorts in the TCGA are the following: BLADDER – BLCA, BRAIN – GBMLGG, BREAST -BRCA, COLON – COAD, ESOPHAGUS – ESCA, HEAD - HNSC, KIDNEY – KIPAN, LIVER – LIHC, LUNG (LUAD) – LUAD, LUNG (LUSC) – LUSC, PANCREAS – PAAD, PROSTATE – PRAD, SKIN – SKCM, THYROID – THCA, UTERUS – UCEC.
Because LADs have been shown to overlap with late-replicating regions (14,70), we asked whether we would observe an association between hypomethylation and replication timing. To answer this question, we used RepliSeq data to sort probes by the cell cycle stage in which their region was replicated. Between 31% and 66% of probes that were previously assigned to LADs were assigned to the S4 and G2 stages (depending on the LAD definition), with an additional 21% to 32% assigned to the S3 stage. In cancer, the enrichment of hypomethylated probes gradually increased from early- to late-replicating regions (Figure 5B, top). A similar trend was observed during ageing; however, it was weak (Figure 5B, bottom). The clear association between replication time point and degree of hypomethylation suggested that the temporal organization of replication contributed to the methylation pattern observed in cancer.
We next wondered whether fast-dividing cells that undergo more rounds of replication would therefore be more hypomethylated in LADs. To test whether cell division rates and loss of methylation in LADs were associated, we approximated the subpopulation of dividing cells in our tissues using an expression-based estimate of the mitotic index. To have a broader distribution of mitotic indices, we generated linear regression models for five additional tissues where at least 15 samples from normal tissue were available. Namely, these were bladder (BLCA), esophagus (ESCA), brain (GBMLGG), pancreas (PAAD), and skin (SKCM). The enrichment of hypomethylation in constitutive LADs in cancer positively correlated with the mitotic index (r = 0.66, P-value = 0.007; Pearson correlation) (Figure 5C). We next asked whether we would also observe such a correlation using data from normal tissues. Mitotic indexes calculated for normal tissues varied slightly more than those for tumours (range 2.64–3.28 versus 3.13–3.54, standard deviation 0.159 versus 0.148). The enrichment of hypomethylation in constitutive LADs during ageing correlated stronger with normal tissue mitotic indexes (r = 0.76, P-value = 0.001; Pearson correlation) (Figure 5D). As an additional confirmation, we calculated the correlation between the expression of Ki67, a well-known proliferation marker (79) and the enrichment of LAD hypomethylation in cancer and ageing. Even though this correlation was weaker and not significant in cancer (r = 0.42, P = 0.11; Pearson correlation), the correlation in ageing and normal tissues slightly increased (r = 0.78, P = 0.0006; Pearson correlation) when compared to the mitotic index. To ensure that the correlations with the mitotic index were specific to LADs, we did 1000 permutations of probes assigned to LADs and calculated the enrichment of hypomethylation and its correlation with mitotic index. Our test showed that both obtained correlations were significant—the P-value was equal to 0.023 for cancer cells and 0.002 for normal tissues (Supplementary Figure S4).
To better understand whether LAD hypomethylation would be associated with differential activity of specific genes, we asked if there were any genes that correlated in their expression with the enrichment of hypomethylation in LADs over individuals. In patients from whom methylation data was available from both the tumour and adjacent normal tissue, we calculated the difference in beta-values between the two samples. These values were used as input to calculate the enrichment of hypomethylation in LADs. We compared the calculated enrichment to the patient's absolute expression of genes in the tumour. The analysis was performed for cancer types which contained more than 10 patients with both methylation and expression data. Namely, these were: BRCA, COAD, HNSC, KIPAN, LIHC, LUAD, LUSC, PRAD and THCA. We performed a GSEA (80) to identify pathways enriched among genes whose expression was correlated with the degree of hypomethylation in LADs in patients. In multiple cancer types, we found several pathways related to cell division that were significantly enriched among the highly correlated genes (e.g. ‘protein localization to kinetochore’ in COAD, KIPAN and PRAD and ‘DNA strand elongation involved in DNA replication’ in COAD, KIPAN, PRAD, LIHC and THCA). The enrichment of both GO terms was independent of each other as no genes were annotated with both terms. Moreover, these genes were not enriched in LAD regions, excluding the possibility of LADs directly contributing to the observed expression values. We concluded that the association between the extent of LAD hypomethylation and the expression of genes participating in cell division within the same cancer type could be a proxy for variation in cell division rates.
Having observed an association between LAD hypomethylation and cell division rates, we wondered if we would also observe LAD hypomethylation in embryonic cells, which are naturally fast-dividing. To test this, we compared the level of methylation in LADs between various foetal tissues and their corresponding adult tissues (adrenal gland, brain, heart, lung, spleen, and stomach) under the assumption that foetal tissues divide faster than their adult counterparts. Excluding the adrenal gland, hypomethylation of LADs and late-replicating regions was generally stronger in foetal tissues, although the enrichment was generally weaker than that observed in cancer (Supplementary Table S5).
Mechanisms of DNA methylation loss
We could think of three scenarios to explain the loss of methylation in LADs in cancer (Figure 6A). In the lamina damage scenario, LADs no longer become associated with the nuclear lamina following dissolution of the nuclear envelope and large structural rearrangements of the genome (Figure 6A, left). Because the nuclear envelope provides an environment for the maintenance of the hypermethylated state of the chromatin (81), the disruption of LAD connection to the nuclear envelope leads to their eventual hypomethylation. In the replication timing effect scenario, methylation maintenance in the late-replicating regions of the genome is less efficient than that in early-replicating regions of the genome (82). For example, this could be due to lack of recruitment of the required methylation maintenance machinery (83). As the cell undergoes multiple divisions, late-replicating regions gradually become less methylated (Figure 6A, centre). Finally, our observed methylation levels could have been the result of a population level effect. When the number of dividing cells in a population is high, the proportion of newly-synthesized unmethylated DNA is also higher. Regions that become methylated later in the course of the cell cycle do not yet have methylation information transferred to the new strand. Under the assumption that LADs are some of these late methylated regions, the hypomethylation in LADs could be a reflection of the higher proportion of dividing cells in a population (Figure 6A, right).
Figure 6.

Mechanisms of DNA methylation loss in LADs. (A) Three proposed scenarios of methylation loss in LADs in cancer. In the lamina damage scenario, hypomethylation of LADs follows detachment of these regions of the genome of the nuclear lamina. In the replication timing effect scenario, methylation in late-replicating regions is gradually lost over the course of multiple cell divisions. In the population level effect scenario, the observed hypomethylation reflects a larger proportion of dividing cells in a population. In these dividing cells, methylation information has not been transferred to all regions of the newly-synthesized DNA strands yet. (B) Concordance of direction of differentially methylated probes across tissues. The percentage of hypermethylated or hypomethylated probes is plotted against the number of tissues in which these probes are hypermethylated or hypomethylated. Slopes used for determining hypermethylation and hypomethylation were taken from the linear regression model of cancer. (C) Pairwise tissue correlations of probes outside LADs versus those of probes inside LADs. Every point corresponds to one pairwise tissue comparison. The correlation coefficient of slopes from probes located outside of LADs is plotted against the correlation coefficient of slopes from probes located in LADs. Slopes used for calculating the correlations were taken from the linear regression model of cancer.
We aimed to rule out the last of the three scenarios by creating a mathematical model of methylation transfer based on strongly conservative estimates in favour of this scenario (see Methods, Supplementary Figure S5). As the enrichment analysis was performed using a fifth of the probes, we set the fraction of hypomethylated probes to 20% in the model. We assumed a mitotic index of 40%, which is considered an upper bound for cell division rate in several cancers (84,85). The maximum expected log2(odds ratio) that could be obtained with these parameters was ∼1.17. In eight tissues (BRCA, COAD, HNSC, LIHC, LUAD, LUSC, PRAD and UCEC), the observed LAD hypomethylation enrichment was higher for the HT1080A, HT1080B1 and constitutive LAD definitions (Figure 5A, Supplementary Figure S3). In BRCA and LIHC, this was also true for the TIG-3 LAD definition (Supplementary Figure S3). Moreover, we observed a chromosome-specific extent of methylation loss both in the array and the bisulphite sequencing data (Supplementary Figure S6). Although there was some disagreement between the two platforms, this could be the result of looking at the average hypomethylation over the whole cohort versus hypomethylation in an individual patient. Taken together, our observations suggest that, at least for certain cancer types, the observed hypomethylation of LADs could not be the result of only a population level effect.
We then directly compared hypomethylation in LADs and in late replicating regions to determine whether the lamina damage or the replication timing scenario would be more likely. We pooled probes assigned to the S4 and G2 stages in the RepliSeq data and treated these as late-replicating region probes. The enrichment of hypomethylation in late-replicating regions in cancer was comparable to the values found for different definitions of LADs (Supplementary Figure S7A). Probes that overlapped between LADs and late-replicating regions displayed the most hypomethylation, while probes located exclusively in LADs or late-replicating regions displayed less but still significant levels of hypomethylation (Supplementary Figure S7B).
To further investigate the association between loss of methylation and structural reorganisation of the genome with respect to the nuclear lamina, we looked at two other phenotypes that were characterized by such rearrangements. Senescence, the state of irreversible cell cycle arrest, has been previously shown to be accompanied by degradation of lamin B1 and loss of nuclear envelope integrity (86–88). Progeria, a premature aging disorder, is known to be caused by a mutation in lamin A. As a result, a truncated version of lamin A is expressed, which disrupts proper connection of the genome to the nuclear lamina (89). In both phenotypes, we observed enrichment of hypomethylation in LADs, although it was not as strong as in cancer (Figure 5A, Supplementary Figure S5A). These observations appeared to be in favour of the lamina damage scenario, however, methylation of late-replicating regions in senescent cells has also been shown to be affected by the replication timing effect (83). As a result, we were unable to distinguish the two scenarios with currently available data and perhaps a combination of the two is what results in the observed hypomethylation.
Having observed an association between hypomethylation in cancer, structural reorganisation of the genome, replication timing and cell division rates, we went back to the original output from our cancer linear regression models. We observed that hypomethylated probes were more consistently affected across different tissues than hypermethylated probes (P-value = 0; Mann–Whitney U test) (Figure 6B). In addition, pairwise differential methylation correlations between different tissues in cancer were higher for probes located in constitutive LADs compared to probes located outside LADs (Figure 6C). Based on our observations, we propose that cancer-associated hypomethylation at least partially arises due to rearrangements in the chromatin structure. Moreover, considering ∼30% of the hypomethylated probes were located in LADs according to at least one LAD definition, these rearrangements contribute to the similarity across tissues and cancer types and are associated to the differences in cell division rates of healthy tissues and cancer cells.
DISCUSSION
In our study, we focused on aberrant methylation during cancer and determined methylation patterns that were shared between several cancer types. Our observations suggested that functional selection for changes in methylation of cancer genes could only be associated with a small fraction of the methylation events in cancer. We therefore studied methylation patterns established during ageing in normal tissues to try to explain aberrant methylation in cancer. After comparing changes in methylation that occur in ageing and cancer, we found a smaller overlap than we expected. To better understand the observed differences in methylation changes during ageing and cancer, we determined which chromatin features could influence the established methylation landscapes. Hypermethylation was associated with regions flanking the transcription start site, enhancers, and regions targeted by the Polycomb complex, suggesting it affects gene expression. Hypomethylation was associated with heterochromatin, repetitive regions, and LADs.
We further investigated the hypomethylation of LADs in ageing and cancer. We observed that regions that were replicated later were more hypomethylated, this effect being more pronounced in cancer than in ageing. Additionally, the extent of hypomethylation in LADs correlated with our estimate of cell division rates. Finally, genes associated with cell division were upregulated in patients displaying more hypomethylation in LADs. We excluded that this association between hypomethylation in LADs and cell division was simply the result of a higher proportion of dividing cells in a population. Taken together, our results suggested that the aberrant methylation landscape in cancer is partially the result of structural reorganisation of the genome with respect to the nuclear lamina that is enhanced by the fast division rates of cancer cells. Therefore, the functional interpretation of such DNA methylation events should be done with caution.
Although the majority of previously annotated cancer genes were not affected by differential methylation, oncogenes were significantly more hypermethylated across the tissues we tested. This finding was in contrast to our expectations. We reasoned that other factors of gene expression regulation could come into play to ensure the expression of these oncogenes in the tumour. Another possible explanation for our observations could be the mutually exclusive nature of oncogenes, with hypermethylation used as a mechanism to prevent expression of certain combinations of oncogenes.
A finding that contrasted our expectations was the low correlation between ageing-associated events and cancer-associated events. Previously, a strong association between the two has been suggested (20,23). Studies where this association was observed, however, heavily focused on methylation in colon, the tissue in which we observed the highest similarity between cancer and ageing. A study investigating the relationship between ageing and cancer in breast tissue found a selection of sites that where common between the two (90). This selection, however, contained slightly more than 10% of all differentially methylated regions in ageing. Thus, we propose that although ageing and cancer share common features in the methylation landscape, only a minority of methylation events that occur during ageing overlap with those that occur during cancer development.
We observed an overlap in methylation patterns established in cancer in multiple tissues. Tissue-independent methylation patterns of cancer have previously been observed using smaller selections of CpG sites (91–93). More recently, a larger scale study used probes of the 450k array located in the vicinity of genes and also showed high correlation in cancer methylation patterns that were common between different tissue types (94).
Our associations between differentially methylated regions in cancer and chromatin features help to better understand previous observations. The prevalence of hypermethylation in gene promoter regions and regions targeted by the Polycomb complex has also been found by others before us (95,96). In addition, multiple enhancer types have been found to be hypermethylated, including bivalent enhancers. Bivalent enhancers are frequently located upstream of developmental genes (97). Hypermethylation of these regions may be an aspect of the cell reusing developmental programs during progression towards a malignant state (98).
By contrast, we found that hypomethylation in cancer was not as strongly associated with chromatin states potentially affecting gene expression. In line with our findings, repetitive regions were previously shown to lose methylation in cancer (99,100). The same was true for heterochromatin and H3K9me3 (15), which were previously linked to elevated mutation rates and copy number aberrations in cancer (101,102). Finally, hypomethylation has been shown to affect long stretches of the genome which coincide with LADs and late-replicating regions (16,103). We have shown that this trend holds true in a variety of cancer and healthy tissues, that this trend is stronger in cancer than in ageing, and that a fraction of the differences between tissues and cancer types are related to particular cell division rates.
We have chosen to focus on the association between hypomethylation and LADs in cancer as it supported the idea of aberrant methylation being influenced by changes in chromatin structure (15). We proposed three possible scenarios by which hypomethylation in LADs could be established during cancer development. We excluded the scenario in which this hypomethylation could be the result of a higher proportion of dividing cells within a population. It has been previously shown that population effects are insufficient to explain differences in the level of methylation observed in early-replicating regions and late-replicating regions (104,105). Moreover, these differences are not pronounced in early passage cells (104), and thus appear to be the result of methylation loss accumulation over multiple cell cycles.
Our findings suggest that hypomethylation in LADs is both the result of the structural reorganisation of the genome with respect to the nuclear lamina and the coincidence of LADs with late-replicating regions. Fast dividing cells in normal embryonic development showed a stronger loss of methylation in LADs when compared to the corresponding adult tissues, suggesting that this effect was reversible. Although we hypothesize that LAD hypomethylation is the consequence of structural rearrangements of the genome and the replication timing effect, we cannot exclude the possibility of this hypomethylation acting as a facilitator for structural reorganisation of the genome and faster cell division.
We were not able to determine whether the replication timing effect or the structural rearrangements of the genome played a stronger role in LAD hypomethylation. In cells approaching senescence, DNMT1, the maintenance methyltransferase, has been shown to fail to localize at late-replicating regions (83). Similarly, a less efficient recruitment of DNMT1 to late-replicating regions combined with the many cell divisions undergone by the cancer cell could result in a major loss of methylation in these regions. However, although DNMT1 is responsible for most of the maintenance methylation, other methyltransferases assist with maintenance after recruitment to specific chromatin states (106). Thus, the observed hypomethylation could have been preceded by histone mark aberrations. In particular, the H3K9me3 mark coincides with LADs and heterochromatin (17), and investigating its distribution in cancer could provide mechanistic insight into whether methyltransferases are indeed efficiently recruited to these areas. Finally, although we propose that hypomethylation of LADs mainly occurs as the result of a passive demethylation process, there is a possibility that active demethylation mechanisms also come into play.
A drawback of our study was that we were not able to obtain tissue-specific data on late-replicating regions and LADs. Although a subset of genomic regions is constitutively late-replicating and located in lamina-associated domains, different cell types noticeably vary in their replication timing program (70) and facultative LADs exist (17). The differences in replication timing are in fact significant enough to identify cell types (107). Moreover, cancer cells display abnormalities in the replication timing program when compared to normal cells of the same type (108) and are known to undergo rearrangements in genome-nuclear lamina interactions. In our data, however, we did not observe many differences between using replication timing data from BJ, BG02es, IMR90, MCF-7 or HepG2 to assign probes to cell cycle stages. The overlap between probe assignment to different LAD definitions was lower, especially between HESCs and other cell lines. In future studies, we therefore advise simultaneous measurement of methylation, lamina-associated domains, and late-replicating regions within the same cell type to gain a better mechanistic understanding of the underlying processes.
In addition, we ruled out some but not all factors that could confound our observations. Specifically, we excluded that copy number variations contributed to the variation in methylation and that LAD hypomethylation was associated with the hypoxia levels of the tumour. However, we did not check for some other potential confounders such as tumour heterogeneity due to lack of immediate tests.
In conclusion, we associated the amount of hypomethylation in LADs to cell division both on the level of different tissues and cancer types and the level of individuals within the same cancer type. The number of stem cell divisions has previously been shown to correlate with lifetime risk of cancer (109,110). Based on this observation, the ‘bad luck’ hypothesis of cancer aetiology has been proposed. According to this hypothesis, faster dividing cells accumulate more mutations due to random errors in DNA replication, thus having a higher chance of malignant transformation. The hypothesis has been met with controversy as it reduces the effect of environmental factors on cancer development (111).
Our observations suggest that the ‘bad luck’ hypothesis has an epigenetic component to it as well. Recently, the average levels of CpG island methylation in a tissue have also been shown to correlate with the tissue's risk to develop cancer (112). In our study, we observed increased hypomethylation of LADs in faster dividing cells. Overall hypomethylation is often linked to increased genome instability (113–115). Therefore, methylation changes could also contribute to the accumulation of mutations in faster dividing cells prior to cancer development. In addition, our results suggest that cell division rates affect the degree of similarity of the methylation landscapes in healthy tissues and cancer. In fast-dividing tissues such as lung and colon, ageing-associated changes in the DNA methylome correlate more with those that occur during cancer development. However, other factors such as the availability and efficiency of DNA repair and methylation maintenance machinery could also influence this similarity, which may be why we do not observe a direct association for all tissues. Up until now, supporters and opponents of the ‘bad luck’ hypothesis have focused on the contribution of genetic factors to carcinogenesis. We would like to emphasize the importance of investigating the contribution of epigenetic factors to carcinogenesis as well.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
German Research Foundation [SCHA 1933/1-1]; European Union Seventh Framework Programme [FP7/2007-2013] under grant agreements [HEALTH-F4-2011-278568 (PRIMES)]; Spanish Ministry of Economy, Industry and Competitiveness (MEIC) [BFU2015-63571-P] and to the EMBL partnership; the European Regional Development Fund (ERDF/FEDER); Centro de Excelencia Severo Ochoa and the support of the CERCA Programme/Generalitat de Catalunya. Funding for open access charge: Spanish Ministry of Economy, Industry and Competitiveness (MEIC) [BFU2015-63571-P].
Conflict of interest statement. None declared.
REFERENCES
- 1. Baylin S.B., Jones P.A.. A decade of exploring the cancer epigenome — biological and translational implications. Nat. Rev. Cancer. 2011; 11:726–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. You J.S., Jones P.A.. Cancer Genetics and epigenetics: two sides of the same coin. Cancer Cell. 2012; 22:9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shen H., Laird P.W.. Interplay between the cancer genome and epigenome. Cell. 2013; 153:38–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Susak H., Zapata L., Friedländer M.R., Drechsel O., Ossowski S., Estivill X.. Signatures of positive selection reveal a universal role of chromatin modifiers as cancer driver genes. Sci. Rep. 2017; 7:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Suvà M.L., Riggi N., Bernstein B.E.. Epigenetic reprogramming in cancer. Science. 2013; 339:1567–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Massie C.E., Mills I.G., Lynch A.G.. The importance of DNA methylation in prostate cancer development. J. Steroid Biochem. Mol. Biol. 2017; 166:1–15. [DOI] [PubMed] [Google Scholar]
- 7. Majumdar S., Buckles E., Estrada J., Koochekpour S.. Aberrant DNA Methylation and Prostate Cancer. Curr. Genomics. 2011; 12:486–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Feinberg A.P., Tycko B.. The history of cancer epigenetics. Nat. Rev. Cancer. 2004; 4:143–153. [DOI] [PubMed] [Google Scholar]
- 9. Herman J.G., Baylin S.B.. Gene silencing in cancer in association with promoter hypermethylation. N. Engl. J. Med. 2003; 349:2042–2054. [DOI] [PubMed] [Google Scholar]
- 10. Cheishvili D., Stefanska B., Yi C., Li C.C., Yu P., Arakelian A., Tanvir I., Khan H.A., Rabbani S., Szyf M.. A common promoter hypomethylation signature in invasive breast, liver and prostate cancer cell lines reveals novel targets involved in cancer invasiveness. Oncotarget. 2015; 6:33253–33268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Upchurch G.M., Haney S.L., Opavsky R.. Aberrant promoter Hypomethylation in CLL: does it matter for disease development. Front. Oncol. 2016; 6:182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Klug M., Heinz S., Gebhard C., Schwarzfischer L., Krause S.W., Andreesen R., Rehli M.. Active DNA demethylation in human postmitotic cells correlates with activating histone modifications, but not transcription levels. Genome Biol. 2010; 11:R63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Calvanese V., Fernández A.F., Urdinguio R.G., Suárez-Alvarez B., Mangas C., Pérez-García V., Bueno C., Montes R., Ramos-Mejía V., Martínez-Camblor P. et al. . A promoter DNA demethylation landscape of human hematopoietic differentiation. Nucleic Acids Res. 2012; 40:116–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bergman Y., Cedar H.. DNA methylation dynamics in health and disease. Nat. Struct. Mol. Biol. 2013; 20:274–281. [DOI] [PubMed] [Google Scholar]
- 15. Madakashira B.P., Sadler K.C.. DNA methylation, nuclear organization, and cancer. Front. Genet. 2017; 8:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhou W., Dinh H.Q., Ramjan Z., Weisenberger D.J., Nicolet C.M., Shen H., Laird P.W., Berman B.P.. DNA methylation loss in late-replicating domains is linked to mitotic cell division. Nat. Genet. 2018; 50:591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van Steensel B., Belmont A.S.. Lamina-associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell. 2017; 169:780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schneider G., Schmidt-Supprian M., Rad R., Saur D.. Tissue-specific tumorigenesis: context matters. Nat. Rev. Cancer. 2017; 17:239–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zane L., Sharma V., Misteli T.. Common features of chromatin in aging and cancer—cause or coincidence. Trends Cell Biol. 2014; 24:686–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Klutstein M., Nejman D., Greenfield R., Cedar H.. DNA methylation in cancer and aging. Cancer Res. 2016; 76:3446–3450. [DOI] [PubMed] [Google Scholar]
- 21. Jung M., Pfeifer G.P.. Aging and DNA methylation. BMC Biol. 2015; 13:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013; 14:3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nejman D., Straussman R., Steinfeld I., Ruvolo M., Roberts D., Yakhini Z., Cedar H.. Molecular rules governing de novo methylation in cancer. Cancer Res. 2014; 74:1475–1483. [DOI] [PubMed] [Google Scholar]
- 24. The Cancer Genome Atlas Research Network Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014; 507:315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. The Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. The Cancer Genome Atlas Network Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487:330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. The Cancer Genome Atlas Research Network Integrated genomic characterization of oesophageal carcinoma. Nature. 2017; 541:169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brennan C.W., Verhaak R.G.W., McKenna A., Campos B., Noushmehr H., Salama S.R., Zheng S., Chakravarty D., Sanborn J.Z., Berman S.H. et al. . The somatic genomic landscape of glioblastoma. Cell. 2013; 155:462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. The Cancer Genome Atlas Research Network Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N. Engl. J. Med. 2015; 372:2481–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. The Cancer Genome Atlas Network Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015; 517:576–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. The Cancer Genome Atlas Research Network Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013; 499:43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Davis C.F., Ricketts C.J., Wang M., Yang L., Cherniack A.D., Shen H., Buhay C., Kang H., Kim S.C., Fahey C.C. et al. . The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell. 2014; 26:319–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. The Cancer Genome Atlas Research Network Comprehensive molecular characterization of papillary renal-cell carcinoma. N. Engl. J. Med. 2016; 374:135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. The Cancer Genome Atlas Research Network Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. 2017; 169:1327–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. The Cancer Genome Atlas Research Network Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014; 511:543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. The Cancer Genome Atlas Research Network Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012; 489:519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Raphael B.J., Hruban R.H., Aguirre A.J., Moffitt R.A., Yeh J.J., Stewart C., Robertson A.G., Cherniack A.D., Gupta M., Getz G. et al. . Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell. 2017; 32:185–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. The Cancer Genome Atlas Network The molecular taxonomy of primary prostate cancer. Cell. 2015; 163:1011–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Akbani R., Akdemir K.C., Aksoy B.A., Albert M., Ally A., Amin S.B., Arachchi H., Arora A., Auman J.T., Ayala B. et al. . Genomic classification of cutaneous melanoma. Cell. 2015; 161:1681–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Agrawal N., Akbani R., Aksoy B.A., Ally A., Arachchi H., Asa S.L., Auman J.T., Balasundaram M., Balu S., Baylin S.B. et al. . Integrated genomic characterization of papillary thyroid carcinoma. Cell. 2014; 159:676–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. The Cancer Genome Atlas Research Network Integrated genomic characterization of endometrial carcinoma. Nature. 2013; 497:67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Harris R.A., Nagy-Szakal D., Mir S.A., Frank E., Szigeti R., Kaplan J.L., Bronsky J., Opekun A., Ferry G.D., Winter H. et al. . DNA methylation-associated colonic mucosal immune and defense responses in treatment-naïve pediatric ulcerative colitis. Epigenetics. 2014; 9:1131–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Huynh J.L., Garg P., Thin T.H., Yoo S., Dutta R., Trapp B.D., Haroutunian V., Zhu J., Donovan M.J., Sharp A.J. et al. . Epigenome-wide differences in pathology-free regions of multiple sclerosis–affected brains. Nat. Neurosci. 2014; 17:121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nones K., Waddell N., Song S., Patch A.-M., Miller D., Johns A., Wu J., Kassahn K.S., Wood D., Bailey P. et al. . Genome-wide DNA methylation patterns in pancreatic ductal adenocarcinoma reveal epigenetic deregulation of SLIT-ROBO, ITGA2 and MET signaling. Int. J. Cancer. 2014; 135:1110–1118. [DOI] [PubMed] [Google Scholar]
- 45. Vandiver A.R., Irizarry R.A., Hansen K.D., Garza L.A., Runarsson A., Li X., Chien A.L., Wang T.S., Leung S.G., Kang S. et al. . Age and sun exposure-related widespread genomic blocks of hypomethylation in nonmalignant skin. Genome Biol. 2015; 16:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wockner L.F., Noble E.P., Lawford B.R., Young R.M., Morris C.P., Whitehall V.L.J., Voisey J.. Genome-wide DNA methylation analysis of human brain tissue from schizophrenia patients. Transl. Psychiatry. 2014; 4:e339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Qu X., Sandmann T., Frierson H., Fu L., Fuentes E., Walter K., Okrah K., Rumpel C., Moskaluk C., Lu S. et al. . Integrated genomic analysis of colorectal cancer progression reveals activation of EGFR through demethylation of the EREG promoter. Oncogene. 2016; 35:6403–6415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Murphy T.M., Crawford B., Dempster E.L., Hannon E., Burrage J., Turecki G., Kaminsky Z., Mill J.. Methylomic profiling of cortex samples from completed suicide cases implicates a role for PSORS1C3 in major depression and suicide. Transl. Psychiatry. 2017; 7:e989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Viana J., Hannon E., Dempster E., Pidsley R., Macdonald R., Knox O., Spiers H., Troakes C., Al-Saraj S., Turecki G. et al. . Schizophrenia-associated methylomic variation: molecular signatures of disease and polygenic risk burden across multiple brain regions. Hum. Mol. Genet. 2017; 26:210–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Roos L., Sandling J.K., Bell C.G., Glass D., Mangino M., Spector T.D., Deloukas P., Bataille V., Bell J.T.. Higher nevus count exhibits a distinct DNA methylation signature in healthy human skin: implications for melanoma. J. Invest. Dermatol. 2017; 137:910–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lowe R., Overhoff M.G., Ramagopalan S.V., Garbe J.C., Koh J., Stampfer M.R., Beach D.H., Rakyan V.K., Bishop C.L.. The senescent methylome and its relationship with cancer, ageing and germline genetic variation in humans. Genome Biol. 2015; 16:194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Heyn H., Moran S., Esteller M.. Aberrant DNA methylation profiles in the premature aging disorders Hutchinson-Gilford Progeria and Werner syndrome. Epigenetics. 2013; 8:28–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nazor K.L., Altun G., Lynch C., Tran H., Harness J.V., Slavin I., Garitaonandia I., Müller F.-J., Wang Y.-C., Boscolo F.S. et al. . Recurrent variations in DNA methylation in human pluripotent stem cells and their differentiated derivatives. Cell Stem Cell. 2012; 10:620–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chen Y., Lemire M., Choufani S., Butcher D.T., Grafodatskaya D., Zanke B.W., Gallinger S., Hudson T.J., Weksberg R.. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics. 2013; 8:203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Du P., Kibbe W.A., Lin S.M.. lumi: a pipeline for processing Illumina microarray. Bioinformatics. 2008; 24:1547–1548. [DOI] [PubMed] [Google Scholar]
- 56. Leek J.T., Johnson E.W., Parker H.S., Fertig E.J., Jaffe A.E., Storey J.D.. sva: Surrogate Variable Analysis. 2012. [Google Scholar]
- 57. Robinson M.D., Stirzaker C., Statham A.L., Coolen M.W., Song J.Z., Nair S.S., Strbenac D., Speed T.P., Clark S.J.. Evaluation of affinity-based genome-wide DNA methylation data: Effects of CpG density, amplification bias, and copy number variation. Genome Res. 2010; 20:1719–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hansen K.D. IlluminaHumanMethylation450kanno. 2015; ilmn12. hg19: annotation for illumina's 450k methylation arrays. [Google Scholar]
- 59. Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K.. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015; 43:e47–e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Smedley D., Haider S., Durinck S., Pandini L., Provero P., Allen J., Arnaiz O., Awedh M.H., Baldock R., Barbiera G. et al. . The BioMart community portal: an innovative alternative to large, centralized data repositories. Nucleic Acids Res. 2015; 43:W589–W598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Forbes S.A., Beare D., Boutselakis H., Bamford S., Bindal N., Tate J., Cole C.G., Ward S., Dawson E., Ponting L. et al. . COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017; 45:D777–D783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Andersson R., Gebhard C., Miguel-Escalada I., Hoof I., Bornholdt J., Boyd M., Chen Y., Zhao X., Schmidl C., Suzuki T. et al. . An atlas of active enhancers across human cell types and tissues. Nature. 2014; 507:455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Aran D., Sabato S., Hellman A.. DNA methylation of distal regulatory sites characterizes dysregulation of cancer genes. Genome Biol. 2013; 14:R21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Heyn H., Vidal E., Ferreira H.J., Vizoso M., Sayols S., Gomez A., Moran S., Boque-Sastre R., Guil S., Martinez-Cardus A. et al. . Epigenomic analysis detects aberrant super-enhancer DNA methylation in human cancer. Genome Biol. 2016; 17:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Roadmap Epigenomics Consortium Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J. et al. . Integrative analysis of 111 reference human epigenomes. Nature. 2015; 518:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ernst J., Kellis M.. ChromHMM: automating chromatin state discovery and characterization. Nat. Methods. 2012; 9:215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Guelen L., Pagie L., Brasset E., Meuleman W., Faza M.B., Talhout W., Eussen B.H., de Klein A., Wessels L., de Laat W. et al. . Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008; 453:948–951. [DOI] [PubMed] [Google Scholar]
- 68. Meuleman W., Peric-Hupkes D., Kind J., Beaudry J.-B., Pagie L., Kellis M., Reinders M., Wessels L., van Steensel B.. Constitutive nuclear lamina–genome interactions are highly conserved and associated with A/T-rich sequence. Genome Res. 2013; 23:270–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hinrichs A.S., Karolchik D., Baertsch R., Barber G.P., Bejerano G., Clawson H., Diekhans M., Furey T.S., Harte R.A., Hsu F. et al. . The UCSC genome browser database: update 2006. Nucleic Acids Res. 2006; 34:D590–D598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hansen R.S., Thomas S., Sandstrom R., Canfield T.K., Thurman R.E., Weaver M., Dorschner M.O., Gartler S.M., Stamatoyannopoulos J.A.. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yang Z., Wong A., Kuh D., Paul D.S., Rakyan V.K., Leslie R.D., Zheng S.C., Widschwendter M., Beck S., Teschendorff A.E.. Correlation of an epigenetic mitotic clock with cancer risk. Genome Biol. 2016; 17:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. The GTEx Consortium The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015; 348:648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Thienpont B., Steinbacher J., Zhao H., D’Anna F., Kuchnio A., Ploumakis A., Ghesquière B., Van Dyck L., Boeckx B., Schoonjans L. et al. . Tumor hypoxia causes DNA hypermethylation by reducing TET activity. Nature. 2016; 537:63–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Eustace A., Mani N., Span P.N., Irlam J.J., Taylor J., Betts G.N., Denley H., Miller C.J., Homer J.J., Rojas A.M. et al. . A 26-gene hypoxia signature predicts benefit from hypoxia-modifying therapy in laryngeal cancer but not bladder cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013; 19:4879–4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Esteller M., Corn P.G., Baylin S.B., Herman J.G.. A Gene Hypermethylation profile of human cancer. Cancer Res. 2001; 61:3225–3229. [PubMed] [Google Scholar]
- 76. Gu Z., Steinmetz L.M., Gu X., Scharfe C., Davis R.W., Li W.-H.. Role of duplicate genes in genetic robustness against null mutations. Nature. 2003; 421:63–66. [DOI] [PubMed] [Google Scholar]
- 77. Wang T., Birsoy K., Hughes N.W., Krupczak K.M., Post Y., Wei J.J., Lander E.S., Sabatini D.M.. Identification and characterization of essential genes in the human genome. Science. 2015; 350:1096–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zapata L., Pich O., Serrano L., Kondrashov F.A., Ossowski S., Schaefer M.H.. Negative selection in tumor genome evolution acts on essential cellular functions and the immunopeptidome. Genome Biol. 2018; 19:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Scholzen T., Gerdes J.. The Ki-67 protein: from the known and the unknown. J. Cell Physiol. 2000; 182:311–322. [DOI] [PubMed] [Google Scholar]
- 80. Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. et al. . Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ciccarone F., Tagliatesta S., Caiafa P., Zampieri M.. DNA methylation dynamics in aging: how far are we from understanding the mechanisms. Mech. Ageing Dev. 2017; doi:10.1016/j.mad.2017.12.002. [DOI] [PubMed] [Google Scholar]
- 82. Aran D., Toperoff G., Rosenberg M., Hellman A.. Replication timing-related and gene body-specific methylation of active human genes. Hum. Mol. Genet. 2011; 20:670–680. [DOI] [PubMed] [Google Scholar]
- 83. Cruickshanks H.A., McBryan T., Nelson D.M., VanderKraats N.D., Shah P.P., van Tuyn J., Rai T.S., Brock C., Donahue G., Dunican D.S. et al. . Senescent cells harbour features of the cancer epigenome. Nat. Cell Biol. 2013; 15:1495–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Bandettini L., Filipponi F., Romagnoli P.. Increase of the mitotic index of colonic mucosa after cholecystectomy. Cancer. 1986; 58:685–687. [DOI] [PubMed] [Google Scholar]
- 85. Grossman D., McNiff J.M., Li F., Altieri D.C.. Expression and targeting of the apoptosis inhibitor, survivin, in human melanoma. J. Invest. Dermatol. 1999; 113:1076–1081. [DOI] [PubMed] [Google Scholar]
- 86. Shimi T., Butin-Israeli V., Adam S.A., Hamanaka R.B., Goldman A.E., Lucas C.A., Shumaker D.K., Kosak S.T., Chandel N.S., Goldman R.D.. The role of nuclear lamin B1 in cell proliferation and senescence. Genes Dev. 2011; 25:2579–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Freund A., Laberge R.-M., Demaria M., Campisi J.. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell. 2012; 23:2066–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ivanov A., Pawlikowski J., Manoharan I., van Tuyn J., Nelson D.M., Rai T.S., Shah P.P., Hewitt G., Korolchuk V.I., Passos J.F. et al. . Lysosome-mediated processing of chromatin in senescence. J. Cell Biol. 2013; 202:129–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ahmed M.S., Ikram S., Bibi N., Mir A.. Hutchinson–gilford progeria syndrome: a premature aging disease. Mol. Neurobiol. 2017; 55:4417–4427. [DOI] [PubMed] [Google Scholar]
- 90. Johnson K.C., Koestler D.C., Cheng C., Christensen B.C.. Age-related DNA methylation in normal breast tissue and its relationship with invasive breast tumor methylation. Epigenetics. 2014; 9:268–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Costello J.F., Frühwald M.C., Smiraglia D.J., Rush L.J., Robertson G.P., Gao X., Wright F.A., Feramisco J.D., Peltomäki P., Lang J.C. et al. . Aberrant CpG-island methylation has non-random and tumour-type–specific patterns. Nat. Genet. 2000; 24:132–138. [DOI] [PubMed] [Google Scholar]
- 92. Sproul D., Kitchen R.R., Nestor C.E., Dixon J.M., Sims A.H., Harrison D.J., Ramsahoye B.H., Meehan R.R.. Tissue of origin determines cancer-associated CpG island promoter hypermethylation patterns. Genome Biol. 2012; 13:R84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Hansen K.D., Timp W., Bravo H.C., Sabunciyan S., Langmead B., McDonald O.G., Wen B., Wu H., Liu Y., Diep D. et al. . Increased methylation variation in epigenetic domains across cancer types. Nat. Genet. 2011; 43:768–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Chen Y., Breeze C.E., Zhen S., Beck S., Teschendorff A.E.. Tissue-independent and tissue-specific patterns of DNA methylation alteration in cancer. Epigenet. Chromatin. 2016; 9:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Schlesinger Y., Straussman R., Keshet I., Farkash S., Hecht M., Zimmerman J., Eden E., Yakhini Z., Ben-Shushan E., Reubinoff B.E. et al. . Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet. 2007; 39:232–236. [DOI] [PubMed] [Google Scholar]
- 96. Widschwendter M., Fiegl H., Egle D., Mueller-Holzner E., Spizzo G., Marth C., Weisenberger D.J., Campan M., Young J., Jacobs I. et al. . Epigenetic stem cell signature in cancer. Nat. Genet. 2007; 39:157–158. [DOI] [PubMed] [Google Scholar]
- 97. Bernstein B.E., Mikkelsen T.S., Xie X., Kamal M., Huebert D.J., Cuff J., Fry B., Meissner A., Wernig M., Plath K. et al. . A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006; 125:315–326. [DOI] [PubMed] [Google Scholar]
- 98. Smith Z.D., Shi J., Gu H., Donaghey J., Clement K., Cacchiarelli D., Gnirke A., Michor F., Meissner A.. Epigenetic restriction of extraembryonic lineages mirrors the somatic transition to cancer. Nature. 2017; 549:543–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Ross J.P., Rand K.N., Molloy P.L.. Hypomethylation of repeated DNA sequences in cancer. Epigenomics. 2010; 2:245–269. [DOI] [PubMed] [Google Scholar]
- 100. Miousse I.R., Koturbash I.. The fine LINE: methylation drawing the cancer landscape. BioMed Res. Int. 2015; 2015:131547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Schuster-Böckler B., Lehner B.. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature. 2012; 488:504–507. [DOI] [PubMed] [Google Scholar]
- 102. Cramer D., Serrano L., Schaefer M.H.. A network of epigenetic modifiers and DNA repair genes controls tissue-specific copy number alteration preference. eLife. 2016; 5:e16519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Berman B.P., Weisenberger D.J., Aman J.F., Hinoue T., Ramjan Z., Liu Y., Noushmehr H., Lange C.P.E., van Dijk C.M., Tollenaar R.A.E.M. et al. . Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat. Genet. 2012; 44:40–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Vandiver A.R., Idrizi A., Rizzardi L., Feinberg A.P., Hansen K.D.. DNA methylation is stable during replication and cell cycle arrest. Sci. Rep. 2015; 5:17911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Desjobert C., Maï M.E., Gérard-Hirne T., Guianvarc’h D., Carrier A., Pottier C., Arimondo P.B., Riond J.. Combined analysis of DNA methylation and cell cycle in cancer cells. Epigenetics. 2015; 10:82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Jones P.A., Liang G.. Rethinking how DNA methylation patterns are maintained. Nat. Rev. Genet. 2009; 10:805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Ryba T., Hiratani I., Sasaki T., Battaglia D., Kulik M., Zhang J., Dalton S., Gilbert D.M.. Replication Timing: A Fingerprint for Cell Identity and Pluripotency. PLOS Comput. Biol. 2011; 7:e1002225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Ryba T., Battaglia D., Chang B.H., Shirley J.W., Buckley Q., Pope B.D., Devidas M., Druker B.J., Gilbert D.M.. Abnormal developmental control of replication-timing domains in pediatric acute lymphoblastic leukemia. Genome Res. 2012; 22:1833–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Tomasetti C., Vogelstein B.. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. 2015; 347:78–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Tomasetti C., Li L., Vogelstein B.. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science. 2017; 355:1330–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Wu S., Powers S., Zhu W., Hannun Y.A.. Substantial contribution of extrinsic risk factors to cancer development. Nature. 2016; 529:43–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Klutstein M., Moss J., Kaplan T., Cedar H.. Contribution of epigenetic mechanisms to variation in cancer risk among tissues. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:2230–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Chen R.Z., Pettersson U., Beard C., Jackson-Grusby L., Jaenisch R.. DNA hypomethylation leads to elevated mutation rates. Nature. 1998; 395:89–93. [DOI] [PubMed] [Google Scholar]
- 114. Gaudet F., Hodgson J.G., Eden A., Jackson-Grusby L., Dausman J., Gray J.W., Leonhardt H., Jaenisch R.. Induction of tumors in mice by genomic hypomethylation. Science. 2003; 300:489–492. [DOI] [PubMed] [Google Scholar]
- 115. Daskalos A., Nikolaidis G., Xinarianos G., Savvari P., Cassidy A., Zakopoulou R., Kotsinas A., Gorgoulis V., Field J.K., Liloglou T.. Hypomethylation of retrotransposable elements correlates with genomic instability in non-small cell lung cancer. Int. J. Cancer. 2009; 124:81–87. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.