


Abstract
Genome replication, transcription and repair require the assembly/disassembly of the nucleosome. Histone chaperones are regulators of this process by preventing formation of non-nucleosomal histone–DNA complexes. Aprataxin and polynucleotide kinase like factor (APLF) is a non-homologous end-joining (NHEJ) DNA repair factor that possesses histone chaperone activity in its acidic domain (APLFAD). Here, we studied the molecular basis of this activity using biochemical and structural methods. We find that APLFAD is intrinsically disordered and binds histone complexes (H3-H4)2 and H2A-H2B specifically and with high affinity. APLFAD prevents unspecific complex formation between H2A-H2B and DNA in a chaperone assay, establishing for the first time its specific histone chaperone function for H2A-H2B. On the basis of a series of nuclear magnetic resonance studies, supported by mutational analysis, we show that the APLFAD histone binding domain uses two aromatic side chains to anchor to the α1–α2 patches on both H2A and H2B, thereby covering most of their DNA-interaction surface. An additional binding site on both APLFAD and H2A-H2B may be involved in the handoff between APLF and DNA or other chaperones. Together, our data support the view that APLF provides not only a scaffold but also generic histone chaperone activity for the NHEJ-complex.
INTRODUCTION
Replication, transcription and repair of the genome are essential for cell division, growth and maintenance of genome integrity (1–3). In order to carry out their function, dedicated molecular machineries have to be able to gain access to the DNA, perform their task, and subsequently restore a functional chromatin state. Thus, these processes are highly dependent on chromatin dynamics down to its smallest organizational level: the nucleosome. The nucleosome is characterized by 146 bp of DNA wrapped around a protein core of two histone H2A-H2B dimers and one (H3-H4)2 tetramer (4). Assembly and disassembly of nucleosomes is coordinated by histone chaperones, a family of histone binding proteins (5,6). These chaperones prevent the formation of non-nucleosomal histone–DNA complexes, mediate histone variant exchange and store histone complexes.
Aprataxin and Polynucleotide kinase Like Factor (APLF) is a DNA repair factor that facilitates repair of DNA single- and double-strand breaks (DSBs) (7–9). In non-homologous end-joining (NHEJ) of DSBs (10,11), it provides a scaffold for the NHEJ complex (12,13), promotes the retention of specific NHEJ-subunits at DSBs in vivo and stimulates the rate of NHEJ-repair (14). APLF is a 57 kDa protein composed of several distinct functional domains (Figure 1A). The N-terminal forkhead-associated (FHA) domain interacts with DNA repair proteins XRCC1 and the XRCC4/DNA Ligase IV complex (9,15). The central, mostly unstructured part of APLF contains Ser116, which is phosphorylated in response to DNA damage (9,16) and a binding motif for DNA repair factor Ku (17). The C-terminal half of the protein contains two poly(ADP-ribose) (PAR) binding zinc (PBZ) domains (18–21). PAR is a post-translational modification attached rapidly and transiently at sites of DNA damage by ADP-ribosyltransferases (ARTs) (22,23). APLF is recruited to DNA damage sites via interaction of its PBZ domains with PAR and in a PAR-independent manner via interactions with XRCC1 and Ku (7–9,18). Recently, the C-terminal acidic domain (CTAD) of APLF was shown to harbor histone chaperone activity based on its capability to mediate chromatin assembly, to bind core histones and to disassemble tetrasomes (24).
Figure 1.

APLF contains an extended HBD in its acidic domain. (A) Schematic representation of the APLF domain architecture. FHA = Forkhead associated domain, P = phosphorylation site Ser116, Ku = Ku-binding motif, PBZ = Poly(ADP)ribose Binding Zinc finger, AD = acidic domain. (B) Sequence alignment of APLFAD, motif analysis and alignment with histone chaperones. APLFAD shows conservation among species and with two motifs from histone chaperones: the NAP1L-motif present in Nap1-like proteins and the H2A-H2B binding cap-anchor motif of histone chaperones Anp32E, Spt16 and YL1. The amino acids are displayed by Seaview with color coding according to amino acid properties. Abbreviations: Hs = Homo sapiens; Mm = Mus musculus; Bt = Bos taurus; Sc = Saccharomyces cerevisiae; Dm = Drosophila melanogaster.
Here, we investigated the molecular basis of the histone chaperone function of the C-terminal APLF acidic domain, APLFAD, using a combination of nuclear magnetic resonance (NMR) spectroscopy, crosslinking, microscale thermophoresis (MST), isothermal titration calorimetry (ITC) and a functional assay.
We show that APLFAD is intrinsically disordered. It binds specifically and with high affinity to both (H3-H4)2 and H2A-H2B histone complexes suggesting that it is a generic histone chaperone. Since the specificity and functionality of the H2A-H2B binding activity of APLF had not been addressed before, we characterized here in detail the molecular basis for the H2A-H2B binding of APLFAD. The histone binding domain (HBD) of APLFAD, which contains two aromatic sidechains, binds specifically to the region of H2A-H2B that interacts with DNA in the nucleosome. APLFAD is shown to interfere with DNA binding to H2A-H2B, proving its H2A-H2B histone chaperone function. To rationalize these findings, we propose a novel double-anchor model in which two aromatic side chains anchor to the α1–α2 patches on H2A and H2B. Furthermore, we found evidence for a secondary binding mode involving the H2B αC helix. Since this helix is an exposed feature on the surface of the nucleosome and many chaperone–histone complexes, this additional binding mode could represent a key step in the transfer of histone complexes from and to DNA or other chaperones. Collectively, our results suggest that APLF provides the NHEJ-machinery with the capacity to bind and transfer histone complexes generically through an interaction mode that presents a novel variation in the recognition of the histone surface.
MATERIALS AND METHODS
Protein expression and purification
APLFAD production
An APLFAD expression plasmid was constructed according to the enzyme free cloning method for bacterial expression of proteins with an N-terminal histidine (His) and glutathione-S-transferase (GST) tag using a plasmid with the DNA sequence of human APLF as DNA template (25). The following primers were used:
5′-GCCGCGCGGCAGCCTGGATGAAGATAATGATAATGTTGGGCAAC-3′ (LICFW) +
5′-CTATTTTCTTTTCATAAACCTTTTTGCTTC-3′ (RV) and
5′-TGGATGAAGATAATGATAATGTTGGGCAAC-3′ (FW) +
5′-CAAGAAGAACCCCCTATTTTCTTTTCATAAACCTTTTTGCTTC-3′ (LICRV)
and the pLIC-His-GST vector. Correct construction of the plasmid was verified by DNA sequencing. APLFAD mutants were generated by site-directed mutagenesis on this vector and verified by sequencing.
APLFAD was expressed as fusion protein with N-terminal His-GST tag in Rosetta2 (DE3) cells (Novagen). All media used contained ampicillin (100 mg/l) and chloramphenicol (34 mg/l). Colonies from a lysogeny broth (LB) agar plate were first grown in liquid LB at 37°C. Cells from the LB culture were transferred to supplemented M9 minimal medium (MM) containing either 14NH4Cl and 12C-glucose or 15NH4Cl and 13C-glucose as the sources of nitrogen and carbon, respectively, and left to grow at 37°C. Expression of recombinant protein was induced at OD600 0.8 with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) and the culture was transferred to 30°C. Cells were harvested 10–14.5 h after induction and frozen for storage. For purification, cells were resuspended in lysis buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 5 mM β-mercaptoethanol (BME), 1 mM ethylenediaminetetraacetic acid (EDTA)) supplemented with 0.2 mM phenylmethylsulfonyl fluoride (PMSF) and in-house made protease inhibitor cocktail (PIC) (100 μM AEBSF, 0.3 μM aprotinin, 1 μM bestatin, 1 μM E-64, 10 μM leupeptin, 1 μM pepstatin A), then treated with lysozyme (Sigma-Aldrich) (2 mg per 1 g of wet cell pellet; 20 min on ice) and benzonase (Merck Millipore), frozen, thawed and sonicated. After insoluble cell material was removed by centrifugation (30 min, 35 000 g, 4°C), His-GST-APLFAD was purified using glutathione agarose beads (Sigma-Aldrich) preincubated in lysis buffer, and, after fusion protein binding, washed with lysis buffer. Bound fusion protein was eluted with 15 mM reduced glutathione (Sigma-Aldrich) in lysis buffer and cleaved with thrombin (Sigma-Aldrich) at room temperature, typically overnight. Cleavage was monitored using sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis and after complete cleavage, the protein mixture was dialyzed extensively against lysis buffer to remove glutathione and reapplied on glutathione agarose beads to remove GST. APLFAD was further purified by anion exchange on a 5 ml HiTrap Q HP column (GE Healthcare Life Sciences) using ion exchange (IEX) low salt buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 5 mM BME, 1 mM EDTA) and IEX high salt buffer (20 mM Tris, pH 7.5, 1 M NaCl, 5 mM BME, 1 mM EDTA). Fractions with APLFAD were pooled and concentrated using a 3 kDa molecular weight cut-off (MWCO) Amicon Ultra Centrifugal Filter Unit (Merck Millipore) and elution buffer was exchanged to assay buffer (25 mM NaPi, pH 7.0, 300 mM NaCl). APLFAD was aliquoted, flash-frozen in liquid nitrogen and stored at −20°C until further use.
Histone production
Histone proteins H2A, H2B, H3 and H4 from Drosophila melanogaster (Dm) were expressed in Rosetta2 (DE3) cells (Novagen) from a pET21b plasmid. All media contained ampicillin (100 mg/l) and chloramphenicol (34 mg/l). Colonies from an LB agar plate were first grown in LB at 37°C. The cell culture was transferred to supplemented M9 MM (unlabeled or containing 13C6D7-glucose, 15NH4Cl and D2O for labeling) and grown at 37°C. Expression of recombinant protein was induced at OD600 0.6–0.8 with 0.5 mM IPTG. Cells with H4 were harvested 3 h after induction, cells with other histones 12.5 h after induction by centrifugation at 4000 g for 20 min. Cells were resuspended in histone lysis buffer (50 mM Tris at pH 7.5, 100 mM NaCl, 5 mM BME, 1 mM EDTA) supplemented with 0.2 mM PMSF and in-house made PIC (as above), treated with lysozyme (as above), frozen, thawed and sonicated. First steps of purification—isolation and solubilization of histone inclusion bodies—were done according to the protocol described by Luger et al. (26). Solubilized histones were first purified on a gel filtration column HiLoad Superdex 75 pg (GE Healthcare Life Sciences) pre-equilibrated with histone gel filtration buffer (50 mM NaPi, pH 7.5, 150 mM NaCl, 5 mM BME, 1 mM EDTA, 7 M urea). Histone-containing fractions were pooled and loaded on a cation exchange chromatography column HiTrap SP HP (GE Healthcare Life Sciences) pre-equilibrated with histone gel filtration buffer. The same buffer was used to wash the column after loading. Histones were eluted with a linear gradient of NaCl (0.15–1 M), dialyzed against water, lyophilized and stored at −20°C.
Preparation of histone complexes
Lyophilized histone proteins were unfolded in unfolding buffer (50 mM Tris, pH 7.5, 100 mM NaCl, 10 mM dithiothreitol (DTT), 6 M guanidine hydrochloride) and mixed (H2A with H2B and H3 with H4) in equimolar ratios to a final protein concentration of 1 mg/ml. Refolding by dialysis and gel filtration of histone complexes were performed as described before for histone octamers (26). The gel filtration buffer was exchanged to assay buffer (see above) using a 3 and 10 kDa MWCO Amicon Ultra Centrifugal Filter Unit (Merck Millipore) for H2A-H2B and (H3-H4)2, respectively. Protein samples were concentrated and used for experiments immediately or flash-frozen in liquid nitrogen and stored at −20°C.
Crosslinking experiments
Crosslinking was performed using dithiobis(succinimidyl-propionate) (DSP). APLFAD (5 μl of 40 μM) was mixed with H2A-H2B (5 μl of 20, 40 or 80 μM). Experiments on individual proteins were done by mixing APLFAD (5 μl of 5 μM) or H2A-H2B (5 μl of 20 μM) with 5 μl of assay buffer. After 15 min of incubation at room temperature (RT), 1 μl of 1 mM DSP in dimethyl sulfoxide (DMSO) was added. Samples were crosslinked for 25 min at RT and then mixed with 4 μl of 4× non-reducing Laemmli sample buffer. After 15 min at RT, the samples were heated to 95°C for 5 min and analyzed by non-reducing SDS-PAGE on a 14% polyacrylamide gel and stained with Coomassie Brilliant Blue G.
Microscale thermophoresis
For MST experiments, APLFAD was labeled with a fluorescent tag (NT-647) on exposed lysine amino groups with the Monolith NT™ Protein Labeling Kit RED-NHS (NanoTemper Technologies) according to the supplied labeling protocol. NT-647 labeled APLFAD was used at a concentration of 25 nM. Unlabeled H2A-H2B was titrated in 1:1 dilutions beginning at 118 μM. Unlabeled (H3-H4)2 was titrated in 1:1 dilutions beginning at 217.5 μM (tetramer concentration). Optimization of experimental conditions is described in ref. (27). Experiments were performed in assay buffer supplemented with 0.5 mg/ml bovine serum albumin (BSA) and 0.05% Tween-20 and measured in hydrophilic capillaries (NanoTemper Technologies). Three independent measurements were done on a NanoTemper Monolith NT.015 instrument at 25°C, 20% MST and 100% LED power with 30/5 s laser-on/off time. Data were fitted to a sequential-binding model using in-house MATLAB 2017a (The MathWorks, Inc.) scripts (available upon request). Error bars were set to the standard deviation of each replicate point or to 0.5 at minimum. Errors in fit parameters were based on statistical F-test with 95% confidence interval (see Supplementary Figure S2).
Isothermal titration calorimetry
A calorimetric titration of APLFAD to H2A-H2B or (H3-H4)2 was performed using a MicroCal VP-ITC microcalorimeter (GE Healthcare) at 25°C. Proteins were buffer exchanged exhaustively at 4°C into assay buffer before use in the titration experiments. All solutions were degassed under vacuum for 5 min with gentle stirring immediately before use. For comparison between histone complexes (Figure 3C and D), H2A-H2B or (H3-H4)2 was used in the sample cell at a concentration of 30 μM and titrated with 450 μM APLFAD in the injection syringe. For mutational analysis (Figure 8B), binding of WT and mutant APLFAD to H2A-H2B was measured using 10 μM H2A-H2B in the cell and 90 μM APLFAD in the syringe. The H2A-H2B and APLFAD cell and syringe components as well as the reaction mixes at the end of the titrations were analyzed by Tris-Tricine SDS PAGE followed by coomassie staining as control of the integrity of the proteins and protein concentrations (Supplementary Figure S4C). Binding isotherms were generated by plotting the heat change of the binding reaction against the ratio of total concentration of APLFAD to total concentration of H2A-H2B or (H3-H4)2. The enthalpy of binding (ΔH, kcal mol−1) was determined by integration of the injection peaks (5 μl ) and correction for heats of dilution were determined from identical experiments without histone complexes. The entropy of binding (ΔS), the stoichiometry of binding (N), and the dissociation constant (KD) were determined by fitting the resulting corrected binding isotherms by nonlinear least-squares analysis to a one set of sites binding model using the Origin software (MicroCal, Inc.). Error in fit parameters are the standard errors derived from the regression analyses as reported by the software.
Figure 3.

APLFAD binds with high and similar affinities to H2A-H2B and (H3-H4)2. (A) SDS-PAGE gel of room temperature crosslinking experiment of APLFAD, H2A-H2B and their mixtures (molar ratios indicated on top of the gel). The molecular size regions for the histone and APLFAD monomers are indicated (M), as well as regions for crosslinked histone dimers (HD), histone tetramers (HT) and histone-APLFAD complexes: the primary and secondary complex (PC/SC). APLFAD (7.7 kDa) migrates as an ∼14 kDa protein (lane 9, control: non-crosslinked), presumably due to the acidic character of the protein. APLFAD stains poorly, in particular in presence of the crosslinking agent (lane 3). (B) MST-derived binding curves of H2A-H2B (red) or (H3-H4)2 (blue) titrated to fluorescently labeled APLFAD at 25°C. Data points are the average from three measurements, error bars are one standard deviation. Best-fit affinities using a sequential-binding model are listed in the table, the 95% confidence interval based on F-test is given in square brackets (see Supplementary Figure S2). Note that the concentration of (H3-H4)2 is expressed in dimer units to allow direct comparison with H2A-H2B. (C) Calorimetric titration of APLFAD to H2A-H2B (red) and (H3-H4)2 (blue) via ITC at 25°C. Experimental heat changes of injections of APLFAD to H2A-H2B or (H3-H4)2 are shown (upper panel) in red and blue, respectively. The resulting binding isotherms (lower panel) were fit to a one-set-of-sites binding mode. Best-fit values and fitting errors are shown in the table (D, top) together with the derived thermodynamic parameters (D, bottom). All data are obtained in 25 mM NaPi, pH 7.0, 300 mM NaCl.
Figure 8.

APLFAD functions as histone chaperone and prevents H2A-H2B-mediated DNA precipitation. (A) Native PAGE analysis of 167 base-pair (bp) DNA (1 μM) with the 601 nucleosome positioning sequence in presence or absence of APLFAD WT or mutants and H2A-H2B. Chaperone assay performed at 37°C. Lane 1: 50 bp DNA ladder (M). Lane 2: free DNA. Lane 3: DNA upon addition of 15 μM H2A-H2B. Lanes 4–15: DNA upon addition of 15 μM H2A-H2B preincubated with increasing concentrations (15, 45 or 90 μM) of APLFAD WT, Y476A, W485A or Y476A/W485A. (B) Calorimetric titration of APLFAD WT or mutants to H2A-H2B via ITC at 25°C. The resulting binding isotherms (upper panel) were fit to a one-set-of-sites binding mode. Best-fit values and fitting errors are shown in the table together with the derived thermodynamic parameters (lower panel). All data are obtained in 25 mM NaPi, pH 7.0, 300 mM NaCl.
NMR experiments, backbone assignments and titration analysis
All NMR experiments were carried out on Bruker Avance III HD spectrometers. All NMR spectra were processed using Bruker TopSpin or NMRPipe (28) and analyzed using Sparky (29).
Samples of free APLFAD typically contained 200–500 μM protein in buffer containing 25 mM NaPi, pH 7, 300 mM NaCl, 5% (v/v) D2O, 0.02% NaN3 and 1× PIC (cOmplete EDTA-free Protease Inhibitor Cocktail (Roche)). Experiments for backbone assignment of APLFAD were performed on a spectrometer operating at 600 MHz 1H Larmor frequency and equipped with a cryoprobe. Spectra were recorded on [13C/15N]APLFAD at 300 K for optimal resolution. Relaxation experiments to measure 15N-T1 and -T2 of free APLFAD, were performed at 750 MHz 1H Larmor frequency on [15N]APLFAD at 298K.
Backbone resonances of APLFAD were assigned to 94% completeness using 3D HNCO, HN(CA)CO, HNCACB, CBCA(CO)NH and HBHA(CO)NH spectra. The program secondary structure probability (SSP) was used to analyze the secondary structure (SS) propensities based on the assigned Cα and Cβ chemical shifts (30).
Samples for assignment of H2B contained 500 μM H2A-[U-2H/13C/15N]-H2B in 95/5% H2O/D2O in NMR assignment buffer (20 mM NaPi, pH 6.5, 50 mM NaCl, 5% D2O, 0.02% NaN3, 1× PIC). Backbone assignments were based on TROSY-based HNCACB, HN(CO)CACB, HNCA, HN(CO)CA, HNCB, HN(CO)CB, HNCO and HN(CA)CO spectra, recorded at 900 MHz 1H Larmor frequency at 308 K. Assignment of H2B backbone resonances was 94.1% complete. Assignments were transferred to assay conditions through a buffer titration. Assignment of H2A (96.1% complete) will be reported elsewhere (manuscript in preparation).
NMR titration of [15N]APLFAD with H2A-H2B was done at 900 MHz 1H Larmor frequency at 308 K using sample containing 200 μM (at start) [15N]APLFAD in NMR titration buffer (25 mM NaPi, pH 7, 300 mM NaCl, 5% D2O, 0.02% NaN3, 1× PIC). 15N-TROSY spectra were measured for the free APLFAD and after each addition of unlabeled H2A-H2B (12 points from 1:0 to 1:4 H2A-H2B).
H2A-H2B refolded with either 15N-labeled H2A or [2H/13C/15N]-labeled H2B was used at a concentration of 200 μM (at start) for NMR titration experiments with unlabeled APLFAD. Both APLFAD and H2A-H2B samples were buffer exchanged to NMR titration buffer (as above). 15N-TROSY spectra were measured for the free H2A-H2B and after each addition of APLFAD at 308 K. The two titrations consisted of eight (12) points in the range of 1:0 and 1:1 (1:4) molar ratio (H2A-H2B:APLFAD) on a 850 (750) MHz spectrometer with H2A(H2B)-labeled H2A-H2B.
Reported peak intensity ratios are corrected for differences in protein concentration (due to dilution) and number of scans. Residue-specific chemical shift perturbations (CSPs) were quantified from the perturbations in the 1H (ΔδH) and 15N (ΔδN) dimensions as the weighted average (composite) CSP in ppm:
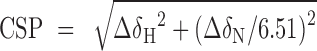 |
2D NMR lineshape analysis
2D NMR lineshape analysis of the H2A-[15N]-H2B NMR titration with APLFAD was done using the program TITAN (31). The experimental data were fitted using a sequential binding model. Fitting was done in three steps: (i) using residues that reported only on the high affinity interaction (A35, Y37, I38, K40, T49, G50, A55, I58 and N60) the parameters KD,1, koff.1 and n1 of the high affinity binding event were determined using a simple 1:1 binding model with flexible binding stoichiometry; (ii) keeping these parameters fixed, data from residues that reported primarily on the second interaction (R89, V95, E102, H106, S109, G111, K113, K117 and S120) were fitted to a sequential-binding model to extract the parameters KD,2 and koff,2 of the low-affinity binding event whilst fixing n2 to 1; (iii) in the final step the thus obtained global parameters were kept fixed and the data for resonances with kinked trajectories that report on both binding events, such as S52 and T119 (see Figure 6C and D), were fitted by optimizing the chemical shifts and line widths for the free and the two bound states. Error estimates for the fit-parameters were obtained using the bootstrap resampling of residuals procedure implemented in TITAN.
Figure 6.

The H2B α3 and αC helices form a secondary binding site for APLFAD. (A) Weighted average CSP per residue in H2B observed between one and four molar equivalents of APLFAD added to H2A-H2B. Color coding, labeling of residues and SS plot as in Figure 5. (B) Observed CSPs color coded on the cartoon representation of H2A-H2B (PDB ID: 2PYO). Color coding: yellow—H2A; gray—residues without titration data; green—DNA. (C and D) Observed and fitted 2D NMR lineshapes for two residues (C: S52 and D: T119) with pronounced kinked trajectories. Color coding of spectra is indicated in the figure. The black (gray) arrows indicate the displacements for the high (low) affinity interactions. (E) Table with best-fit values for binding parameters. N = number of binding sites.
Chaperone assay
A high-copy number plasmid containing 12 tandem repeats of a 167 base pair strong positioning DNA sequence (Widom’s 601; (32,33)) was transformed into DH5α cells. The plasmid was purified using a Qiagen Plasmid Giga Kit. The 167-bp fragment was released from the vector by ScaI digestion and purified by anion exchange.
The ratio of H2A-H2B to DNA that caused complete precipitation was determined experimentally at a ratio of 15 molar equivalents of histone dimer to DNA (Supplementary Figure S4A). Controls containing assay buffer or WT or mutant APLFAD at the concentration corresponding to the highest titration point of the assay were also carried out with DNA alone (Supplementary Figure S4A). For the assay, histone dimer (final reaction concentration: 15 μM) was pre-incubated alone or with 1, 3 or 6 molar equivalents of APLFAD wild-type (WT) or its mutants. Binding of chaperone to histone was allowed to proceed at 37°C for 15 min before the addition of DNA to a final concentration of 1 μM in a total reaction volume of 20 μl. Precipitation was carried out at 37°C for 15 min before the addition of 5 μl native PAGE loading buffer (10 mM Tris–HCl, pH 7.5, 1 mM EDTA, 1 mM DTT, 0.1 mM PMSF, 0.1 mg/ml BSA, 25% sucrose, 0.1% bromophenol blue), removal of precipitates by centrifugation and separation of the remaining soluble complexes on a 5% polyacrylamide gel run in 0.2 × TBE (17.8 mM Tris, 17.8 mM boric acid, 0.4 mM EDTA) buffer at 4°C. The gels were stained with DNA stain G (SERVA) before visualization using a Molecular Imager Gel Doc XR System (Bio-Rad). The histone dimer-APLFAD reaction mixes were analyzed by Tris-Tricine SDS-PAGE followed by coomassie staining as control of the integrity of the proteins and protein concentrations (Supplementary Figure S4B).
Structural modeling
A structural model for APLF (residues 471–490) was built using the H2A.Z-H2B bound state of YL1 (residues 39–59, PDB ID: 5CHL (34)) as template in the program MODELLER (http://salilab.org/modeller/; last accessed: 17 May 2018) (35). The sequences were aligned as in Figure 7A. This structure was used as input for docking to the H2A-H2B dimer using the program HADDOCK (36). Structure of the H2A-H2B dimer was taken from the Dm. nucleosome, PDB ID: 2PYO (37). Docking was driven using unambiguous distance restraints to impose the cap-anchor interaction with the H2B chaperone region. These restraints were defined between side chain heavy atoms of E473/Y476 and heavy atoms of H2B Y39, I51, S52, K54 and M56 as average distance observed in the structures of histone chaperones Anp32E, Spt16 and YL1 bound to H2A(.Z)-H2B (PDB 4CAY, 4WNN, 5CHL (34,38,39)). No active or passive residues were defined, otherwise the default HADDOCK protocol was followed. Out of the 200 final water-refined structures, all but one clustered in a single cluster. The structure with lowest HADDOCK score was used for display. Validation statistics are reported in Supplementary Table S1.
Figure 7.

Structural model of APLFHBD bound to H2A-H2B using two aromatic anchors. (A) Sequence alignment of APLFHBD with YL1 used as template for modeling. Cap (c) and anchor (a) residues are indicated, second aromatic residue is indicated with an arrow. (B) Double-anchor model for APLFHBD bound to H2A-H2B. APLFHBD in cartoon representation with sidechains shown as sticks. Electrostatic potential is color coded on the Van-der-Waals surface of H2A-H2B. (C) APLFHBD (surface representation) binds to NMR-derived binding interface and interferes with DNA binding to H2A-H2B. Color coding of CSPs as in Figure 5. (D and E) Close-up views on the two aromatic anchor interactions. Cap-anchor bound to H2B chaperone region in (D), second anchor bound to H2A α1–α2 patch in (E). APLFHBD in cartoon representation with sidechains shown as sticks. Selected residues and histone SS elements are labeled. Hydrogen bonds indicated with dashes. Green—DNA; cyan—APLF; yellow—H2A; red—H2B.
RESULTS
The C-terminal acidic domain of APLF is conserved and unstructured
The APLF protein, including its CTAD, is highly conserved in a broad range of organisms (8,24). Further sequence analysis shows that the APLF acidic domain (APLFAD, Homo sapiens (Hs) APLF residues 450–511) is homologous to various known histone chaperones in two partly overlapping regions (Figure 1B). The first region, residues 476–499, is conserved in the nucleosome assembly protein 1-like 1 (NAP1L1) protein and this region has been shown to be important for interaction with histone proteins and DNA repair functionality (24). The second region is a negatively charged region (residues 471–477, starting just before the NAP1L1 motif) that aligns well with the conserved cap-anchor peptide motifs used by other histone chaperones for binding the H2A-H2B or H2A.Z-H2B histone dimer (34,38–42).The region of similarity with other chaperones is followed by a conserved positively charged C-terminal region, here termed KR-motif (with K/R as single-letter codes of amino acids lysine/arginine), which could have functional relevance. The presence of an extended, highly conserved region with homology to known histone chaperones suggests that the entire conserved region in APLF may be involved in histone binding. We therefore define residues 471–499 as the (putative) HBD of APLF.
The low complexity of the primary sequence and the sparsity of hydrophobic residues suggest that APLFAD is unstructured (43–46). To characterize the structural properties of APLFAD experimentally, we used NMR spectroscopy and recorded the fingerprint 1H-15N HSQC NMR spectrum of free APLFAD (Figure 2A and Supplementary Figure S1). All backbone amide protons resonate in a narrow spectral region between 7.8 and 8.7 ppm, indicating that APLFAD is mostly unstructured in solution. To allow more detailed analysis, the backbone chemical shifts were assigned and analyzed for SS propensities using the program SSP (30). The entire domain, including the HBD, has a very low probability of being in either β-strand or α-helical conformation, implying that APLFAD is mostly unstructured (Figure 2B, upper panel). This is in agreement with a recent study that showed that full length APLF is largely an intrinsically disordered protein (13). Noteworthy, the stretch of residues 496–506 at the C-terminal edge of the HBD shows a probability of up to ∼40% of being in an α-helical conformation as found in our SSP analysis. Based on relaxation experiments by NMR spectroscopy, it is found that residues in this same stretch have significantly higher T1/T2 values compared to the rest of the acidic domain, which is indicative of reduced local flexibility and in agreement with transient formation of SS (Figure 2B, lower panel).
Figure 2.

APLFAD is an unstructured protein domain. (A) Zoomed 1H-15N HSQC spectrum of APLFAD showing all backbone amide resonances with their assignments. Spectrum recorded at 22°C, 25 mM NaPi, pH 6.6, 300 mM NaCl, 900 MHz 1H Larmor frequency. (B) SSPs derived from NMR Cα and Cβ chemical shifts (upper panel) and experimental T1/T2 ratios from NMR relaxation measurements (lower panel) plotted against the sequence of APLFAD. In the SSP diagram, negative and positive values indicate the probability of β-strand and α-helical conformation, respectively. In the T1/T2 plot, the solid (dashed) lines represent the average (average + one standard deviation) value. HBD is indicated in blue, the boundaries of the region with helical propensity are indicated with red lines.
APLFAD interacts with high affinity with histones and forms specific complexes with H2A-H2B
In order to obtain the binding properties of APLFAD and histone complexes, we studied their interactions by crosslinking, MST and ITC (Figure 3). All experiments were done at 300 mM salt in order to shield non-specific interactions.
The crosslinking agent dithiobis(succinimidyl-propionate) (DSP), which reacts with primary amine groups at the N-terminus of peptides and in lysine side chains, was used to trap complexes of APLFAD with histone complexes. While mixtures of (H3-H4)2 and APLFAD precipitated (data not shown), mixtures of APLFAD and H2A-H2B were readily crosslinked (Figure 3A). In the absence of APLFAD, crosslinking of H2A-H2B yields dimers (∼25 kDa) and a small population of (H2A-H2B)2 tetramers (∼50 kDa) (Figure 3A, lane 4), consistent with the ability of H2A-H2B to form transient tetramers (47). When APLFAD is titrated with H2A-H2B, a primary complex with an apparent molecular weight of ∼40 kDa is formed, corresponding to one H2A-H2B dimer bound to one APLFAD (Figure 3A, lanes 5–7). In the presence of 2-fold excess of H2A-H2B, additional bands corresponding to free H2A-H2B and secondary, higher-order complexes appear. The dominant band at ∼70 kDa could correspond to one or two APLFAD bound to two H2A-H2B dimers (Figure 3A, lane 7).
Next, we set out to determine the affinity of binding between APLFAD and histone complexes using MST. Addition of H2A-H2B or (H3-H4)2 to fluorescently labeled APLFAD resulted in clear changes in thermophoresis, reflecting binding of both types of histone complexes to the acidic domain (Figure 3B). Interestingly, in both cases two binding events were observed, one with a KD in the sub-micromolar and one in the high micromolar range. This is consistent with the observation of a primary and a secondary complex formation in the crosslinking experiment described above that had the same overall titration setup, namely titration of histone complex to APLFAD. To extract the corresponding binding affinities, the data were fit to a sequential-binding model in which it was assumed that (H3-H4)2, given its 2-fold symmetry, presents two binding sites to APLFAD. The KD values for the high-affinity binding events are 0.8 μM and 1.4 μM for H2A-H2B and (H3-H4)2, respectively (Figure 3B and Supplementary Figure S2). For the secondary binding, the best-fit KD value is ca. 50-fold higher, although it cannot be determined precisely because of lack of saturation (see Supplementary Figure S2).
In order to determine the stoichiometry and thermodynamic parameters of binding, we investigated the binding between APLFAD and histones by ITC (Figure 3C). APLFAD binds to H2A-H2B with a KD of 0.94 μM to form an enthalpically and entropically favorable complex (Figure 3D). APLFAD binds to (H3-H4)2 with a KD of 3.81 μM to form an enthalpically favorable but entropically unfavorable complex. The numbers of binding sites determined by ITC are ∼0.8 and ∼1.7 on H2A-H2B and (H3-H4)2, respectively. This is consistent with one APLFAD binding to one H2A-H2B or H3-H4 dimer. The ITC-derived KD value for H2A-H2B is very close to the high affinity binding value (KD,1) obtained by MST, while the KD value for (H3-H4)2 is somewhat higher but close to the upper limit of the MST-derived KD,1. Since high affinity and high enthalpy interactions dominate the ITC curve, no evidence for additional binding modes with lower affinity can be retrieved from the ITC experiments. Overall, MST and ITC data consistently show high-affinity interactions between APLFAD and both H2A-H2B and (H3-H4)2 with low- to sub-micromolar dissociation constants, while crosslinking and MST data point to multiple binding modes at excess of histones.
H2A-H2B binds to the HBD of APLFAD
Since previous work had suggested that APLF preferentially chaperones (H3-H4)2 and that the interaction with H2A-H2B is less salt-tolerant (24), we next asked whether the high-affinity interaction with H2A-H2B is specific or merely driven by unspecific electrostatic interactions. To determine the H2A-H2B interaction site on APLFAD, we titrated unlabeled H2A-H2B dimers to 15N labeled APLFAD and monitored spectral changes by NMR spectroscopy. Several APLFAD backbone resonances show a change in peak intensity and/or a change in their chemical shift (CSP), indicating that these residues are involved in binding and/or undergo structural reorganization upon binding (Figure 4A). In contrast, the N-terminal part of APLFAD up to residue number 468 is largely unaffected by the interaction (Figure 4B). Some of the affected resonances, such as of E477 and W485, disappear at low molar ratios of added histone dimer, reflecting a large exchange-induced line broadening (Figure 4A). This indicates that for these resonances the exchange between free and bound states is intermediate on the NMR chemical shift time scale, signifying a large chemical shift difference between these states. In a typical 1:1 binding model the bound state resonances should be visible at the end of the titration, but most resonances in the stretch from D475 to E486 were not detected at high molar equivalents of H2A-H2B where APLFAD should be fully bound. The increased size of the complex, in combination with residual exchange dynamics or additional dynamics in the bound states, may cause such loss of NMR signals. This has been demonstrated before for other unstructured histone chaperone domains binding to a histone complex (34,40,48). The overall pattern of observations is consistent with a low- to sub-micromolar affinity of APLFAD for H2A-H2B.
Figure 4.

APLFAD interacts with H2A-H2B through its HBD. (A) Zoomed region of overlaid 1H-15N-TROSY spectra of APLFAD with increasing concentrations of H2A-H2B. The inset shows the region containing Y476 and W485 backbone resonances. Color coding indicated in the figure. Direction of peak shifts for disappearing resonances is indicated with dashed arrows. Peak shifts of selected resonances up to one (four) equivalents added are indicated with orange (black) arrows. Recorded at 900 MHz 1H Larmor frequency at 35°C in 25 mM NaPi buffer, pH 7.0 with 300 mM NaCl. (B) Analysis of APLFAD 1H-15N peak intensity ratios (upper panel) and weighted average CSPs (lower panel) between H2A-H2B bound (4:1 molar ratio of H2A-H2B:APLFAD) and free APLFAD. Resonances that disappear during the titration are indicated by arrows and labeled. Resonances with CSPs more than two (one) standard deviation (SD) (orange (yellow) line) from the 10% trimmed mean (green dashed line) are highlighted in orange (yellow) and labeled. Position of the HBD and KR-motifs are indicated above the plot, white stripes denote the helical region. Residues without titration data due to overlap or missing resonances are indicated with gray bars. Residues with strongly overlapped resonances in the bound-state are indicated with black bars.
To map the H2A-H2B binding region on APLFAD, CSPs and peak intensity ratios were calculated from spectra without or with four molar equivalents of H2A-H2B (Figure 4B). Resonances corresponding to the central part of the putative HBD, residues D475 to E486, either show a substantial decrease in intensity or completely disappear. Resonances from adjacent residues near the termini of the HBD, including the C-terminal helical element, have low signal intensity and high CSPs. These data demonstrate that the primary region to interact with H2A-H2B is in fact the HBD of APLF. Importantly, this region also includes hydrophobic residues, in particular W485, which was previously found to be required for interaction with (H3-H4)2 (24) and Y476 which is part of the cap-anchor motif (see Figure 1B). Additionally, several resonances display curved peak trajectories when adding more than one equivalent of histone dimer, see for instance L464, L469, K494 and A504 in Figure 4A. Together, our data indicate that the conserved HBD of APLFAD is responsible for a direct and high-affinity interaction with H2A-H2B, and that regions either N- or C-terminal to the HBD may play a role in secondary binding modes of APLF and H2A-H2B.
APLFAD interacts with the DNA and chaperone binding region of H2A-H2B
To further investigate the nature of the interaction between APLFAD and H2A-H2B, we mapped where the HBD contacts the H2A-H2B dimer. For this purpose, we titrated unlabeled APLFAD into the H2A-H2B dimer with either H2A or H2B 15N-labeled ([15N]-H2A-H2B and H2A-[15N]-H2B, respectively) (Figure 5A and B). Backbone assignments of H2A and H2B in the H2A-H2B dimer were performed and transferred to assay conditions. Addition of APLFAD to [15N]-H2A-H2B causes significant CSPs for several residues (Figure 5A). Including resonances that disappear during the titration, such as R28, the affected residues cluster in and around the α1 helix and the L2 loop of H2A, which contain DNA binding residues (Figure 5C). In particular, the largest CSP is observed for R76, which anchors the L2 loop into the minor-groove of DNA in the nucleosome (4,37). Additionally, the region around G27 and R28 is part of the α1–α2 patch formed by the α1 helix, L1 loop and α2 helix, which is the contact point for DNA at the next superhelical location (Figure 5F).
Figure 5.

APLFAD binds to the DNA and histone chaperone binding region of H2A-H2B. (A and B) Zoomed region of overlaid 1H-15N-TROSY spectra of [15N]-H2A-H2B (A) and H2A-[15N]-H2B (B) with increasing concentrations of APLFAD. Color coding of spectra is indicated in the figure. Data recorded at 850 MHz (A) or 750 MHz (B) 1H Larmor frequency in 25 mM NaPi buffer, pH 7.0 with 300 mM NaCl, 35°C. Direction of peak shifts for disappearing resonances is indicated with dashed arrows in (A). Peak shifts of selected resonances up to one (four) equivalents added are indicated with orange (pink) arrows in (B). (C and D) Weighted average CSP per residue in H2A (C) and H2B (D) upon addition of one molar equivalent of APLFAD. Resonances that disappear during the titration are indicated by blue arrows and labeled in (C). Resonances with CSPs more than two (one) SD (orange (yellow) line) from the 10% trimmed mean (green dashed line) are highlighted in orange (yellow) and labeled. Residues without titration data due to overlap or missing resonances are indicated with gray bars. SS of H2A/H2B as in the nucleosome (PDB ID: 2PYO) (37)) indicated below the plot (line = loop, rectangle = α-helix, triangle = β-strand) with naming of SS-elements as in Ref. (60). Residues that interact with nucleosomal DNA (PDB ID: 2PYO) (37)) are indicated by red arrows. (E and F) CSPs color coded on the surface (E) and cartoon (F) representation of H2A-H2B. H2A residues of which resonances disappear during the titration are colored blue and labeled. Surface residues with significant CSPs (more than two SD from the 10% trimmed mean, see panels C and D) are labeled in yellow (red) for H2A (H2B). Gray: residues without titration data; green: DNA.
In the titration experiment with H2A-[15N]-H2B, very pronounced CSPs were observed such as for A55, I58 and K117 shown in Figure 5B. Strikingly, the trajectories of the A55 and I58 resonances approach the bound state after addition of one molar equivalent APLFAD whereas the K117 resonance shifts throughout the titration up to four equivalents. Together with kinked or curved CSP trajectories for several resonances (see Figure 6C and D; Supplementary Figure S3), these observations point to the presence of two binding sites on the H2A-H2B dimer, one with high affinity and a second with lower affinity. Using the CSPs observed up to one equivalent APLFAD added, we identified the α1–α2 patch of H2B as the main binding region for APLFAD (Figure 5D). This region was previously identified as chaperone binding region for H2A(.Z)-H2B chaperones Anp32E, Swr1, Spt16 and YL1 (34,38–41,49). In addition, the H2B αC helix is also involved in the binding of APLFAD as can be seen from the significant CSPs of H2B residues T116-T119 (Figure 5D). Using the CSPs between one and four added molar equivalents of APLFAD, the secondary binding site can be mapped to the α3 and αC helix of H2B, while no such rigorous conclusion can be drawn for H2A (Figure 6A). The most obvious indication of secondary binding in the titration data of [15N]-H2A-H2B is found for H2A G21, located close to the H2B αC helix. The resonances of this residue display a curved peak trajectory, thus supporting the secondary binding to this site (Supplementary Figure S3D and E). Interestingly, the H2B αC helix is on the accessible surface of the nucleosome (Figure 6B).
To compare the affinities of the primary and secondary APLFAD-binding sites on H2A-H2B, the NMR lineshapes were quantitatively analyzed using the program TITAN (31). NMR lineshapes are very sensitive to the off-rate, koff, and thus reasonably accurate KD values can be determined even when the protein concentration exceeds the KD. An excellent fit to the data was obtained using a sequential binding model in which binding affinity to the primary binding site (formed by the chaperone-binding region and the H2A α1-patch) is in the low micromolar range and ∼40-fold higher than the affinity for the secondary site on the α3 and αC helix of H2B (Figure 6E and Supplementary Figure S3). The difference in affinity can be attributed to a difference in dissociation rates (koff), with best-fit values for the koff of 6⋅102 s−1 and 4⋅103 s−1 for the high- and low-affinity interaction, respectively (Figure 6E). This translates to an average lifetime of 1.7 ms for APLFAD bound to the primary interaction region. The highly dynamic nature of the complex underscores the important role of electrostatics in the interface.
We thus find that the negatively charged HBD of APLFAD binds with high affinity to the DNA and chaperone-binding region of the H2A-H2B dimer. In addition, there is strong evidence for additional interaction modes between APLFAD and H2A-H2B from the observed secondary binding sites on both APLFAD and H2A-H2B.
A double-anchor model for APLFAD binding to H2A-H2B
Having mapped the binding interfaces of APLFAD and H2A-H2B, we sought to understand the interaction in more detail. Since APLFAD is disordered and we have no detailed structural information on the bound-state conformation of APLFAD, we decided to make use of homology to other known histone chaperones to build a structural model. In the procedure detailed below we make use of the following two assumptions.
First, we assume that the cap-anchor motif in APLF binds to H2A-H2B in the same way as seen in recent structures of chaperone–histone complexes. These structures show that the aromatic anchor residue of this motif is buried in the H2B chaperone-binding region, while the cap residue forms a hydrogen bond to the N-terminus of the H2B α2 helix (34,38–41). This assumption is based on the conservation of the cap-anchor motif in APLFAD (see Figure 1B) and the observation that this region (residues E473/Y476) as well as the chaperone-binding region in H2A-H2B (H2B residues Y34-I58) show the strongest effects in the NMR titrations (see Figures 4B and 5D).
Second, we assume that the bound-state conformation of the APLFHBD resembles that of YL1. The structure of this chaperone bound to H2A.Z-H2B shows that YL1 binds through a cap-anchor interaction and a second aromatic side chain that interacts with the H2A α1–α2 patch (34,41). Similarly, the H2A-H2B binding region of APLF contains two aromatic residues (Y476 and W485, see Figure 7A), and the H2A α1–α2 patch is involved in binding APLFAD (Figure 5E). Notably, while in YL1 the aromatic anchors are spaced 14 residues apart, their separation in APLFAD is only 9 residues (Figure 7A), illustrating the need for an atomistic model of the APLFHBD–H2A-H2B complex to verify this assumption.
To model the APLFHBD–H2A-H2B complex, we thus first constructed a model of APLFHBD based on the bound-state structure of histone chaperone YL1 using the program MODELLER (35) and the alignment in Figure 7A. The resulting APLFHBD model was subsequently docked onto the structure of H2A-H2B using HADDOCK (36) driven only by distance restraints enforcing the cap-anchor binding motif. This procedure allows to verify whether both Y476 and W485 can simultaneously interact with H2A-H2B and allows validation using the NMR-derived binding interfaces.
The resulting model for the APLFHBD–H2A-H2B complex shows that both aromatic residues are positioned close to the surface of H2A-H2B, connected by the negatively charged linker that runs over the positively charged surface of H2A-H2B (Figure 7B). The model has good physico-chemical properties, as indicated from excellent electrostatic match between the HBD and the dimer surface, the absence of clashes and Ramachandran plot statistics (see Supplementary Table S1). The HBD covers the H2A-H2B surface identified in the NMR titration studies and effectively replaces the nucleosomal DNA (Figure 7C). Importantly, this structure shows that, despite different spacing compared to YL1, both aromatic residues can anchor to the dimer. The modeled cap–anchor interaction at the H2B α1–α2 patch is shown in Figure 7D. At the H2A α1–α2 patch, W485 of APLF is buried in the H2A patch with π-stacking to R16 in H2A, with additional stabilization from a salt bridge between APLF E490 and R41 in H2A (Figure 7E), although the precise details of this interaction depend strongly on the conformation of APLFHBD submitted for docking (data not shown). In short, our model is in line with the experimental evidence and suggests a novel interaction mode between a histone chaperone and the H2A-H2B dimer with APLFAD binding the H2B hydrophobic pocket with a conserved tyrosine anchor and the H2A patch with a tryptophan anchor.
Two aromatic anchors in APLFAD are essential to prevent aggregation of H2A-H2B–DNA complexes
To test our structural model of the APLF–H2A-H2B interaction and to validate its functional relevance, we assayed the histone chaperone activity of APLFAD in vitro. In absence of H3-H4, H2A-H2B binds in non-native manner to DNA, resulting in aggregation and precipitation at high concentrations of histone dimer (50). As a true histone chaperone, APLFAD should be able to prevent such incorrect histone–DNA interactions (51). We thus used a native PAGE assay to detect the chaperone-mediated rescue of DNA precipitation (52). Control incubations of DNA with APLFAD WT or its mutants showed no effect, while incubation of DNA with H2A-H2B dimer led to loss of free DNA due to precipitation (Supplementary Figure S4A), culminating in complete precipitation of DNA at a 15:1 H2A-H2B:DNA ratio (Figure 8A, lanes 2–3). However, when H2A-H2B was pre-incubated with different amounts of APLFAD WT, DNA precipitation was prevented and free DNA as well as soluble DNA complexes were observed (Figure 8A, lanes 4–6). Mutating the aromatic amino acids that we identified as key anchor residues in APLFAD according to our structural model reduces its ability to prevent histone–DNA aggregates. While in APLFAD Y476A chaperone activity is partially abolished (Figure 8A, lanes 7–9), the mutation W485A greatly reduces chaperone activity (Figure 8A, lanes 10–12), which is almost completely lost in the double mutant Y476A/W485A (Figure 8A, lanes 13–15). This reveals that APLFAD interferes with DNA binding to H2A-H2B and that the two aromatic sidechains in the HBD of APLFAD are essential for chaperone function. These data imply that the interaction between APLFAD and H2A-H2B is specific and functional, thus strongly supporting the role of APLFAD as a domain with H2A-H2B chaperone function. In support of this result, we compared the affinities of H2A-H2B to the APLFAD mutants used in the chaperone assay by ITC. While the single mutants Y476A and W485A bound with slightly reduced affinities compared to WT APLFAD to H2A-H2B, no binding was detected for the double mutant Y476A/W485A (Figure 8B). This suggests that the linker region retains a high degree of flexibility such that the two anchors behave independently. Furthermore, whereas the binding of APLFAD WT and Y476A is mostly enthalpy driven, binding of W485A is entropy-driven. This suggests a higher conformational flexibility of the histone–chaperone complex with W485A, reducing its capability to screen the H2A-H2B DNA binding surface, leaving it open for interaction with DNA.
DISCUSSION
We describe here a detailed characterization of the interaction between the acidic domain of DNA repair factor APLF with core histone complexes to understand its histone chaperone activity. We have performed structural investigation on the recognition of H2A-H2B dimers and we present evidence that APLF can function as a true H2A-H2B chaperone. For this function, APLF makes use of a double-aromatic-anchor binding motif, a model supported by mutational analysis.
We find that APLFAD binds comparably to both (H3-H4)2 and H2A-H2B, implying that APLF is a generic histone chaperone without specificity for either type of histone complex. While Mehrotra et al. (ref. (24)), who first characterized APLF as a histone chaperone, focused on the interaction with the H3-H4 tetramer, our data show consistently, quantitatively and using a variety of techniques, that APLFAD binds H2A-H2B with very similar affinity as (H3-H4)2 (Figure 3). The NMR data indicate that the HBD binds generic elements of the histone fold in H2A-H2B, namely the α1–α2 patches of each histone that form contact points with nucleosomal DNA (Figure 5). Based on homology with YL1 and supported by mutational analysis, our structural model indicates that the APLFHBD anchors to these patches via two aromatic anchors: W485 binds the H2A-patch (potentially via H2A R16) and Y476 binds the H2B-patch (most likely via H2B Y39). The unstructured nature of APLFAD may provide the HBD with the required conformational flexibility to adapt to the surfaces of either (H3-H4)2 or H2A-H2B (53,54).
The double-anchor model of APLFAD bound to H2A-H2B presented here constitutes a novel interaction mode that highlights adaptability in recognition of the H2A-H2B surface. Comparison of the model and the H2A.Z-H2B-YL1 structure suggests that the change in spacing between the two anchor residues (from 14 in YL1 to 9 residues in APLF) prevents anchor W485 in APLF from deep burial in the H2A-patch and instead forces it to form a different set of interactions.
Sequence analysis of histone chaperones Spt16, Pob3 and hNap1 (Nap1L1) shows that these also contain a second aromatic residue in their histone binding regions. Therefore, we hypothesize that the double-anchor interaction mode is also present in these H2A-H2B chaperones. The minimal binding domains (MBD) of both Spt16 and Pob3 contain a second aromatic residue, spaced 13 and 15 residues, respectively, C-terminal to the tyrosine H2B-patch anchor (39). While not present in the crystal structure of Spt16MBD with H2A-H2B, mutagenesis data show a strong contribution of this C-terminal region in the MBD to binding affinity. In case of hNap1, its CTAD has been shown to facilitate binding of two H2A-H2B dimers to the core of hNap1 (42). There are two cap-anchor motifs in the CTAD with their aromatic anchors spaced 23 residues apart. Since it was found that both motifs bind to one H2A-H2B dimer, it is likely that the CTAD is able to bind in a similar double-anchor mode as APLF. Differences in the spacer length and the identity of anchor and surrounding residues may account for differences in histone specificity.
We showed that APLFAD binds both H2A-H2B dimers and H3-H4 tetramers with similar, low- to sub-micromolar affinities at high ionic strength, comparable to other chaperones under similar conditions. The results of our chaperone assay suggest that APLFAD is able to compete with DNA to bind the H2A-H2B DNA-interaction surface. This is in line with thermodynamic studies indicating that the chaperone–histone binding affinity must be high enough to prevent nonnative histone–DNA complexes but low enough to allow handoff to the DNA when proper nucleosomal contacts are made (50,55). Thus, APLFAD may promote proper nucleosome formation by preventing the formation of unspecific histone–DNA complexes.
In this regard, the additional weak interaction to the αC helix of H2B, which is accessible in the nucleosome, could play a role in facilitating nucleosome assembly/disassembly or in the exchange of histone variants by providing a contact point compatible with DNA or chaperone binding (Figure 9A). In particular, APLFAD is essential for the recruitment of macroH2A to sites of DNA damage and thus likely plays a role in the exchange of this variant (24,56). We speculate that the C-terminal KR-motif of APLF is involved in this interaction, based on the NMR data and the structural model showing that this region is closest to the H2B αC helix. More investigation is needed to delineate the functional role of the secondary binding as well as its structural basis.
Figure 9.

Schematic model of APLF chaperone function in NHEJ. (A) APLF binds to the nucleosome via its acidic domain to the H2B αC helix. After nucleosome disassembly, a conformational rearrangement leads to APLF binding to the DNA binding surface of the histones to prevent non-nucleosomal interactions and store the core histones. (B) Schematic model of the NHEJ complex, based on ref. (13) and this work. Broken DNA strands are held in place by the XRCC4 (X4)/XRCC4-like factor (XLF) protein filaments. The DNA ends are bound by Ku and DNA ligase IV (L4). APLF is bound to X4/L4 via its FHA domain at the DNA break site and stores core histone complexes for later reassembly of nucleosomes. Color coding: gray—DNA; green—H3-H4; yellow/orange—H2A-H2B.
It was recently shown that full-length APLF scaffolds DNA repair factors in an extended and flexible DNA repair complex for NHEJ (13). NHEJ involves formation of long protein filaments on naked DNA to capture the broken DNA strands (57–59), requiring disassembly of nucleosomes. The presence of APLF at the DNA end-sites as in the model of Hammel et al. (13) suggests that APLF could function to temporarily store histone complexes and mediate their transfer to and from DNA or other chaperones at the break site (Figure 9B). After ligation, APLF may promote formation of a nucleosome to ‘seal’ the repaired break. Overall, APLF could facilitate NHEJ with the required recognition of DNA damage, histone eviction, DNA repair, histone variant exchange and nucleosome assembly in one extended multi-protein DNA repair complex.
CONCLUSION
In conclusion, we show that the acidic domain of APLF is a histone chaperone that can bind both the histone H2A-H2B dimer and H3-H4 tetramer. We provide experimental proof for the chaperone activity for H2A-H2B and show that this can be rationalized by the screening of the DNA-binding surface by APLFAD. APLFAD binds H2A-H2B through electrostatic interactions and two aromatic residues that form anchors to the α1–α2 patches on both histones. The recognition of these generic histone-fold elements combined with the unstructured nature of APLFAD suggests that APLF has the capability to temporarily store histone complexes at the DNA damage site for later nucleosome reassembly. Chaperone activity may be facilitated by the observed binding to the exposed H2B αC helix as a key step in nucleosome (dis)assembly and histone transfer. Together, our study extends the assigned functions of APLF and underscores its important role in NHEJ. At last, the case of the APLF acidic domain highlights that such domains are more than simple polyanionic unstructured polymers: their sequences encode specific recognition of the histone surface and may be tuned to provide the required specificity and affinity.
DATA AVAILABILITY
NMR backbone assignment of Hs. APLFAD and Dm. H2B are deposited in the Biological Magnetic Resonance database BMRB under accession codes 27186 and 27187, respectively.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Prof. Marcellus Ubbink (Macromolecular Biochemistry, Leiden Institute of Chemistry, Leiden University, The Netherlands) for providing access to the VP-ITC MicroCalorimeter. We thank Jelmer Eerland (Leiden University, The Netherlands) for the NMR backbone assignment of Dm. H2A (manuscript in preparation). We thank Dr Ivan Ahel (The Sir William Dunn School of Pathology, University of Oxford, The United Kingdom) for plasmids of APLF.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Marie Curie Initial Training Network Innovative Doctoral Programme ManiFold (317371) European Union’s Seventh Framework Programme [FP7/2007-201].
Conflict of interest statement. None declared.
REFERENCES
- 1. MacAlpine D.M., Almouzni G.. Chromatin and DNA replication. Cold Spring Harb. Perspect. Biol. 2013; 5:a010207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Li B., Carey M., Workman J.L.. The role of chromatin during transcription. Cell. 2007; 128:707–719. [DOI] [PubMed] [Google Scholar]
- 3. Papamichos-Chronakis M., Peterson C.L.. Chromatin and the genome integrity network. Nat. Rev. Genet. 2013; 14:62–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Luger K., Mader A.W., Richmond R.K., Sargent D.F., Richmond T.J.. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997; 389:251–260. [DOI] [PubMed] [Google Scholar]
- 5. Hammond C.M., Stromme C.B., Huang H., Patel D.J., Groth A.. Histone chaperone networks shaping chromatin function. Nat. Rev. Mol. Cell Biol. 2017; 18:141–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mattiroli F., D’Arcy S., Luger K.. The right place at the right time: chaperoning core histone variants. EMBO Rep. 2015; 16:1454–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bekker-Jensen S., Fugger K., Danielsen J.R., Gromova I., Sehested M., Celis J., Bartek J., Lukas J., Mailand N.. Human Xip1 (C2orf13) is a novel regulator of cellular responses to DNA strand breaks. J. Biol. Chem. 2007; 282:19638–19643. [DOI] [PubMed] [Google Scholar]
- 8. Iles N., Rulten S., El-Khamisy S.F., Caldecott K.W.. APLF (C2orf13) is a novel human protein involved in the cellular response to chromosomal DNA strand breaks. Mol. Cell Biol. 2007; 27:3793–3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Macrae C.J., McCulloch R.D., Ylanko J., Durocher D., Koch C.A.. APLF (C2orf13) facilitates nonhomologous end-joining and undergoes ATM-dependent hyperphosphorylation following ionizing radiation. DNA Repair (Amst). 2008; 7:292–302. [DOI] [PubMed] [Google Scholar]
- 10. Williams G.J., Hammel M., Radhakrishnan S.K., Ramsden D., Lees-Miller S.P., Tainer J.A.. Structural insights into NHEJ: building up an integrated picture of the dynamic DSB repair super complex, one component and interaction at a time. DNA Repair (Amst). 2014; 17:110–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Radhakrishnan S.K., Jette N., Lees-Miller S.P.. Non-homologous end joining: emerging themes and unanswered questions. DNA Repair (Amst). 2014; 17:2–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grundy G.J., Rulten S.L., Zeng Z., Arribas-Bosacoma R., Iles N., Manley K., Oliver A., Caldecott K.W.. APLF promotes the assembly and activity of non-homologous end joining protein complexes. EMBO J. 2013; 32:112–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hammel M., Yu Y., Radhakrishnan S.K., Chokshi C., Tsai M.S., Matsumoto Y., Kuzdovich M., Remesh S.G., Fang S., Tomkinson A.E. et al. . An intrinsically disordered APLF links Ku, DNA-PKcs, and XRCC4-DNA ligase IV in an extended flexible non-homologous end joining complex. J. Biol. Chem. 2016; 291:26987–27006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rulten S.L., Fisher A.E., Robert I., Zuma M.C., Rouleau M., Ju L., Poirier G., Reina-San-Martin B., Caldecott K.W.. PARP-3 and APLF function together to accelerate nonhomologous end-joining. Mol. Cell. 2011; 41:33–45. [DOI] [PubMed] [Google Scholar]
- 15. Ali A.A., Jukes R.M., Pearl L.H., Oliver A.W.. Specific recognition of a multiply phosphorylated motif in the DNA repair scaffold XRCC1 by the FHA domain of human PNK. Nucleic Acids Res. 2009; 37:1701–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fenton A.L., Shirodkar P., Macrae C.J., Meng L., Koch C.A.. The PARP3- and ATM-dependent phosphorylation of APLF facilitates DNA double-strand break repair. Nucleic Acids Res. 2013; 41:4080–4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shirodkar P., Fenton A.L., Meng L., Koch C.A.. Identification and functional characterization of a Ku-binding motif in aprataxin polynucleotide kinase/phosphatase-like factor (APLF). J. Biol. Chem. 2013; 288:19604–19613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rulten S.L., Cortes-Ledesma F., Guo L., Iles N.J., Caldecott K.W.. APLF (C2orf13) is a novel component of poly(ADP-ribose) signaling in mammalian cells. Mol. Cell. Biol. 2008; 28:4620–4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eustermann S., Brockmann C., Mehrotra P.V., Yang J.C., Loakes D., West S.C., Ahel I., Neuhaus D.. Solution structures of the two PBZ domains from human APLF and their interaction with poly(ADP-ribose). Nat. Struct. Mol. Biol. 2010; 17:241–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li G.Y., McCulloch R.D., Fenton A.L., Cheung M., Meng L., Ikura M., Koch C.A.. Structure and identification of ADP-ribose recognition motifs of APLF and role in the DNA damage response. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:9129–9134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ahel I., Ahel D., Matsusaka T., Clark A.J., Pines J., Boulton S.J., West S.C.. Poly(ADP-ribose)-binding zinc finger motifs in DNA repair/checkpoint proteins. Nature. 2008; 451:81–85. [DOI] [PubMed] [Google Scholar]
- 22. Wei H., Yu X.. Functions of PARylation in DNA damage repair pathways. Genomics Proteomics Bioinformatics. 2016; 14:131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Palazzo L., Mikoc A., Ahel I.. ADP-ribosylation: new facets of an ancient modification. FEBS J. 2017; 284:2932–2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mehrotra P.V., Ahel D., Ryan D.P., Weston R., Wiechens N., Kraehenbuehl R., Owen-Hughes T., Ahel I.. DNA repair factor APLF is a histone chaperone. Mol. Cell. 2011; 41:46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. de Jong R.N., Daniels M.A., Kaptein R., Folkers G.E.. Enzyme free cloning for high throughput gene cloning and expression. J. Struct. Funct. Genomics. 2006; 7:109–118. [DOI] [PubMed] [Google Scholar]
- 26. Luger K., Rechsteiner T.J., Richmond T.J.. Preparation of nucleosome core particle from recombinant histones. Methods Enzymol. 1999; 304:3–19. [DOI] [PubMed] [Google Scholar]
- 27. Corbeski I., Horn V., van der Valk R.A., le Paige U., Dame R.T., van Ingen H.. Microscale thermophoresis analysis of chromatin interactions. Methods Mol. Biol. 2018; doi:10.1007/978-1-4939-8674-3. [DOI] [PubMed] [Google Scholar]
- 28. Delaglio F., Grzesiek S., Vuister G.W., Zhu G., Pfeifer J., Bax A.. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995; 6:277–293. [DOI] [PubMed] [Google Scholar]
- 29. Lee W., Tonelli M., Markley J.L.. NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics. 2015; 31:1325–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Marsh J.A., Singh V.K., Jia Z., Forman-Kay J.D.. Sensitivity of secondary structure propensities to sequence differences between alpha- and gamma-synuclein: implications for fibrillation. Protein Sci. 2006; 15:2795–2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Waudby C.A., Ramos A., Cabrita L.D., Christodoulou J.. Two-dimensional NMR lineshape analysis. Sci. Rep. 2016; 6:24826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thastrom A., Bingham L.M., Widom J.. Nucleosomal locations of dominant DNA sequence motifs for histone-DNA interactions and nucleosome positioning. J. Mol. Biol. 2004; 338:695–709. [DOI] [PubMed] [Google Scholar]
- 33. Lowary P.T., Widom J.. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 1998; 276:19–42. [DOI] [PubMed] [Google Scholar]
- 34. Liang X., Shan S., Pan L., Zhao J., Ranjan A., Wang F., Zhang Z., Huang Y., Feng H., Wei D. et al. . Structural basis of H2A.Z recognition by SRCAP chromatin-remodeling subunit YL1. Nat. Struct. Mol. Biol. 2016; 23:317–323. [DOI] [PubMed] [Google Scholar]
- 35. Webb B., Sali A.. Comparative protein structure modeling using MODELLER. Curr. Protoc. Protein Sci. 2016; 86:1–37. [DOI] [PubMed] [Google Scholar]
- 36. van Zundert G.C., Rodrigues J.P., Trellet M., Schmitz C., Kastritis P.L., Karaca E., Melquiond A.S., van Dijk M., de Vries S.J., Bonvin A.M.. The HADDOCK2.2 web server: user-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2016; 428:720–725. [DOI] [PubMed] [Google Scholar]
- 37. Clapier C.R., Chakravarthy S., Petosa C., Fernandez-Tornero C., Luger K., Muller C.W.. Structure of the Drosophila nucleosome core particle highlights evolutionary constraints on the H2A-H2B histone dimer. Proteins. 2008; 71:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Obri A., Ouararhni K., Papin C., Diebold M.L., Padmanabhan K., Marek M., Stoll I., Roy L., Reilly P.T., Mak T.W. et al. . ANP32E is a histone chaperone that removes H2A.Z from chromatin. Nature. 2014; 505:648–653. [DOI] [PubMed] [Google Scholar]
- 39. Kemble D.J., McCullough L.L., Whitby F.G., Formosa T., Hill C.P.. FACT disrupts nucleosome structure by binding H2A-H2B with conserved peptide motifs. Mol. Cell. 2015; 60:294–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hong J., Feng H., Wang F., Ranjan A., Chen J., Jiang J., Ghirlando R., Xiao T.S., Wu C., Bai Y.. The catalytic subunit of the SWR1 remodeler is a histone chaperone for the H2A.Z-H2B dimer. Mol. Cell. 2014; 53:498–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Latrick C.M., Marek M., Ouararhni K., Papin C., Stoll I., Ignatyeva M., Obri A., Ennifar E., Dimitrov S., Romier C. et al. . Molecular basis and specificity of H2A.Z-H2B recognition and deposition by the histone chaperone YL1. Nat. Struct. Mol. Biol. 2016; 23:309–316. [DOI] [PubMed] [Google Scholar]
- 42. Ohtomo H., Akashi S., Moriwaki Y., Okuwaki M., Osakabe A., Nagata K., Kurumizaka H., Nishimura Y.. C-terminal acidic domain of histone chaperone human NAP1 is an efficient binding assistant for histone H2A-H2B, but not H3-H4. Genes Cells. 2016; 21:252–263. [DOI] [PubMed] [Google Scholar]
- 43. Romero P., Obradovic Z., Li X., Garner E.C., Brown C.J., Dunker A.K.. Sequence complexity of disordered protein. Proteins. 2001; 42:38–48. [DOI] [PubMed] [Google Scholar]
- 44. Coeytaux K., Poupon A.. Prediction of unfolded segments in a protein sequence based on amino acid composition. Bioinformatics. 2005; 21:1891–1900. [DOI] [PubMed] [Google Scholar]
- 45. Oldfield C.J., Dunker A.K.. Intrinsically disordered proteins and intrinsically disordered protein regions. Annu. Rev. Biochem. 2014; 83:553–584. [DOI] [PubMed] [Google Scholar]
- 46. Wright P.E., Dyson H.J.. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015; 16:18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. D’Arcy S., Martin K.W., Panchenko T., Chen X., Bergeron S., Stargell L.A., Black B.E., Luger K.. Chaperone Nap1 shields histone surfaces used in a nucleosome and can put H2A-H2B in an unconventional tetrameric form. Mol. Cell. 2013; 51:662–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhou Z., Feng H., Hansen D.F., Kato H., Luk E., Freedberg D.I., Kay L.E., Wu C., Bai Y.. NMR structure of chaperone Chz1 complexed with histones H2A.Z-H2B. Nat. Struct. Mol. Biol. 2008; 15:868–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Moriwaki Y., Yamane T., Ohtomo H., Ikeguchi M., Kurita J., Sato M., Nagadoi A., Shimojo H., Nishimura Y.. Solution structure of the isolated histone H2A-H2B heterodimer. Sci. Rep. 2016; 6:24999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Andrews A.J., Chen X., Zevin A., Stargell L.A., Luger K.. The histone chaperone Nap1 promotes nucleosome assembly by eliminating nonnucleosomal histone DNA interactions. Mol. Cell. 2010; 37:834–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Laskey R.A., Honda B.M., Mills A.D., Finch J.T.. Nucleosomes are assembled by an acidic protein which binds histones and transfers them to DNA. Nature. 1978; 275:416–420. [DOI] [PubMed] [Google Scholar]
- 52. Hondele M., Stuwe T., Hassler M., Halbach F., Bowman A., Zhang E.T., Nijmeijer B., Kotthoff C., Rybin V., Amlacher S. et al. . Structural basis of histone H2A-H2B recognition by the essential chaperone FACT. Nature. 2013; 499:111–114. [DOI] [PubMed] [Google Scholar]
- 53. Liu Z., Huang Y.. Advantages of proteins being disordered. Protein Sci. 2014; 23:539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Berlow R.B., Dyson H.J., Wright P.E.. Functional advantages of dynamic protein disorder. FEBS Lett. 2015; 589:2433–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Andrews A.J., Downing G., Brown K., Park Y.J., Luger K.. A thermodynamic model for Nap1-histone interactions. J. Biol. Chem. 2008; 283:32412–32418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Timinszky G., Till S., Hassa P.O., Hothorn M., Kustatscher G., Nijmeijer B., Colombelli J., Altmeyer M., Stelzer E.H., Scheffzek K. et al. . A macrodomain-containing histone rearranges chromatin upon sensing PARP1 activation. Nat. Struct. Mol. Biol. 2009; 16:923–929. [DOI] [PubMed] [Google Scholar]
- 57. Hammel M., Yu Y., Fang S., Lees-Miller S.P., Tainer J.A.. XLF regulates filament architecture of the XRCC4.ligase IV complex. Structure. 2010; 18:1431–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hammel M., Rey M., Yu Y., Mani R.S., Classen S., Liu M., Pique M.E., Fang S., Mahaney B.L., Weinfeld M. et al. . XRCC4 protein interactions with XRCC4-like factor (XLF) create an extended grooved scaffold for DNA ligation and double strand break repair. J. Biol. Chem. 2011; 286:32638–32650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Reid D.A., Keegan S., Leo-Macias A., Watanabe G., Strande N.T., Chang H.H., Oksuz B.A., Fenyo D., Lieber M.R., Ramsden D.A. et al. . Organization and dynamics of the nonhomologous end-joining machinery during DNA double-strand break repair. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:E2575–E2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chantalat L., Nicholson J.M., Lambert S.J., Reid A.J., Donovan M.J., Reynolds C.D., Wood C.M., Baldwin J.P.. Structure of the histone-core octamer in KCl/phosphate crystals at 2.15 A resolution. Acta Crystallogr. D Biol. Crystallogr. 2003; 59:1395–1407. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
NMR backbone assignment of Hs. APLFAD and Dm. H2B are deposited in the Biological Magnetic Resonance database BMRB under accession codes 27186 and 27187, respectively.