


Abstract
High-throughput methylation sequencing enables genome-wide detection of differentially methylated sites (DMS) or regions (DMR). Increasing evidence suggests that treatment-induced DMS can be transmitted across generations, but the analysis of induced methylation changes across multiple generations is complicated by the lack of sound statistical methods to evaluate significance levels. Due to software design, DMS detection was usually made on each generation separately, thus disregarding stochastic effects expected when a large number of DMS is detected in each generation. Here, we present a novel method based on Monte Carlo sampling, methylInheritance, to evaluate that the number of conserved DMS between several generations is associated to an effect inherited from a treatment and not randomness. Moreover, we developed an inheritance simulation package, methInheritSim, to demonstrate the performance of the methylInheritance method and to evaluate the power of different experimental designs. Finally, we applied methylInheritance to a DNA methylation dataset obtained from early-life persistent organic pollutants (POPs) exposed Sprague-Dawley female rats and their descendants through a paternal transmission. The results show that metylInheritance can efficiently identify treatment-induced inherited methylation changes. Specifically, we identified two intergenerationally conserved DMS at transcription start site (TSS); one of those persisted transgenerationally. Three transgenerationally conserved DMR were found at intra or integenic regions.
INTRODUCTION
DNA methylation is an important epigenetic modification that plays a fundamental role in cell differentiation and in the development of multicellular organisms (1), and in disease states like carcinogenesis (2,3).
In vertebrates, methylation occurs primarily at cytosines (C) in cytosine–phosphate–guanine dinucleotides (CpG) enriched regions, called CpG islands. This modification allows the regulation of gene expression and is essential for cell differentiation and tissue integrity (4). The effect of methylation on gene expression depends on where methylation occurs: while high levels of methylation in promoter regions are strongly associated with transcriptional repression, low levels of methylation show a more nuanced and context-dependent relationship with transcriptional activity (4,5). Recent technological advances such as whole-genome bisulphite sequencing (WGBS) and reduced representation bisulphite sequencing (RRBS) provide the opportunity to interrogate DNA methylation modifications (6). These technologies can now be used to detect differentially methylated elements (DME) (including differentially methylated sites (DMS) or regions (DMR)) between pathologies or to help decipher the impact of exercise (7), nutrition (8) and environmental exposure (9) and interindividual differences (10).
Recently, studies have shown that environmental exposure, particularly exposure to toxic metals (11), air population (12) and toxins found in tobacco smoke (13) are associated with DNA methylation modifications and changes in gene expression that influence human health. For example, changes in methylation patterns were observed in the blood of individuals exposed to benzene, a potent carcinogen, and could be linked to an increased risk of acute myeloid leukemia (14). Preliminary human studies have provided evidence that epigenetic modifications associated with such environmental factors can be transmitted from the parents to their offspring (15,16).
For instance, prenatal exposure to tobacco smoke is associated with reproducible DNA methylation changes at a global and gene-specific level in the newborn that persist well in childhood and adolescence (17). In 2015, Sen et al. (18) showed that DNA methylation patterns in the children could be traced back to their grand-mother’s lead exposure.
Transmission of DNA methylation change in at least one generation that had no direct exposure to treatment (including gametes and in utero exposure) are called transgenerational epigenetic inheritance (TEI) and are due to germ-line transmission (19). When a gestating female (F0) has been exposed to an environmental factor, both the embryo (F1) and their germ-line that will become the second generation (F2) are directly exposed. Another generation (F3) is thus required to investigate for the presence of TEI (20). Statistically infering transgenerational epigenetic inheritance can be a challenging analysis when only few DMR are found in the intersection of all studied generations. Permutation analysis can overcome the experimental design limitation caused by small size samples (21,22) and is an attractive alternative to statistical tests based on standard distribution. That being said, the number of samples must be high enough to enable a large number of unique permutations. In a transgenerational case–control analysis, already >1.37 × 1011 unique permutations can be generated with only three individuals per group of identical size (21). A statistical framework to identify significant transgenerational methylation modification have already been proposed. The genome-wide Identification of Significant Methylation Alteration (GISAIM) framework uses permutation tests to identify inherited differential methylation patterns across multiple generations (23). In the GISAIM procedure, a methylation score is calculated for each promoter by summing the logarithm of the fold change of each generation. The methylation patterns in promoters that have consistently larger methylation scores than what would be obtained randomly are selected as inherited DMR. A possible limitation of the methylation score is that an important change in one generation with no variation in the others could be confounded with a multigenerational change. Moreover, Aiken et al. (24) noted that only few studies with three generations or more had significant results. For this reason, it could be useful to conduct inheritance simulation studies under different conditions to optimize the experimental design.
Also, only a limited number of software simulating methylation data (either WGBS (25,26) or RRBS (27–30)) are available to the scientific community (31). Those existing methods, while highly valuable, lack the capacity to simulate methylation inheritance.
In the present study, we developed a novel permutation analysis to evaluate the significance level of the number of conserved DMS or DMR over multiple generations. Furthermore, an inheritance simulation model was produced to generate simulated RRBS dataset over several generations. We conducted inheritance simulation studies to show the performance of the proposed permutation analysis under various conditions. Finally, the permutation analysis was applied to a RRBS dataset from early-life POPs exposed Sprague-Dawley female rats and their descendants through a paternal transmission. The results show that the proposed permutation analysis is able to infer that the number of conserved DMS between generations is related to the inherited effect of the environmental exposure.
MATERIALS AND METHODS
Permutation analysis
We developed a novel permutation method called methylInheritance to enable the evaluation of the significance level of the number of conserved differentially methylated sites or regions over multiple generations. The method follows a statistical hypothesis testing procedure. The number of conserved sites or regions between generations is tested against the following null hypothesis: the number of conserved sites or regions corresponds to a value that can be obtained through a randomness analysis. In order to simplify text, sites will be used in this section although the method could also apply to regions.
The significance level is calculated by comparing the experimental results with the reference distribution obtained by Monte Carlo sampling.
Figure 1 displays the workflow of the permutation analysis. Most of those steps have been implemented in the R/Bioconductor methylInheritance package. The main steps to realize this workflow are described in greater detail below.
Figure 1.

Flow chart of the permutation analysis. The flow chart depicts a summary of the most important steps of the permutation analysis workflow. Data are shown in light orange while analysis steps are shown in light blue.
Differential methylation analysis
Using the real dataset, the number of conserved DMS between cases and controls is calculated for each generation. The intersections of the DMS between all generations and between two consecutive generations are then identified to gather the observed number of conserved DMS. This number of conserved DMS (the observed value) is then compared against the reference distribution obtained by randomization (statistic inference).
Permutation cycle
The number of permutation to run within a permutation cycle is set. Executing a permutation cycle implies running a differential methylation analysis and extracting the number of conserved DMS between generations for each permuted dataset. Permuted dataset are created by exchanging labels of all datasets (cases and controls) on all generations.
Statistics
At the end of each cycle, the statistic is retrieved by calculating the number of permutations that have obtained an equal or a greater number of conserved DMS than the observed value. By adding the observed value to the distribution, we ensure that there will always be at least one case that is as extreme as the observed value in the calculation.
Convergence
To ensure the stability of the probability estimates, convergence must be evaluated. This can be done by plotting statistics calculated at the end of each cycle as a function of the number of Carlo permutations performed. Convergence is assessed qualitatively as the point where the plot reach a more or less flat region.
Significant level
The number of conserved DMS obtained in each permutation is gathered to create the reference distribution. The observed number of conserved DMS is also included to the reference distribution. The significance level is the proportion of values that are at least as extreme as the observed value in the reference distribution.
When the significant level is lower than the threshold (traditionally set at 0.05), the null hypothesis is rejected and the alternative hypothesis is considered to be more plausible to explain the data. The latter implies that the number of conserved DMS is a result of the treatment or environmental exposure.
Simulation method
A simulation method was developed to produce simulated RRBS dataset over multiple generations. The simulation method, called methInheritSim, is broken down into five major steps: (i) construction of a synthetic chromosome from a biological datasets; (ii) selecting the differentially methylated sites (DMS) for the first generation; (iii) modeling the intergenerational DMS inheritance; (iv) assigning methylation level for the first generation and (v) for the following generations. Figure 2 displays the steps of the methInheritSim pipeline.
Figure 2.

Schematic representation of the synthetic chromosome creation and differentially methylated sites (DMS) inheritance modeling by the methInheritSim method. (A) The genome of a biological dataset must be provided by the user. (B) Chromosome regions, containing all the same number of CpG sites, are sampled and assembled. (C) The CpGs sites present in the sampled regions are establishing the sites of the synthetic chromosome. (D) CpG sites are randomly selected as seed and labeled as DMS using an exponential distribution. The seeds mark the beginning of a differentially methylated region. (E) Following a specific algorithm, some CpG sites located in close range of the seeds are selected as DMS (in light blue). (F) All CpG sites, labeled or not as DMS, form the synthetic chromosome for the F1 generation. (G) Only a fraction of the differentially methylated regions of the F1 generation are inherited by the following generation. Those inherited regions are randomly selected. This synthetic chromosome is the reference chromosome for all generations following F1.
Step 1: Synthetic chromosome creation
An important section of the simulation consists in generating the distribution of the CpG sites on a synthetic chromosome as summarized in Figure 2. The distribution of CpG sites is characterized by clusters of CpG dinucleotides in close proximity separated by larger gaps (27). The simulation method must approximate this distribution with the highest possible accuracy. A reference dataset, provided by the user, is used to sample CpG sites and to approximate methylation level distribution while differentially methylated regions and inheritance are generated using parameterisable models. The input reference dataset requires only one generation of untreated controls.
The synthetic chromosome is created by assembling randomly selected regions (having the same number of CpG sites) from the reference genome (Figure 2A–C). The number of sampled regions, as well as the number of CpG sites per region are two parameters specified by the user. All sampled chromosome regions are assembled into the synthetic chromosome in the same order they are sampled as shown in Figure 2B. The methInheritSim method enable the creation of multiple chromosomes and the number of chromosomes is also a parameter specified by the user. The simulated cases and controls are all based on this synthetic chromosome and all contained the same CpG sites.
Step 2: Selection of differentially methylated sites for the F1 generation
Once the synthetic chromosome is generated, a subset of CpG sites have to be labeled as differentially methylated for the F1 generation. The DMS labeling is done through a multiple steps algorithm, as shown in Figure 2D and E. To mimic the widely held observation that sites in close proximity exhibit similar levels of methylation (27), the algorithm enables the creation of differentially methylated sites clusters that are flanked with lower DMS density regions. Thus, the methylation status of a CpG site is highly dependent of the surrounding CpGs (26).
There are four steps in the iterative DMS labeling algorithm:
To enable the creation of a low density DMS zone in between differentially methylated regions, a jump is required to separate DMR. A temporary size for the jump (s) is obtain using an exponential distribution with a λ parameter corresponding to the mean probability of being in presence of a differentially methylated site. The final size of the jump (s) is the highest rounded value between s and 1. In the methInheritSim package, the λ value is assigned through the rateDiff parameter which can be modified by the user.
The notion of seed refers to the first CpG of a differentially methylated region. From the current position, which is the beginning of the chromosome for the first iteration and the last tested site in step (iv) for the other iterations, the sth following CpG site is selected as a seed and labeled as DMS (as shown by the dotted arrows located above the chromosome in Figure 2D).
-
To reproduce differentially methylated region, all following CpG sites within 1000 bases paired of the preceding site are assigned as differentially methylated with a probability p. The probability p is calculated using an exponential function:
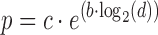
where c = 1.0, b = −1e−01 (both empirical values inspired from WGBSSuite (26)) and d is the distance from the prior site. Each site is also assigned a q value using an U(0, 1) uniform distribution. In the end, only sites that respect the condition q < p are labeled as DMS to enable the creation of DMS clusters as shown in Figure 2E.
When the next CpG site is located at >1000 bases of the previous site, the algorithm goes back to step (i) using this current CpG site as the starting point of a new cycle, such as shown by dotted arrows linking Figure 2D and E.
The final result is a synthetic chromosome with labeled DMS for the F1 generation, as shown in Figure 2F.
Step 3: Modeling intergenerational differentially methylated sites inheritance
Not all differentially methylated sites are transmitted to the following generations. Methylation state are reversible by nature (32) and imprinted regions undergo DNA demethylation during early germ-line development (33). In the methInheritSim method, the propInherite parameter controls the proportion of differentially methylated regions that is inheritable to the following generation. The propInherite parameter has a default value of 0.3 that can be modified by the user. The inheritable regions are randomly selected and all DMS present within a selected region are labeled as inheritable DMS. The result is a synthetic chromosome with labeled inheritable DMS for all generations following F1, as shown in Figure 2G.
Step 4: Methylation level assignation for F1 generation
An important stage of the methInheritSim method is the assignation of the methylation level to all CpG sites of the cases and controls for the F1 generation. First, each CpG site of the controls is assigned a methylation value using a Beta(α, β) distribution. Beta distribution often appears as a reasonable choice in several studies of genomic data (34), mainly when data ∈ (0, 1) such as methylation data (28). The α and β parameters are approximated using the mean and variance of the input control dataset at that same site (see Supplementary Material, section 1).
The methylation level is then assigned to the CpG sites of the cases of the F1 generation. Poulsen et al. (35) have found that in monozygotic twins exposed to famine during their gestation, the smaller twin is more likely to develop diabetes. Since stress-induced methylation responses are heterogeneous across individuals (36), a penetrance factor is used to enable the user to select the percentage of cases affected by the treatment. The penetrance mimics individual variability in environmental sensitivity of the epigenome, thus giving the possibility to separate the probability of the treatment to affect a patient from the treatment effects on methylation level.
Cases affected by the treatment are randomly selected following a truncated normal distribution with user-specified mean and standard deviation of the penetrance factor (vpDiff and vpDiffsd parameters in the methInheritSim package). For the cases affected by the treatment, all sites that are not labeled as differentially methylated are automatically assigned a methylation level using a Beta(α, β) distribution such as the simulated controls. Sites labeled as differentially methylated are assigned a methylation level using a Beta(α + vDiff, β) distribution where α is shifted relatively to the controls. The value of the shift (vDiff parameter) is fixed by the user. The cases not affected by the treatment are assigned methylation levels on all their CpG sites using the same protocol as for controls. All CpG sites of the simulated cases and controls of the F1 generation are now assigned methylation values as shown in Figure 2G.
Step 5: Methylation level assignation following F1 generation
The methylation level assignation method for the following generation is similar to that of F1, but with three differences. The first difference is the use of the synthetic chromosome with labeled inheritable DMS to identify sites that should follow a shifted Beta(α + vDiff, β) distribution. The second is the use of an updated value of vDiff in the shifted Beta distribution. The shift value vDiff is multiplied by a ratio called propHetero that is specified by the user. The propHetero ratio mimics the side effect of mating cases with controls. Its default value is 0.5 because half of the chromosomes are inherited from the control (when cases are mated with controls). The third difference is the use of a modified penetrance mean to identify the ratio of cases that are not affected by the treatment. The modified penetrance mean (vpDiff parameter) is calculated using a function that depends on the generation as well as the vInheritance parameter that is specified by the user:
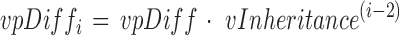 |
where vpDiff is the penetrance mean and i is the generation (2 or above). The default value of vInheritance is 0.5 that represent the situation where cases, in each generation, are mated with controls.
All five steps of the simulation method have been implemented into a R package to facilitate its use and the package is distributed through Bioconductor.
Simulation schemes
Several three-generation datasets were simulated, using the methInheritSim package, to evaluate the inheritance of induced methylation modification over various conditions. F1 control methylation data from Sprague–Dawley male rats (see following section) was used as reference dataset. For each simulated dataset, only one synthetic chromosome was created. The number of sampled regions and the number of CpG sites per region were respectively set to 400 regions (nbBlock parameter) and 50 sites (nbCpG parameter) to generate a synthetic chromosome of 20 000 CpGs. The rateDiff parameter that affects the size of the low-density DMS zones between differentially methylated regions was fixed to 0.01.
To test sample size effect, we first defined three groups of simulations with different total number of individuals (vNbSample = 6, 12 and 18 per generation). The number of cases and controls remained even in all groups. We also created groups with different effect sizes representing weak, median and strong induced treatment methylation effects that are controlled by the DMS shift (vDiff = 0.5, 0.7 and 0.8). The penetrance that fixes the ratio of cases affected by the treatment was set to 0.9 (vpDiff parameter). The standard deviation of the penetrance (vpDiffsd parameter) was fixed to 0.1 (default value). Hundred simulated dataset were generated for each of the nine schemes.
All parameters related to inheritance remained constant. Thus, the vInheritance was fixed to 0.5 which represents the situation where cases are mated with controls in each generation. The propInherite also remained constant to 0.3 (default value).
Finally, a permutation analysis was run on each simulation with 1000 permutations (nbrPermutations parameter) using the methylInheritance package. The minimum percentage of methylation change between cases and controls was fixed to 20 (minMethDiff parameter). All the other parameters remained at their default values. The number of detected DMS obtained from each simulated dataset was extracted from the first step of the permutation analysis. The power of detection and false discovery rate (FDR) has been calculated for each simulation using custom Perl scripts. A set of 100 null simulations (vpDiff = 0) for vNbSample = 6 was also performed.
Transgenerational methylation data application
We applied our permutation analysis to a methylation dataset of Sprague–Dawley male rats and their descendants exposed in early-life to a persistent organic pollutants (POPs) mixture. This POPs mixture was designed to mimic contaminants in ringed seal blubber, a traditional Inuit meal (37). Briefly, the POPs mixture was dissolved in corn oil (Aldrich-Sigma, Oakville, ON, Canada) to form a 5 mg PCB/ml stock solution and diluted to 500 μg PCB/ml before gavage. The dosage was administered twice weekly for 5 weeks (500 μg/kg body weight) to the founder F0 female rats in order to simulate levels observed in blood of Québec Inuit population (38). A schematic representation of the experimental design is shown in Figure 3.
Figure 3.

Schematic experimental design representation. The founder F0 female was exposed to a POPs mixture. Intergenerational (F1 and F2) and transgenerational (F1, F2 and F3) inheritance of treatment-induced methylation profiles were evaluated using the methylInheriance method.
Genome-wide methylation profiles of rat epididymal sperm across F1, F2 and F3 generations were obtained through reduced representation bisulfite sequencing (RRBS) (39). A total of 36 adult male spermatozoa profiles (six cases and six controls per generation) were sequenced on an Illumina HiSeq 2000 sequencer (Illumina, San Diego) at Génome Québec Innovation Centre (McGill University, Montréal), using 100 bp paired-end reads (statistics for the sequencing quality in Supplementary Material, section 3.1). Reads were sorted using SAMtools v1.2 (40), transformed in FASTQ files using BEDTools v2.17 (41) and trimmed using Trim Galore! v0.4.0. The cleaned reads were aligned on bisulfite-seq rattus norvegicus genome (Rnor_5.0) using Bismark v0.14.5 (42) and indexed with SAMtools v1.2. (statistics for the quality of alignment in Supplementary Material, section 3.2). The CpG sites for each sample have been detected using R package methylKit v0.9.4 (43). Only the filtered CpG sites with a minimum coverage of 15 reads have been retained for the downstream analyses (statistics for the CpG sites, as well as coverage distribution, in Supplementary Material, section 3.3). All RRBS dataset are available through Expression Omnibus accession numbers GSE109056.
RESULTS
Simulation results
We studied the power of detection of treatment-induced DMS across multiple generations using simulation studies performed on one synthetic chromosome generated by methInheritSim package. To simulate different experimental settings, we varied the sample size and the strength of the treatment-induced methylation effect. Hundred simulations were generated for each settings, to the distribution of the results could be investigated. Figure 4A shows the power of detection across three generations for different strength of treatment induced methylation effects (treatment effect = 0.5, 0.7 and 0.8) and a fixed sample size of 6 individuals par generation. The treatment-induced methylation effect has a mild impact on the power of detection for the first generation. Besides, all results were >94% of detection in the first generation. However, the impact of the strength of induced treatment methylation effect sharpens across generations. At the second generation, the weaker treatment effect (treatment effect = 0.5) is noticeably below the power of detection of the two other treatment effects and it drops almost to zero in the third generation with a maximum value <3%. For the third generation, with the strongest treatment-induced methylation effect (treatment effect=0.8), we can observe a small peak of simulations with a high power of detection, another small peak of simulations with a power of detection around 50% and the rest of the simulation with low power of detection.
Figure 4.

Comparison of (A) power of detection and (B) false discovery rate (FDR) across generations using 100 simulation studies with different strengths of treatment-induced methylation effects (treatment effect=0.5, 0.7 and 0.8) on a fixed sample size of six individuals per generation.
The false discovery rate is shown in Figure 4B and was produced using the same settings used for the power of detection (Figure 4A). The FDR markedly escalates with the generation analyzed. While FDR is well below 5% for the first generation, it reaches 90% for the second generation (treatment effect = 0.5) and hits 100% for the third one. The strength of treatment-induced methylation has mainly an impact on the third generation where the FDR is inversely proportional the strength of the treatment-induced methylation effect.
The population size has mild impact on the first generation, but for second and third generations, the power of detection decreases for the simulation with the weaker treatment-induced effect (treatment effect = 0.5) (Supplementary Figures S1–S3). A similar simulation analysis was done using the differentially methylated regions (DMR), the results for a fixed sample size of 6 and 12 individuals are in Supplementary Material (section 2.2, Supplementary Figures S4 and S5).
The power of detection of transgenerational inheritance of DMS was calculated by running methylInheritance on the simulation studies (Figure 5). The ratio of transgenerationally conserved methylation changes increases with the strength of the treatment-induced methylation effect.
Figure 5.

Distribution of the ratio of inheritable induced methylation changes that are transgenerationally conserved for three strength of treatment effects: (A) treatment effect = 0.5, (B) treatment effect = 0.7 and (C) treatment effect = 0.8. The simulations have been run 100 times for each treatment effect. The red distribution represents the significant transgenerational inheritance of DMS obtained by methylInheritance and the blue the non-significant results.
At last, a set of 100 null simulations (treatment effect = 0) was performed for validation. Interestingly, 40% of the null simulation had one or more significant DMS in common in all three generations. Nonetheless, the P-values obtain by methylInheritance for those control simulations were not significant.
Paternal exposure to arctic contaminants exposure in early life
The permutation analysis was applied on the full methylation dataset from early life POPs exposed Sprague–Dawley males and their descendants. First, significant DMS were identified as sites having false discovery rate <1% and a minimum methylation difference of 20% using methylKit (43) through the methylInheritance package. Respectively, 502, 377 and 736 hypo DMS were detected in F1, F2 and F3 generations. While F1 and F2 generations (intergenerational) share 99 hypo DMS, only 44 DMS are common to all generations (transgenerational) (Figure 6B and D). Similarly, 719, 452 and 658 hyper DMS were detected in F1, F2 and F3 generations. Solely 118 hyper DMS are common to F1 and F2 and 39 to all generations (Figure 6A and C). Globally, the number of conserved DMS is low compared to the total number of DMS detected. The distribution on the genome of the significant DMS for each generation, as well as the intergenerational and transgenerational sites is shown in Supplementary Material (section 3.4 and Supplementary Figure S11). Annotation of the DMS have bright out the limited number of significant DMS assigned to a transcription start site region (TSS). However, those sites are present in higher proportion in the intergenerational and transgenerational analyses (section 3.5 in Supplementary Material).
Figure 6.

Sperm DNA methylation was analyzed in F1, F2 and F3 adults according to paternal lineage. Venn diagram of differentially methylated sites (DMS) detected in each generation. (A) Intergenerational Hyper DMS. (B) Intergenerational Hypo DMS. (C) Transgenerational Hyper DMS. (D) Transgenerational Hypo DMS.
Permutation analysis of the methylation data from early life POPs exposed Sprague–Dawley male rats and their descendants was performed with the methylInheritance package. A total of 4,000 permutations were run and the threshold was fixed to 0.05. For each permutation, DMS detection was done for each generation separately using the same procedure and parameters than previously described. A high variability of the number of DMS detected was observed in each generation between permutations, as shown in Figure 7A. Statistics have been calculated at every 250 permutations during the permutation analysis to ensure that convergence has been reached (Figure 7B). Intergenerational hypo and hyper-methylated analysis both did not exceed significance thresholds (Figure 7C) with respectively a significant level of 0.0252 and 0.0185. While transgenerational hypo and hyper-methylated analysis reached even lower significance levels of 0.0032 and 0.0032 (Figure 7D). A similar permutation analysis was done using the differentially methylated regions (DMR), the results are in Supplementary Material (sections 3.6 and 3.7).
Figure 7.

Summary of the results of the permutation analysis done on the POPs exposed Sprague-Dawley males and their descendants. A total of 4000 permutations have been processed. (A) Distribution of the number of DMS, in each generation, using all permutations. Red values are the observed values. (B) Convergence of the permutation analysis. Statistics have been calculated at each cycle (250 permutations). The orange line represent the intergenerational results while the green line represent the transgenerational results. (C) Distribution of the number of conserved DMS obtained between F1 and F2 generations. Hyper and hypo conserved DMS are shown separately. The observed results are included in the distribution. The red dotted lines represent the observed numbers. (D) Distribution of the number of transgenerationally conserved DMS obtained in all permutations. The observed results are included in the distribution. The red dotted lines represent the observed numbers.
A total number of two intergenerational and one transgenerational DMS was detected at TSS and these were all hypo-methylated sites (Supplementary Table S4). Some DMS corresponded to DMR but most DMS were at solo CpG sites. No intergenerational or transgenerational DMS was found within a CpG island. Four intergenerational DMS, with two conserved transgenerationally, were found within CpG shore. These intergenerational DMS were found in an exon of the Plppr3 gene and an intron of the Chat gene. The transgenerational DMS were found in an intron of Tctn2. The biological significance of these DMS is not known. In the whole dataset, three transgenerational DMRs were detected. These were located in intronic or intergenic regions (Supplementary Figure S16). There was no intergenerational or transgenerational DMR at TSS or exon sequences (Supplementary Figure S16). The biological significance of these DMRs is not known.
DISCUSSION
Transgenerational epigenetic inheritance is described as the germline transmission of epigenetic marks, such as DNA methylation, across generations in the absence of continued direct environmental exposure or genetic manipulation (44). In this article, we proposed a permutation analysis based on Monte Carlo sampling to study the persistence of induced DNA methylation changes over multiple generations. The method infers a relation between the number of conserved DMS from one generation to the next to the inheritance effect of treatment. To assists in the study of DNA methylation changes inheritance and to test the permutation analysis we have also provided a new methods to simulate multigenerational DNA methylation in control and case datasets.
Simulation of induced methylation changes inheritance
Simulators based on next-generation sequencing (NGS) technologies play an extensive role in the evaluation of new sequencing methods and the development of suitable experimental workflows (31). The R/Bioconductor methInheritSim package fosters the study of DNA methylation inheritance by providing a way to simulate datasets of inherited DNA induced methylation changes. Parameters can be used to produce simulations that reflect different experimental settings.
The method developed in this paper can model the treatment-induced methylation changes over multiple generations. Furthermore, the simulations can characterize the impact of the introduction of unexposed mates at each generation. A comprehensive simulation study was conducted to test the treatment-induced methylation effect. In a first step, we have examined the statistical behavior of the simulation cases. The performed simulations have shown a rapid decrease of the power of detection and increase of FDR across generations. The third generation is the most affected one with almost no power of detection left. Those results are consistent with the limited number of studies across three generations or more that have had significant results (24). As expected, the strength of the methylation changes induced by the treatment only had a mild impact on the power of detection for the first generation. However, the impact of the strength of treatment-induced methylation effect increases across generations.
Detection of induced methylation changes inheritance
Simulation studies
We then apply the proposed permutation analysis to identify intergenerational and transgenerational inheritance of DMS for each simulation cases. The power of detection was over 80% for every intergenerationally conserved DMS (F1 and F2) with an treatment effect of 0.7 or 0.8. Not only the false discovery rate declines when the number of samples per group (case and control) increases, from 6 to 12 and to 18, there is also a decline in the power of detection. This effect could be explained by the better capacity to detect the real multigenerational DMS with a large sample size which would be accompanied by a diminution of the detection of some DMS obtained by chance with smaller groups.
In the case of a treatment effect of 0.5, the simulations show a lower power of detection than the two stronger treatment effects but still >70%. The simulations with a treatment effect of 0.5 are more affected by the introduction of the first control parent. As the child of F2 receives only one chromosome with inherited DMS, the average treatment effect falls to 0.25 which is near the minimal threshold that we have chosen (minimum of 0.20) to be declared differentially methylated.
In the case of transgenerational inheritance (the intersection between generations F1, F2 and F3), the power of detection dramatically decrease and is <31% for all treatment effects. The principal explanation of the decline of the power of detection is the introduction of the second control parent. In this case, the F3 generation had one heterozygote parent for the inherited DMS and the other is wild type; so each member of the F3 generation has 50% chance to receive two wild type chromosomes. Under this design, the power of detection must be low because, on average, half of the cases are wild type and all the others have, on average, half of their chromosome wild type. This situation should put into perspective the lack of significant results from some transgenerational studies (20).
We have also shown the importance of having a cutoff for the size of the intersection, as the simulations with only noise (null simulations) generate a non-empty transgenerational intersection for 40% of the simulations with six cases and six controls per generation. None of those null simulations (containing only controls) obtained significant methylInheritance p-value for the transgenerational intersection. The methylInheritance method defines the probability to obtain the size of the intersection by shuffling the DMS from the three generations.
Paternal exposure to arctic contaminants exposure in early life
The permutation analysis was used on data of Sprague–Dawley male rats exposed to POPs contaminant and their descendants. With this analysis, we confirmed that the number of intergenerational hyper and hypo-methylated DMS is significantly larger than what could be expected by chance. Unambiguously positive results were also obtained for the transgenerational analysis.
Furthermore, in a previous publication we have shown that both F1 males and their F2 sons that were exposed to the POPs mixture early in life were subfertile (45). In addition, F2 and F3 generations demonstrated significant placental defects, reduced fetal growth, neonatal and postnatal death and other congenital anomalies. Then, the phenotypes observed in the adult population reinforced the concept of inter- (F1 and F2) and transgenerational (F3) effect after a POPs exposition. The risks of neonatal and postnatal infant death are elevated in Inuit communities (46), so is stillbirth rates due to poor fetal growth, placental and congenital disorders (47). Also, exposure to PCBs in utero was shown to be related to a lower quality of alertness in Inuit children aged 11 months (48), poorer emotional development (49) and more respiratory infections in preschoolers from these communities (50). These observations highlight that Inuit health problems and the differential methylated genes associated with human diseases could be related to early life paternal POPs exposure.
Our results tend to support transgenerational epigenetic inheritance of POPs exposure via sperm. Recent findings suggest that paternal transmission of environmental information can occur via the sperm epigenome such as changes in the DNA methylation profile. Lambrot et al. (51) showed that the sperm of mice contains environment sensitive epigenomic regions that respond to diet. Those regions can be transmitted and influence offspring health. The transmission involves histone methylation or DNA methylation. High-fat diet has been shown to transgenerationally reprograms the epigenome of rat sperm cells and to affect metabolic tissues of offspring throughout two generations (52). Gametic DNA methylation may play a role in transgenerational inheritance of metabolic dysfunction as 18 loci differentially methylated in sperm from F0 rats fed a high-fat diet where also identified in their F1 offspring. However, no altered DMR were identified in F2 offspring adipose tissue.
As observed in the simulations, the number of conserved sites is more than 2.5 times lower in the transgenerational analysis than in the intergenerational analysis. This rapid decrease of transmitted altered methylation sites could be linked to the natural dissolution caused by the introduction of untreated mates in each generation. This decrease could also be explained by reported observations suggesting that transgenerational epigenetic inheritance is a soft inheritance mechanism that is reversible changes (53,54). In any case, the gradual disappearance of altered methylation sites from F1 to F3 increases the difficulty of finding significant measurements.
Strength and limitation
The methylInheritance tool is useful for the research community to evaluate the significance level of the number of conserved DMS or DMR over multiple generations. It does so by using an appropriate statistical approach to verify if the number of methylation changes are an effect of inheritance from a treatment and not randomness. To our knowledge, the number of available tools to study methylation inheritance is limited and the methylInheritance package contributes to enrich choices for researchers. The tool can be applied to experimental design to study methylation changes?across multiple generations in response to either nutritional or pharmacological interventions or exposure to environmental stressors. Simulation software, such as R/Bioconductor methInheritSim, have multiple utilities. One of the most common uses of power analysis during the planning of an experiment is to calculate sample size and this can be done using simulations (55). This kind of knowledge can be quite helpful in designing proper treatment protocols with increased chances to find answers to specific hypothesis. Furthermore, simulations provide well characterized dataset to compare the performance of existing and novel statistical methods (26).
Another statistical approach has been developed to identifying significant transgenerational methylation changes. The Genome-wide Identification of Significant Methylation Alteration (GISAIM) method (23), has been specifically developed to detect gene promoter regions with significant methylation changes conserved in three generations. The GISAIM methylation score is calculated by summing the logarithm of the fold changes of each generation. By doing so, it does not account for the dilution of the treatment effect through mating and therefore does not give different weights to generations in the calculation of the methylation score. The fundamental distinction between the two methods is: GISAIM tests if a promoter has transgenerational change while methylInheritance evaluates if the number of common DMS to the multiple generations is higher than the number expected by chance; methylInheritance does not infer if a specific element has transgenerational change. Strengths of the methylInheritance method are that (i) it has broader applicability, as it is not limited to promoter regions, (ii) it analyses the relationship between generations and can thus identify the generation where the treatment effect is lost and 3- contrarily to GISAIM, it doesn’t need to be implemented as it is available and ready to use through a R/Bioconductor package. The principal limitation of methylInheritance is that it does not have its own method to detect if a specific element is a differentially methylated change.
Summary
In this paper, we developed a method to test the hypothesis that the number of conserved DMS between several generations is associated with exposure of the F0 generation to POPs, and that the amount of conserved DMS is significantly different from what could be expected from stochastic changes. To make possible the computation of a significance level for epigenetic inheritance, we implemented methylInheritance, a R package that uses permutation analysis to evaluate if the number of conserved DMS or DMR from one generation to the next is significantly different from what would be expected from a randomness analysis. We also developed methInheritSim, a R package that simulates datasets of inherited treatment-induced DNA methylation changes. This package bases the simulation on a biological dataset provided by the user and it includes a large number of parameters to produce simulations that reflect various experimental settings. The simulator provides a precious help to evaluate potential experimental designs in function of the model of inheritance. It also enables power analysis to assess the capacity to detect a given treatment effect with a given design and sample size. A good strategy to generate simulations that reflect in vivo situation consists in testing a range of realistic parameter values.
DATA AVAILABILITY
The methylInheritance and methInheritSim methods are implemented in two distinct R packages which are freely available, under the Artistic license 2.0, through Bioconductor at http://bioconductor.org/packages/methylInheritance/ (DOI:10.18129/B9.bioc.methylInheritance) and http://bioconductor.org/packages/methInheritSim/ (DOI:10.18129/B9.bioc.methInheritSim).
Supplementary Material
ACKNOWLEDGEMENTS
Computations were made on the supercomputer Colosse Université Laval, managed by Calcul Québec and Compute Canada. The operation of this supercomputer is funded by the Canada Foundation for Innovation (CFI), Ministère de l’Économie, des Sciences et de l’Innovation du Québec (MESI) and le Fonds de recherche du Québec Nature et technologies (FRQ-NT).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Canadian Institutes of Health Research ‘Father’s lasting influence: Molecular foundations of intergenerational transmission of the paternal environment’ [2015-022-ACF]; Fonds Québécois de Recherche sur la Nature et les Technologies [133831]. Funding for open access charge: Father's lasting influence: Molecular foundations of intergenerational transmission of the paternal environment [TE1-138294].
Conflict of interest statement. None declared.
REFERENCES
- 1. Jaenisch R., Bird A.. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 2003; 33(Suppl.):245–254. [DOI] [PubMed] [Google Scholar]
- 2. Baylin S.B., Jones P.A.. A decade of exploring the cancer epigenome - biological and translational implications. Nat. Rev. Cancer. 2011; 11:726–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hansen K.D., Timp W., Bravo H.C., Sabunciyan S., Langmead B., Mcdonald O.G., Wen B., Wu H., Liu Y., Diep D. et al. . Increased methylation variation in epigenetic domains across cancer types. Nat. Genet. 2011; 43:768–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jones P.A. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012; 13:484–492. [DOI] [PubMed] [Google Scholar]
- 5. Bock C. Analysing and interpreting DNA methylation data. Nat. Rev. Genet. 2012; 13:705–719. [DOI] [PubMed] [Google Scholar]
- 6. Yong W.S., Hsu F.M., Chen P.Y.. Profiling genome-wide DNA methylation. Epigenet. Chromatin. 2016; 9:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Barrès R., Zierath J.R.. The role of diet and exercise in the transgenerational epigenetic landscape of T2DM. Nat. Rev. Endocrinol. 2016; 12:441–451. [DOI] [PubMed] [Google Scholar]
- 8. Tobi E.W., Goeman J.J., Monajemi R., Gu H., Putter H., Zhang Y., Slieker R.C., Stok A.P., Thijssen P.E., Müller F. et al. . DNA methylation signatures link prenatal famine exposure to growth and metabolism. Nat. Commun. 2014; 5:5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bauer T., Trump S., Ishaque N., Thu rmann L., Gu L., Bauer M., Bieg M., Gu Z., Weichenhan D., Mallm J.P. et al. . Environment-induced epigenetic reprogramming in genomic regulatory elements in smoking mothers and their children. Mol. Syst. Biol. 2016; 12:861–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chatterjee A., Stockwell P.A., Rodger E.J., Duncan E.J., Parry M.F., Weeks R.J., Morison I.M.. Genome-wide DNA methylation map of human neutrophils reveals widespread inter-individual epigenetic variation. Scientific Rep. 2015; 5:17328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bailey K.A., Fry R.C.. Arsenic-Associated changes to the Epigenome: What are the functional consequences. Curr. Environ. Health Rep. 2014; 1:22–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Breton C.V., Marutani A.N.. Air Pollution and Epigenetics: Recent Findings. Curr. Environ. Health Rep. 2014; 1:35–45. [Google Scholar]
- 13. Wan E.S., Qiu W., Baccarelli A., Carey V.J., Bacherman H., Rennard S.I., Agusti A., Anderson W., Lomas D.A., DeMeo D.L.. Cigarette smoking behaviors and time since quitting are associated with differential DNA methylation across the human genome. Hum. Mol. Genet. 2012; 21:3073–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bollati V., Baccarelli A., Hou L., Bonzini M., Fustinoni S., Cavallo D., Byun H.M., Jiang, J., Marinelli B., Pesatori A.C. et al. . Changes in DNA methylation patterns in subjects exposed to Low-Dose Benzene. Cancer Res. 2007; 67:876–880. [DOI] [PubMed] [Google Scholar]
- 15. Daxinger L., Whitelaw E.. Transgenerational epigenetic inheritance: more questions than answers. Genome Res. 2010; 20:1623–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Blake G.E., Watson E.D.. Unravelling the complex mechanisms of transgenerational epigenetic inheritance. Curr. Opin. Chem. Biol. 2016; 33:101–107. [DOI] [PubMed] [Google Scholar]
- 17. Joubert B.R., Håberg S.E., Nilsen R.M., Wang X., Vollset S.E., Murphy S.K., Huang Z., Hoyo C., Midttun Ø., Cupul-Uicab L.A. et al. . 450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environmental health perspectives. 2012; 120:1425–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sen A., Heredia N., Senut M.C., Land S., Hollocher K., Lu X., Dereski M.O., Ruden D.M.. Multigenerational epigenetic inheritance in humans: DNA methylation changes associated with maternal exposure to lead can be transmitted to the grandchildren. Scientific Rep. 2015; 5:14466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Heard E., Martienssen R.A.. Transgenerational epigenetic inheritance: myths and mechanisms. Cell. 2014; 157:95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Van Otterdijk S.D., Michels K.B.. Transgenerational epigenetic inheritance in mammals: How good is the evidence. FASEB J. 2016; 30:2457–2465. [DOI] [PubMed] [Google Scholar]
- 21. Ludbrook J. Advantages of permutation (Randomization) tests in clinical and experimental pharmacology and physiology. Clin. Exp. Pharmacol. Physiol. 1994; 21:673–686. [DOI] [PubMed] [Google Scholar]
- 22. Legendre P., Legendre L.. Statistical Testing by Permutation. 1998; 2:Amsterdam: Elsevier; 2nd English edn. [Google Scholar]
- 23. Tian Y., Zhang B., Fu Y., Yu G., Wang Y.. A statistical approach to identifying significant transgenerational methylation changes. 2014 IEEE Global Conference on Signal and Information Processing (GlobalSIP). 2014; IEEE; 1394–1397. [Google Scholar]
- 24. Aiken C.E., Ozanne S.E.. Transgenerational developmental programming. Hum. Reprod. Update. 2014; 20:63–75. [DOI] [PubMed] [Google Scholar]
- 25. Liang F., Tang B., Wang Y., Wang J., Yu C., Chen X., Zhu J., Yan J., Zhao W., Li R.. WBSA: web service for bisulfite sequencing data analysis. PLoS ONE. 2014; 9:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rackham O.J.L., Dellaportas P., Petretto E., Bottolo L.. WGBS Suite: Simulating whole-genome bisulphite sequencing data and benchmarking differential DNA methylation analysis tools. Bioinformatics. 2015; 31:2371–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lacey M.R., Baribault C., Ehrlich M.. Modeling, simulation and analysis of methylation profiles from reduced representation bisulfite sequencing experiments. Stat. Applic. Genet. Mol. Biol. 2013; 12:723–742. [DOI] [PubMed] [Google Scholar]
- 28. Hebestreit K., Dugas M., Klein H.U.. Detection of significantly differentially methylated regions in targeted bisulfite sequencing data. Bioinformatics. 2013; 29:1647–1653. [DOI] [PubMed] [Google Scholar]
- 29. Klein H.U., Hebestreit K.. An evaluation of methods to test predefined genomic regions for differential methylation in bisulfite sequencing data. Brief. Bioinform. 2016; 17:796–807. [DOI] [PubMed] [Google Scholar]
- 30. Yu X., Sun S.. Comparing five statistical methods of differential methylation identification using bisulfite sequencing data. Stat. Applic. Genet. Mol. Biol. 2016; 15:173–191. [DOI] [PubMed] [Google Scholar]
- 31. Zhao M., Liu D., Qu H.. Systematic review of next-generation sequencing simulators: computational tools, features and perspectives. Brief. Funct. Genomics. 2016; elw012. [DOI] [PubMed] [Google Scholar]
- 32. Ramchandani S., Bhattacharya S.K., Cervoni N., Szyf M.. DNA methylation is a reversible biological signal. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:6107–6112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hajkova P., Erhardt S., Lane N., Haaf T., El-Maarri O., Reik W., Walter J., Surani M.A.. Epigenetic reprogramming in mouse primordial germ cells. Mech. Dev. 2002; 117:15–23. [DOI] [PubMed] [Google Scholar]
- 34. Raineri E., Dabad M., Heath S.. A note on exact differences between beta distributions in genomic (methylation) studies. PLoS ONE. 2014; 9:5–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Poulsen P., Vaag A.A., Kyvik K.O., Møller Jensen D., Beck-Nielsen H.. Low birth weight is associated with NIDDM in discordant monozygotic and dizygotic twin pairs. Diabetologia. 1997; 40:439–446. [DOI] [PubMed] [Google Scholar]
- 36. Lauria M., Echegoyen-Nava R.A., Rodríguez-Ríos D., Zaina S., Lund G.. Inter-individual variation in DNA methylation is largely restricted to tissue-specific differentially methylated regions in maize. BMC Plant Biol. 2017; 17:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Muir D., Wagemann R., Hargrave B., Thomas D., Peakall D., Norstrom R.. Arctic marine ecosystem contamination. Sci. Total Environ. 1992; 122:75–134. [DOI] [PubMed] [Google Scholar]
- 38. Anas M.K.I., Guillemette C., Ayotte P., Pereg D., Giguère F., Bailey J.L.. In utero and lactational exposure to an environmentally relevant organochlorine mixture disrupts reproductive development and function in male rats. Biol. Reprod. 2005; 73:414–426. [DOI] [PubMed] [Google Scholar]
- 39. Gu H., Smith Z.D., Bock C., Boyle P., Gnirke A., Meissner A.. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat. Protoc. 2011; 6:468–481. [DOI] [PubMed] [Google Scholar]
- 40. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.. The Sequence Alignment/Map format and SAMtools. Bioinformatics (Oxford, England). 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics (Oxford, England). 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Krueger F., Andrews S.R.. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011; 27:1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Akalin A., Kormaksson M., Li S., Garrett-Bakelman F.E., Figueroa M.E., Melnick A., Mason C.E.. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012; 13:R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McBirney M., King S.E., Pappalardo M., Houser E., Unkefer M., Nilsson E., Sadler-Riggleman I., Beck D., Winchester P., Skinner M.K.. Atrazine induced epigenetic transgenerational inheritance of disease, lean phenotype and sperm epimutation pathology biomarkers. PLoS One. 2017; 12:e0184306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bailey J., Maurice C., McGraw S., Lambrot R., Cote N., Droit A., Chan D., Trasler J., Kimmins S.. Multigenerational effects of paternal exposure to environmental contaminants. In: 9th World Congress on Developmental Origins of Health and Disease. J. Dev. Origins Health Dis. 2015; 6:S54–S196. [Google Scholar]
- 46. Luo Z.C., Senécal S., Simonet F., Guimond E., Penney C., Wilkins R.. Birth outcomes in the Inuit-inhabited areas of Canada. CMAJ: Can. Med. Assoc. J. 2010; 182:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Auger N., Park A.L., Zoungrana H., McHugh N.G.L., Luo Z.C.. Rates of stillbirth by gestational age and cause in Inuit and First Nations populations in Quebec. CMAJ: Can. Med. Assoc. J. 2013; 185:E256–E262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Verner M.A., Plusquellec P., Muckle G., Ayotte P., Dewailly E., Jacobson S.W., Jacobson J.L., Charbonneau M., Haddad S.. Alteration of infant attention and activity by polychlorinated biphenyls: unravelling critical windows of susceptibility using physiologically based pharmacokinetic modeling. Neurotoxicology. 2010; 31:424–431. [DOI] [PubMed] [Google Scholar]
- 49. Plusquellec P., Muckle G., Dewailly E., Ayotte P., Bégin G., Desrosiers C., Després C., Saint-Amour D., Poitras K.. The relation of environmental contaminants exposure to behavioral indicators in Inuit preschoolers in Arctic Quebec. Neurotoxicology. 2010; 31:17–25. [DOI] [PubMed] [Google Scholar]
- 50. Dallaire F., Dewailly E., Vézina C., Muckle G., Weber J.P., Bruneau S., Ayotte P.. Effect of prenatal exposure to polychlorinated biphenyls on incidence of acute respiratory infections in preschool Inuit children. Environ. Health Perspect. 2006; 114:1301–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lambrot R., Xu C., Saint-Phar S., Chountalos G., Cohen T., Paquet M., Suderman M., Hallett M., Kimmins S.. Low paternal dietary folate alters the mouse sperm epigenome and is associated with negative pregnancy outcomes. Nat. Commun. 2013; 4:2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. de Castro Barbosa T., Ingerslev L.R., Alm P.S., Versteyhe S., Massart J., Rasmussen M., Donkin I., Sjögren R., Mudry J.M., Vetterli L. et al. . High-fat diet reprograms the epigenome of rat spermatozoa and transgenerationally affects metabolism of the offspring. Mol. Metab. 2016; 5:184–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kubota T., Mochizuki K.. Epigenetic effect of environmental factors on autism spectrum disorders. Int.J. Environ. Res. Public Health. 2016; 13:504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hanson M.A., Low F.M., Gluckman P.D.. Epigenetic epidemiology: the rebirth of soft inheritance. Ann. Nutr. Metab. 2011; 58:8–15. [DOI] [PubMed] [Google Scholar]
- 55. Quinn G.P., Keough M.J.. Experimental Design and Data Analysis for Biologists. 2002; NY: Cambridge University Press. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The methylInheritance and methInheritSim methods are implemented in two distinct R packages which are freely available, under the Artistic license 2.0, through Bioconductor at http://bioconductor.org/packages/methylInheritance/ (DOI:10.18129/B9.bioc.methylInheritance) and http://bioconductor.org/packages/methInheritSim/ (DOI:10.18129/B9.bioc.methInheritSim).