


Abstract
Cell-specific patterns of gene expression are determined by combinatorial actions of sequence-specific transcription factors at cis-regulatory elements. Studies indicate that relatively simple combinations of lineage-determining transcription factors (LDTFs) play dominant roles in the selection of enhancers that establish cell identities and functions. LDTFs require collaborative interactions with additional transcription factors to mediate enhancer function, but the identities of these factors are often unknown. We have shown that natural genetic variation between individuals has great utility for discovering collaborative transcription factors. Here, we introduce MMARGE (Motif Mutation Analysis of Regulatory Genomic Elements), the first publicly available suite of software tools that integrates genome-wide genetic variation with epigenetic data to identify collaborative transcription factor pairs. MMARGE is optimized to work with chromatin accessibility assays (such as ATAC-seq or DNase I hypersensitivity), as well as transcription factor binding data collected by ChIP-seq. Herein, we provide investigators with rationale for each step in the MMARGE pipeline and key differences for analysis of datasets with different experimental designs. We demonstrate the utility of MMARGE using mouse peritoneal macrophages, liver cells, and human lymphoblastoid cells. MMARGE provides a powerful tool to identify combinations of cell type-specific transcription factors while simultaneously interpreting functional effects of non-coding genetic variation.
INTRODUCTION
Molecular mechanisms enabling cell-specific transcriptional responses to intra- and extra-cellular signals remain poorly understood. Genome-wide studies of most lineage-determining (LDTF) and signal-dependent transcription factors (SDTF) indicate that the vast majority of their binding sites are in distal intra- and intergenic locations that frequently exhibit epigenomic features associated with enhancers (1–6) and are evolutionary well conserved (7–9). The complement of active cis-regulatory elements bound by LDTFs changes across cell types, whereas promoters stay the same. Therefore, these findings introduced the notion that enhancers are largely responsible for cell type-specific gene expression (10–12). The ENCODE consortium annotated epigenetic features associated with enhancers in several different cell lines, primary cells and tissues providing evidence for hundreds of thousands of such elements in the human genome (13), greatly exceeding the number of promoters.
Previous studies of macrophages and B cells provided the basis for a collaborative and hierarchical model (14–16). In this model, collaborative binding of two or more LDTFs opens up chromatin to establish enhancers (1), enabling cell-specific actions of broadly expressed SDTFs (17) (reviewed in (18)). The collaborative nature of LDTFs was further demonstrated by analysis of effects of genetic variation in macrophages provided by two inbred strains of mice (19).
Genome-wide association studies, or GWAS (20) have shown that most complex trait-associated genetic variation is located in non-gene/protein regions of the genome. Such non-coding variants have the potential to change conserved sequences recognized by LDTFs and thereby alter enhancer landscapes between different alleles. These differences could manifest between individuals (i.e., between individuals that are each homozygous for opposite alleles), or within an individual that is heterozygous for a functional enhancer variant. A straightforward mechanism by which enhancer function would be altered by genetic variation is where alleles alter the affinity of transcription factors to bind their motifs. Consistent with the enhancer model whereby transcription factors collaborate with each other to bind DNA motifs, reports have found that allelic variation that mutates DNA binding motifs reduces binding of the respective factor while at the same time reducing binding of collaborating factors within 100 base pairs (19,21,22). Since the DNA binding motif of the partner factor is not mutated, these examples demonstrate a coordinated action of transcription factors in accessing DNA. The implication for cell-specific gene regulation is that genetic variants altering collaborative factor binding at enhancers will only be functional in the appropriate cell type where the correct combinations of transcription factors are expressed. The practical implication of these observations is that the particular combinations of factors may be discovered with the general strategy in any cell type. In addition to the discovery of transcription factors, this method identifies the precise genomic loci where genetic variation has a functional role in factor binding that may influence higher order biological processes.
To facilitate discovery of novel collaborating transcription factors using the genetic variation approach, we developed MMARGE (Motif Mutation Analysis for Regulatory Genomic Elements). MMARGE is a suite of software tools to analyze ChIP-seq, ATAC-seq, DNase I Hypersensitivity or other next generation sequencing (NGS) assays where genotyping or DNA sequence data is available.
MMARGE requires two data types: (i) genetic variation, and (ii) high-throughput sequencing data (ChIP-seq, ATAC-seq, DNaseI-seq). It then integrates these data and provides visualization tools to interpret the results. Importantly, MMARGE was built to test for functional effects of alternate alleles at single nucleotide polymorphisms (SNPs) as well as short insertion-deletions (InDels). It performs traditional de-novo motif analysis on genomic sequence for each polymorphic allele to identify DNA binding motifs that potentially affect transcription factor binding based on sequence analysis alone. The next step is to test whether the set of potential variants that mutate a single DNA binding motif are enriched in a set of loci where differential binding/accessibility is observed. For this step, MMARGE associates quantitative measures of binding or accessibility from the ChIP/ATAC/DNaseI-seq data with the list of potential mutations in motifs. It analyzes differences in two genotypes by comparing the transcription factor binding distribution in relation to motif mutations between both genotypes, and also takes advantages of a Linear Mixed Model (LMM) (23,24) to compare many different individuals at the same time
In this report, we apply MMARGE and demonstrate its ability to reliably identify known key regulators of macrophage lineage. We further apply MMARGE to three different ChIP-seq datasets from mouse liver cells and also show that MMARGE can identify important B cell factors in a human PU.1 ChIP-seq dataset from lymphoblastoid cell lines (25). In conclusion, MMARGE is the first publicly available tool that is created to identify combinations of collaborating transcription factors. This approach is agnostic to cell type and can be applied to any dataset where genotypes and epigenetic signatures are measured.
MMARGE is based on the ChIP-seq analysis tool HOMER (1) (http://homer.ucsd.edu/homer/) and it is an extension to the software used in (19,21). Furthermore, MMARGE was used to analyze ChIP-seq and ATAC-seq data from five different strains of mice in (26). The source code and installation package are freely available on GitHub (https://github.com/vlink/marge/blob/master/MMARGE_v1.0.tar.gz) or Zenodo (10.5281/zenodo.1245209).
MATERIALS AND METHODS
Overview
A schematic outlining the major steps of MMARGE is shown in Figure 1. First, MMARGE offers a complete pipeline to process VCF (Variant Call Format) files (27) and generate individualized diploid genomes by extrapolating genetic variants from VCF files and swapping in alternate alleles into a reference genome (Figure 1A and B). Importantly, MMARGE is able to analyze sequencing data from homozygous (e.g., inbred mouse strains) and heterozygous (e.g. human) genomes and includes analysis for Single Nucleotide Polymorphisms (SNPs) as well as short Insertion-Deletions (InDels). Because MMARGE generates genomes for each individual in the VCF file, the investigator can map their sequencing data to the genome with all genetic variations by using user-defined mapping software (e.g. bowtie2 (28) or STAR (29)) (Figure 1C). MMARGE shifts positions of individual sequence to their corresponding reference coordinates for motif analysis and visualization (Figure 1D–G). It offers de-novo motif analysis for individualized genomes (Figure 1G), as well as a new algorithm to identify transcription factor binding motifs associated with allele-specific transcription factor binding or open chromatin (Figure 1H). Each step of the MMARGE pipeline is discussed below.
Figure 1.

Overview of the MMARGE pipeline. (A) MMARGE merges VCF files for SNPs and InDels, offers some basic filtering and split the merged VCF file into separate genotype-specific mutation files. (B) It then generates individual genomes by inserting the annotated mutations in the reference genome per genotype and (C) allows mapping of the experimental data sets to the individualized genomes. (D) The data mapped to the individualized genomes is then shifted back to the reference coordinates. (E) In case of heterozygous data additional processing is necessary. MMARGE offers (F) scripts for data visualization including BED files for genetic variation per genotype. It further offers (G) de-novo motif analysis for the individual genomes to make sure the enrichment analysis is performed on the correct sequence instead of the reference. MMARGE also offers a new algorithm (H) to associated TF binding motifs with genotype-specific binding for pairwise comparisons, as well as comparisons for many different individuals (all-versus-all comparison). Taken all of that together MMARGE is able to identify TF binding motifs that are functionally associated with TF binding.
Merge, filter and pre-process VCF files
The initial step of the MMARGE pipeline is to generate a set of high-confidence sequence differences between the alleles of interest (Figure 1A). MMARGE allows some basic filtering of VCF files by quality scores, however VCFtools (27) provides more sophisticated tools for this purpose. For some sequencing projects like the mouse genome project (30), SNPs and InDels are annotated in separate files, whereas other projects like the 1000 Genome project (31) provides one large file with SNPs and InDels. When SNPs and InDels are provided separately, MMARGE merges them as a first step. If a combined file is provided then the first processing step is skipped. In cases where SNPs overlap deletions or insertions within one genomic background the SNP is filtered out and the longer mutation is kept. MMARGE also simplifies the annotation of the variants per genotype (Figure 2A). In cases where more than one possible mutation occurs in a particular genomic location (e.g. two different genotypes have two different mutations in comparison to the reference genome), the mutation is not always annotated as the shortest mutation per genotype. As shown in Figure 2A the genetic variant for genotype2 is annotated as GTT → GTTGTT. MMARGE processes each genotype separately and therefore calculates the shortest genetic variation for each genotype (in this case T → TGTT).
Figure 2.

Details of pipeline: (A) MMARGE merges SNP and InDel VCF files and then splits the merged file. It finds the shortest annotation for each mutation, changing the original annotation from the VCF file. (B) Comparison of the overall mapping efficiency. There is a small decrease in overall mappability when data is mapped to the reference. (C) Comparison of mapping efficiency for uniquely mapped reads after mapping to different genomes. There is an increase in mapping performance when mapped to individualized genomes. (D) Percentage of peaks uniquely called to dataset mapped to one genotype versus another. Up to 12% of peaks are unique to one genotype. (E) Pipeline for processing heterozygous data: Data is mapped to both alleles and shifted back to the reference coordinates. Reads that do not uniquely align to the genome are filtered out. Perfectly aligned reads, as well as perfectly aligned reads overlapping mutations are filtered out and peaks are called on perfectly aligned reads. For each locus without any mutations, the peaks for both alleles are annotated with half the reads that mapped to this locus. For each locus with mutations a ratio is calculated based on the reads overlapping mutations and then the locus is annotated with the number of perfectly aligned reads multiplied by the corresponding ratio. (F) Schematic of the shifting process: Genomic coordinates of the individual genomes do not concur with the reference due to InDels. MMARGE shifts the individual coordinates to the reference without changing the length of the sequence. (G) Shifting peak coordinates leads to minor loss of peaks. 34 PU.1 ChIP-seq data sets were mapped and peaks were called before and after shifting. Even with 2 million genetic variants between the reference and the individualized genomes only up to 11 peaks are different. (H) UCSC genome browser shot showing PU.1 ChIP-seq data in large peritoneal macrophages in 3 different inbred strains of mice (C57, NOD, and SPRET). Bed graphs generated by MMARGE show genetic differences between the strains. The red rectangle shows a zoomed-in area of the UCSC genome browser.
Generating individualized genomes
MMARGE produces individualized genomes by inserting the alleles from the VCF file into the reference genome and generating fasta files, which then can be used to make indices for mapping software. For homozygous data, only one genomic sequence is generated. Generation of individualized genomes and interpretation of allele-specific mapping for heterozygous data requires an additional step. Specifically, alleles at heterozygous sites need to be assigned on the same chromosome as neighboring heterozygous alleles. In genetics, this is called knowing the phase of the genotypes. Phase is especially important for MMARGE when variants are in close proximity, because most sequencing reads are between 50 and 200 bp in length. When multiple SNPs reside in the same read, the correct combination of alleles in the genomic index is essential for accurate mapping and downstream interpretation. MMARGE inherently assumes that all heterozygous data is phased. There are good resources for phasing genotypes in human populations. For example, phasing can be achieved using BEAGLE (32) or SHAPEIT (33) in conjunction with known haplotype structure of large reference populations such as the 1000 Genomes Project. In cases were phasing is not easily possible (e.g. F2 generation of inbred mice) loci where mutations overlap within the read length should be excluded from the analysis.
Mapping data to individualized genomes
Mapping of sequencing experiments to the individualized genome provides better results and decreases the possibility of incorrect mapping due to technical bias (Figure 1C) (34–37). This is especially true in datasets with a large number of differences to the reference. In these cases, mapping to the reference can introduce bias and in the case of datasets containing heterozygous genotypes can lead to overestimation of allele-specific expression or binding (36–38). To assess the effect of individualized genomes on mapping, we used a ChIP-seq dataset from inbred strains of mice. This provided a simplified situation since their genomes are entirely homozygous and all sequence reads originated from a genome of known sequence. Specifically, we used a PU.1 ChIP-seq dataset from three strains of mice (C57BL/6J, NOD/ShiLtJ, and SPRET/EiJ) (21). C57BL/6J (C57) is the commonly used reference genome and differs to NOD/ShiLtJ (NOD) in about 5 million genetic variants (89% SNPs, 11% InDels), whereas SPRET/EiJ (SPRET) provides about 43 million variants (89% SNPs, 11% InDels). Mapping of the ChIP-seq data to their respective genomes affected the overall mappability of the reads (Figure 2B) and the percentage of uniquely mapped reads (Figure 2C). The difference in mapping is directly correlated to the number of differences between the genomes. After removing all reads that map to multiple locations, peaks were called on all datasets separately and compared. Peaks from the C57 ChIP-Seq mapped to C57 and NOD genomes show only small differences (Figure 2D) (about 1% of peaks are unique to either genotype), but increasing the number of variation between the genotypes lead to many peaks uniquely called in one of the mapped datasets (up to 12%). Also when comparing a PU.1 ChIP-seq dataset in human lymphoblastoid cell lines (25) mapped to the reference versus the individual genomes only ∼90% of reads where mapped to the same loci (Supplementary Figure S1A). The number of differences between the hg19 reference genome and the individualized genomes is smaller than for the mouse data, but still up to 4% of peaks were uniquely called on either the dataset mapped to the reference or the individual genome (Supplementary Figure S1B). Therefore, mapping the data to the correct individualized genome increases the mapping accuracy substantially, leading to a more precise downstream analysis.
Additional processing for heterozygous data
Many studies in mice use hybrid mouse strains (F1) generating heterozygous mice from two homozygous parents (Figure 1D). Furthermore, all human genomes are heterozygous in many loci and due to the advantages in sequencing technology, have become more realistic to study genome-wide. To improve mapping for heterozygous data, statistical methods have been developed (e.g. WASP (39), AlleleSeq (40)). Unfortunately, all of these methods have some downsides. WASP can only handle SNPs, whereas AlleleSeq is able to integrate all kinds of variation, but only reports heterozygous sites with allele-specific binding after processing. For MMARGE to correctly calculate the association of a motif with binding, however, information about homozygous, as well as not allele-specific bound heterozygous sites is required. In order to analyze heterozygous data with InDels and run MMARGE’s downstream analysis, we map our data to two reference genomes corresponding to alternative parental alleles. To effectively analyze heterozygous data, allele-specific expression or binding needs to be calculated. For this step, MMARGE filters all reads with perfect alignment followed by filtering of all reads spanning a variant between the two parental strains (Figure 2E). If the heterozygous data is not phased, all regions that contain more than one mutation within the length of one read should be excluded from the analysis. This procedure makes sure that it is possible to confidently identify the allele of origin. To assign allele-specific reads correctly, all loci without any variation are annotated with half of the perfectly aligned reads, because half of the reads that are sequenced originate from allele 1 and the other half from allele 2. For loci with allele-specific sequences, the ratio of reads per allele is calculated based on the reads spanning variations. Then the loci are annotated with the corresponding ratio of all perfectly aligned reads mapped to this locus. MMARGE does not use a statistical method to assign allele-specific reads. AlleleSeq uses a sophisticated model for this. It is therefore possible to map the data with AlleleSeq and get all allele-specific heterozygous variants. From this data, the user can generate a HOMER peak file, which contains all allele-specific as well as non allele-specific bound heterozygous sites, as well as all homozygous sites. It is important to keep in mind that AlleleSeq uses a statistical model and some allele-specific binding sites might not reach significance. This might influence the MMARGE results. Furthermore, it is important to make sure that the genome used for AlleleSeq corresponds exactly with the MMARGE shifting vectors to get accurate results.
Shifting to reference coordinates
A major challenge of mapping data to individual genomes is that the experiments cannot be easily compared because of insertions and deletions (Figure 1E). For example, the chromosomal locations between individuals (and across homologous chromosomes within heterozygous individuals) do not correspond to each other anymore. Therefore, to be able to use external analysis software and to visualize the data in the UCSC genome browser (41), we designed MMARGE to shift mapped data back to reference coordinates (Figure 2F). To accomplish this, MMARGE generates shifting vectors for each genome (or haploid genome in the case of human/heterozygous data). Motifs can overlap insertions (M2) and deletions (M1) in the reference genome (Figure 2F). The M2 motif consists of 6 bases, but after shifting the length shrank to 3 bases due to the deletion. Therefore, positional shifting has the potential to introduce problems. For example, InDels can cause potential TF binding motifs to disappear or appear, which is of interest because these cases likely have functional consequence. Another complication of shifting coordinates occurs in the identification of ChIP-seq peaks from variable chromosomal sequences (i.e. shifting can cause a loss of peaks). This is because ChIP-seq peak calling tools often require a minimum length in order to identify peaks and this might not be reached after shifting. To check how frequently a peak was lost, each PU.1 ChIP-Seq dataset performed in human lymphoblastoid cell lines (25) was mapped to its individual genome and peaks were called with HOMER both before and after shifting (MMARGE documentation for more details). There are up to 2 million genetic differences between the reference genome (hg19) and the allele-specific genomes per individuals, but only up to 11 peaks are lost after shifting (which corresponds to <0.1% of all peaks) (Figure 2G, Supplementary Table S1). Also when repeating this procedure for diverse mouse strains (with >40 million genetic differences) only ∼0.8% of all peaks were lost (Supplementary Table S2). These peaks show weaker binding than the peaks that are present before and after shifting according to the ChIP-seq signal (Supplementary Figure S1C). Therefore, despite an opportunity for difference to emerge in peak calling, we conclude that this phenomenon is very rare and does not offset the advantages from more accurate mapping.
Data visualization
Tools like the Integrative Genomics Viewer (IGV) (42,43) allow visualization of individual genomes, but require the user to install the software locally, which is not preferable for data sharing. Furthermore, MMARGE is based on HOMER, which mainly uses the UCSC genome browser (38) as visualization software. To build on the powerful tools HOMER already provides, to allow easy sharing of ChIP-seq, RNA-seq and natural genetic variation data, and to take advantage of the many additional resources the UCSC genome browser provides, MMARGE offers some software to directly visualize the data in the UCSC genome browser. Although a powerful tool, the browser does not allow the usage of other genomes than the references. To account for this, after shifting the genomic coordinates from the individualized genomes to the reference genome, MMARGE can generate UCSC genome browser files (e.g. bedGraphs and bigWig files) that take into account individual genomic features (Figure 1F). In addition, it can generate BED (Browser Extensible Data) (44) files with all alternate alleles relative to the reference coordinates for upload to the genome browser (Figure 2H). We also provide basic tools to interact with the different individual genomes. For example, we make it possible to directly compare the number of polymorphisms between different datasets in a table format for either all variants (Table 1) or for all private variants (those which can only be found in a particular individual compared to all others) (Table 2). More importantly however, MMARGE can align nucleotide sequences from different individuals or chromosome sequences such as nucleotides or protein sequences. This application integrates RefSeq (45) or common gene name information to provide alignment for genes of interest, but is also able to extract the sequence for every genomic location of interest. This provides a fast and easy way to check for differences in genes or non-coding regions for different genetic backgrounds. It also simplifies the design of primers or other constructs, because differences can be checked by simple alignments of VCF files. To enable some more user-specific analysis, MMARGE annotates files containing genomic coordinates with all genetic variants and generates files with genotype-specific sequences.
Table 1.
Overview of all natural genetic variation found in all strain-wise comparisons
| Strain comparison | #SNPs | #InDels |
|---|---|---|
| C57BL/6J versus NOD/ShiLtJ | 4 734 324 | 272 463 |
| C57BL/6J versus SPRET/EiJ | 40 757 582 | 2 206 269 |
| NOD/ShiLtJ versus SPRET/EiJ | 41 033 145 | 2 302 767 |
Table 2.
Overview of all private genetic variation found in in this strain versus all other strains
| Private variation per strain | #SNPs | #InDels |
|---|---|---|
| NOD/ShiLtJ | 2 474 126 | 160 882 |
| SPRET/EiJ | 38 490 407 | 2 101 665 |
De novo motif analysis
One of the first steps in downstream analysis of ChIP-seq data is motif analysis. The de novo motif analysis software from HOMER (1) was adapted to allow the integration of the individual genomes (Figure 1G). We extended the de novo motif finding algorithm (1) with a function to extract the sequences of the different genotypes as inputs to make sure that the motif finding algorithm is applied to the correct sequences and finds the motifs enriched in the sequence of the genotype not of the reference. It is possible to use different genotypes for the foreground sequences and the background sequences when unique peaks in two different genotypes are compared as foreground and background. These extensions make MMARGE a powerful tool in comparing enriched motifs in two different genotypes.
Motif mutation analysis
MMARGE was primarily developed to determine importance of various nearby transcription factor motifs on the binding of a given transcription factor (Figure 1H). It can analyze transcription factor binding profiles for two genomes in a pairwise fashion, but is also able to analyze the binding profiles of many different genomes together (Figure 3). The first case is preferable when two datasets have many genetic differences (e.g. two diverse mouse strains), as it may be more cost effective experimentally (pairwise comparison). For the analysis of human samples, however, it may be preferable to have more individuals, as the number of differences between two human genomes is fewer. In this scenario, a larger sample size may be required to achieve statistical power (all-versus-all comparison). MMARGE uses a list of hand-curated motifs from the JASPAR motif database (46) as default, but also allows user-defined input. Previous studies (21,35,47) showed that this and similar strategies allow the identification of transcription factor binding motifs associated with binding. However, MMARGE is the first tool available to implement this analysis. Furthermore, it is the first tool to allow comparison of more than two individuals.
Figure 3.

Schematic showing the algorithm for the motif mutation analysis for pairwise comparisons or comparisons of a big group of individuals. (A) Pairwise comparison: Data is mapped to individual genomes and shifted to reference coordinates. Peaks are called per genotype and are subsequentially merged and annotated with the tag counts from the tag directories with HOMER. The merged file is iteratively scanned for the TF binding motifs of interest. For all peaks containing the current TF motif of interest (marked in green) the binding difference between the two genotypes is calculated (fold change). For each TF the fold change distribution of all peaks is plotted (more information Supplementary Figure S2A) and a Student's t-test is performed on the fold change distribution of all peaks versus all peaks containing a mutation in genotype1 (red) (genotype2 (blue), respectively). Further a t-test is performed comparing the fold change distribution of all peaks missing the motif of interest in genotype1 versus genotype2 (purple) and corrected for multiple testing. (B) Motif mutation analysis on more than two genotypes: Data is mapped to the individual genomes, shifted back to the reference coordinates and peaks are called on each genotype separately and subsequentially merged and annotated. Heterozygous data should be annotated with MMARGE’s annotation function. The merged file is iteratively scanned for the TF motif of interest (marked in green). Per TF an output file is generated containing the locus, the binary existence of a motif, the motif score and the read counts. This output file is then inserted into a linear mixed model (LMM) implemented in R with the package lme4 modeling the binding as dependency of the motif score (or motif existence) with random factors Strain and Locus. A p-value is generated using the R command drop1 and corrected for multiple testing.
Pairwise comparison
For the pairwise comparisons, peak files of both genotype alignments are merged and annotated with read counts (Figure 3A). To account for differences between the alleles, the individual genome sequence is extracted and scanned with the motif-scanning algorithm provided by HOMER. Each motif is analyzed separately. Peaks without the motif that is currently scanned for are excluded from the analysis of this particular motif, but are considered for other motifs. Therefore, the analysis of every transcription factor motif is done on a different number of peaks. The fold change of the normalized read counts between the two alleles is calculated. Finally, the distribution of the fold change is calculated for all peaks, all peaks with a mutation in the motif of interest in allele1 and all peaks with a mutation in the motif of interest in allele2. To ensure that a motif is not just considered allele-specific because its log-odds score was slightly below the arbitrarily defined threshold in one of the alleles, MMARGE extracts the sequence of the potential motif from each allele and calculates the log-odds score based on the provided position weight matrix (PWM). By default a motif is considered missing when the log-odds score is smaller or equal to zero, but the user can change this value to whatever seems suitable. MMARGE also provides the possibility to define a motif as missing when its log-odds score in one allele is <n% of the log-odds score in the other allele. To determine the significance of every motif a Student's t-test is performed between the general fold change distribution and the fold change distribution of allele1 and allele2, respectively.
Furthermore, the p-value between the distributions of the two alleles is calculated. This procedure is repeated for all transcription factors of interest. All p-values are multiplied by the number of comparisons to correct for multiple testing.
Allele-specific binding can be observed due to the loss of the binding site for the collaborative factors or the measured transcription factor itself. In addition to analyzing every peak with the motif of interest, MMARGE can analyze only peaks where all loci with differences in the motif of the measured TF between genotypes are filtered out. A Student's t-test is performed on the remaining distributions and the p-values are multiplied by the number of comparisons. MMARGE outputs a motif mutation plot showing the distribution of mutations in relation to the fold change for each transcription factor (bottom Figure 3A, Supplementary Figure S2A). It further outputs a density distribution plot for the fold change distribution of all peaks with changes in the motif in allele1, allele2 and the background (Supplementary Figure S2B).
All-versus-all comparison
In order to perform an all-versus-all comparison on more than two genotypes, peaks are called for all genotypes individually (Figure 3B) and annotated with read counts. In case of heterozygous genotypes, peaks should be called on alleles separately and also be annotated with allele-specific reads (Figure 2E). Both alleles are then analyzed as if they were independent genotypes. Therefore, when comparing for example three heterozygous genotypes, MMARGE actually analyzes 6 independent samples. All sequences of all genotypes are scanned for the motifs of interest. To model the impact of the motif on the binding of the measured factor a Linear Mixed Model (LMM) is used. The binding of the measured factor is modeled as the fixed effect motif existence or motif score (defined by the user) with random effects locus and genotype (Formula 1) with the lme4 package (48) in R (49).
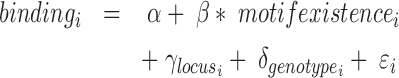 |
or
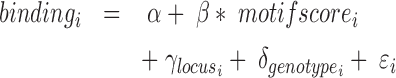 |
with
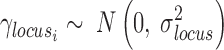 |
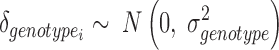 |
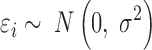 |
To calculate significance for each motif, the drop1 command is used. It compares a model including motif score (motif existence, respectively) with a model without motif score (motif existence, respectively) and reports the Akaike information criterion (AIC) (50) for the difference. To keep the run time reasonable, MMARGE implements threading for this procedure.
Data mapping
All data was mapped using bowtie2 (28) with default parameters. The data for the different inbred strain of mice and the human data were mapped to the individualized genomes. The individualized genomes were generated using bowtie2-build with default parameters. The data for C57 was mapped to the mm10 reference genome from the UCSC genome browser (41). The human reference genome was hg19. Uniquely mapped reads are all reads that were mapped to only one unique region of the genome.
To analyze the impact of the genome on the accuracy of the mapping, all mouse ChIP-seq data sets in LPMs (21) were mapped to the three strain genomes C57, NOD, and SPRET. For the human data (25), all data was mapped against the individualized genome for allele 1, allele 2 and the hg19 reference genome. To assess the impact of the mapping on peak calling all reads that were mapped to more than one region of the genome were removed.
ChIP-seq analysis
All ChIP-seq data sets were analyzed with HOMER after being shifted to reference coordinates. Peaks were called using findPeaks with default parameters and –style factor. For the LPM data set inputs were used for the peak calling. In case of the liver data and the human data no input was available and peaks were called without inputs. After running MMARGE on the data, the list of significant motifs was reduced and summarized using HOMER’s compareMotifs.pl. To compare the binding strength of peaks before and after shifting, tag directories were made with the shifted and unshifted data. Peaks then were called and the peaks from the unshifted data set were shifted towards the reference. After that, peaks were merged and annotated with the shifted tag counts.
Simulation of a data set
MMARGE is based on the model of collaborative binding for TFs and important collaborative TF binding motifs therefore should be identified as significant. According to this model a TF can only bind if the collaborative factor can bind, too. Applying this idea to two different genotypes means that if the motif is missing in genotype1 the binding of the measured factor should be lost in genotype1 and be not affected in genotype2 (genotype-specific binding). It further means if the motif is found in both genotypes binding should be similar between them (genotype-similar binding).
For the synthetic dataset, ten motifs were randomly chosen and defined as important collaborative TF for PU.1 (Tead3, Ventx and Zic1), somewhat collaborative (Rora, Znf354c and Plag1) and not collaborative (Pax6, Nr4a2, Lin54 and Bhlha15) (Figure 4A). The genomes from three mouse strains (C57BL/6J (C57), BALB/cJ (BALB), and SPRET/EiJ (SPRET)) were scanned for the occurrence of all motifs (including PU.1). Next a peak file was generated for all genomic locations where the motif of interest was within 200 bp of the PU.1 motif. These files were merged between two strains (C57 and BALB, C57 and SPRET, BALB and SPRET). To model genotype-specific binding, the fold change was randomly chosen to be between 2- and 10-fold. For genotype-similar binding the fold change between the strains was within 1.5-fold. In all cases the read counts were randomly chosen between 0 and 500. To include biological noise in this dataset 85% of peaks with genotype-specific TF binding motifs follow the genotype-specific binding for highly collaborative motifs. For somewhat collaborative motifs 50% follow this pattern, whereas in the case of not collaborative motifs only 10% of peaks with genotype-specific TF motifs also show genotype-specific binding. To model genotype-similar binding for all highly collaborative motifs 85% of all peaks with the same motif show genotype-similar binding, for somewhat collaborative motifs 50% of the peaks have genotype-similar binding, whereas for not collaborative motifs only 10% show genotype-similar binding. The rest of the peaks show genotype-specific binding randomly assigned to one of the two strains.
Figure 4.

Analysis of a simulated dataset (A) Motifs were defined as important collaborative TF (Tead3, Ventx, Zic1), somewhat collaborative (Rora, Znf354a, Plag1) and not collaborative (Pax6, Nr4a2, lin54, Bhlha15). Peak files were generated for all loci where PU.1 and one of the TF are within 200bp to each other for three mouse strains (C57, BALB, and SPRET) and consecutively merged between two strains. For highly collaborative TF 85% of the strain specific peaks show strain specific binding (somewhat collaborative: 50%, not collaborative: 10%). Fold change was randomly chosen to be between 2- and 10-fold for differently and to be between 1- and 1.5-fold for similarly bound peaks. Read counts were randomly chosen to be between 0 and 500. (B) MMARGE correctly identifies the association between motif and binding data. Motif mutation distribution plot (Supplementary Figure S2A) for one collaborative motif (Tead3) shows a highly significant association between motif mutation and binding data (medium significance for Plag1 (somewhat collaborative), no significant for Nr4a2 (not collaborative)). (C) Summary heat map for all analysis on the simulated datasets. MMARGE showed high significance for the collaborative TF and less or no significance for non-collaborative TF binding motifs. (D) Motif mutation position plot for Tead3, showing which positions are mutated and associated with different binding (more information Supplementary Figure S3A). It furthermore shows that in most cases InDels and multiple SNPs cause significant change in binding. (E) TF binding motif distribution of PU.1 and Ventx. Motifs for Ventx are closely distributed around the PU.1 binding site (more information Supplementary Figure S3B).
RESULTS
MMARGE recognizes collaborative motifs in synthetic dataset
To test the accuracy of the method, a synthetic dataset was generated simulating a ChIP-seq experiment using an antibody against PU.1 (for more details see Material and Methods, Figure 4A). Ten motifs were randomly chosen and defined as important collaborative TF for PU.1 (Tead3, Ventx and Zic1), somewhat collaborative (Rora, Znf354c and Plag1) and not collaborative (Pax6, Nr4a2, Lin54 and Bhlha15) (Figure 4A). Data was simulated for three different homozygous mouse strains (C57, BALB, and SPRET). Comparing one representative of the different motif categories shows that the algorithm is able to detect very high significance for Tead3 (defined as highly collaborative), medium significant for Plag1 (defined as somewhat collaborative) and no significance for Nr4a2 (defined as not collaborative) (Figure 4B, Supplementary Figure S2A). In all three comparisons ∼50% of all peaks had the motif of interest, so the significance is not dependent on the percentage of peaks having the motif. The algorithm is able to detect significance for all motifs that were collaborative and showed lower or no significance for all non-collaborative motifs (Figure 4C). PU.1 was almost always recognized as a significant motif, which is expected as the peaks were modeled according to a PU.1 ChIP-seq experiment.
MMARGE analysis output
In order to learn more about important position in the motif of the candidate transcription factor, MMARGE offers a motif mutation position analysis (Figure 4D, Supplementary Figure S3A). Figure 4D shows an example for mutations within the PU.1 motif for the comparison C57 versus BALB on the simulated data set for Ventx. Mutations with significant effects on binding are marked by dots, whereas stars mark mutations with non-significant effects. Each base is colored differently, so it is not only possible to see which positions are mutated (significantly and non-significantly), but also to which other base. In the simulated dataset, even highly conserved residues in the motif can have mutations without an effect on binding (e.g. Figure 4D, the highly conserved guanine at position 8 has 21 mutations from G→A that are significant but also 5 mutations from G→A with no effect). In the simulated data this was inherently part of it due to the modeling of biological noise (15% of genotype-specific peaks did show genotype-similar binding). It also should be noted that most differences that could be found were InDels (63 significant versus 10 not significant) or multiple SNPs within one motif (27 significant versus 3 not significant). MMARGE also provides a plot that shows the distribution of the Ventx motif around the anchor transcription factor motif PU.1 (Figure 4E, Supplementary Figure S3B) to see if the motif overlaps the anchor TF motif or if it is only randomly distributed within the peak. This plot allows the user to explore how the motifs of interest are distributed around the center of the peak to get a better understanding of the effect of this motif on the binding of the anchor TF.
Pairwise analysis of mouse data
To show that the method also works on real data we analyzed data previously published in (21) and (51). We assessed PU.1 (a macrophage LDTF) binding in large peritoneal macrophages (LPM) in three different inbred mouse strains C57BL/6J (C57), NOD/ShiLtJ (NOD), and SPRET/EiJ (SPRET). These strains differ substantially in mutations to each other (Table 1). To show the correctness of the method we generated a list of motifs that were previously discovered (21) to be involved in the establishment of PU.1 binding in macrophage (PU.1, PU.1-IRF, ETS1, SpiB, CEBP, AP-1, Arid3a). Additionally, we chose some transcription factors not expressed in LPMs or with known binding patterns different from PU.1 in macrophages. We chose the motifs of Bcl6 (not expressed in LPM, with a known function in B cells (52)), NeuroD1 (not expressed in LPM, associated with neurons (53) and diabetes (54)), RORgt (not expressed in LPM and mainly associated with thymocytes (55,56)), and Gfi1b (not expressed in LPM and associated mainly with neutrophil differentiation (57)).
MMARGE could reliably detect motifs that are significantly associated with PU.1 binding, independent of the number of peaks containing the motif, or the number of mutations in these peaks. For example mutations in CEBP, an important LDTF in macrophages, were detected as significantly associated with PU.1 binding (Figure 5A). The plot showing the positions of mutations within the motif shows enrichment for mutations in the conserved bases T (bases 2 and 3) and A (bases 8 and 9) in comparison to the rest of the bases in the motif (Figure 5B). Most causal mutations are due to multiple SNPs or InDels, not merely one single SNP. The CEBP motif is distributed closely around the PU.1 motif (where PU.1 is bound) without any motifs overlapping the PU.1 binding site (Figure 5C). Although the peaks are 200 bp with regard to the reference genome, the sequences analyzed can be longer due to long insertions in the different strains resulting in peaks with a size of 300 in this case. Figure 5D shows two examples of how SNPs can influence observed PU.1 binding. In the left panel PU.1 is only bound in SPRET. A SNP in SPRET in comparison to C57 and NOD adds a PU.1 binding motif adjacent to an existing CEBP motif resulting in the observed genotype-specific binding. The right panel shows how loosing a CEBP binding motif in C57 and SPRET close to a PU.1 binding motif existing in all three strains can cause PU.1 binding to be lost. MMARGE could not find any significant association between motif existence and binding for the motifs chosen to provide negative controls (Figure 5E). Although the number of mutations between two genotypes correlates with the significance of the analysis result (due to a bigger sample size), even with a low number of genetic variations MMARGE was able to detect almost all significant motifs. To further test MMARGE, we applied it to ChIP-seq experiments in four different mouse strains (C57BL/6J (C57), A/J (AJ), CAST/EiJ (CAST) and SPRET/EiJ (SPRET)) for three different factors (CEBPa, FOXA1, and HNF4A) in whole liver from (51). CEBPa is an important TF in hepatocytes (58,59) (which make up ∼70% of all cells in the liver (60)) and macrophages. FOXA1 plays important roles for the development and maintenance of the liver, mainly in hepatocytes (15,61) and HNF4A is an important liver TF mainly associated with hepatocytes (reviewed in (62)). Figure 5F shows an example where the TF binding motifs for all three factors were found, but binding could only be observed in AJ, C57 and CAST. Binding in SPRET was lost due to the loss of an adjacent RORA motif. After applying MMARGE to the data, all significant motifs were compared to each other and summarized (compare Materials and Methods). In almost all pairwise comparisons for the three different factors the measured factor and the two collaborative factors were found as highly significant (Figure 5G). Nuclear receptors, which play important roles in the liver (reviewed in (63)), were found as significant in all three comparisons.
Figure 5.

Analysis using MMARGE’s pairwise-comparison (A) Motif mutation plot (Supplementary Figure S2A) for PU.1 data in LPMs analyzing the impact of mutations in the CEBP binding motif on PU.1 binding. Red ticks show mutations in the CEBP motif in C57 (blue for SPRET). Loss of the CEBP motifs is significantly associated with strain-specific PU.1 binding. (B) Motif position mutation plot (Supplementary Figure S3A) for CEBP motif showing the position and effect of mutations in the CEBP motif in relation to PU.1 binding. The most conserved positions in the CEBP motif are associated with a loss of PU.1 binding. (C) The CEBP motif is distributed closely around the PU.1 motif with a depletion of the CEBP motif at the PU.1 binding site (Supplementary Figure S3B). (D) UCSC genome browser shot - Left panel: The gain of a PU.1 motif in SPRET adjacent to a CEBP motif results in PU.1 binding only in SPRET, but not in C57 or NOD. Right panel: The gain of a CEBP motif in NOD in close vicinity to a PU.1 motif results in PU.1 binding only in NOD. (E) Summary heat map of multiple testing corrected p-values for TF motifs associated with PU.1 binding. The heat map includes some negative control motifs that are not associated with macrophage biology which were not identified as significant. (F) UCSC genome browser shot. The loss of a RORA TF motif in SPRET causes loss of binding of CEBPa, FOXA1, and HNF4A in SPRET, but not in AJ, C57 and CAST. (G) Summary heat map of multiple testing corrected p-values of TF binding motifs associated with CEBPa, FOXA1, and HNF4A binding in whole liver. All factors reached significance in every pairwise comparison. Nuclear receptors were significantly associated with binding of the different factors.
All-versus-all analysis of homozygous mouse data
To show the correctness of the all-versus-all analysis, we reanalyzed the mouse ChIP-seq datasets for CEBPa, FOXA1, and HNF4A from whole liver (Figure 6A). Almost all motifs that were found significant in at least one pairwise comparison were detected as significant in the all-versus-all comparisons (compare Figures 5G and 6A). Applying the motif score or the motif existence in the LMM produced almost the same results, with some motifs differing. The motif existence approach should be used with caution since adjusting the threshold that defines a sequence as motif can have large impacts on the results. Therefore, the all-versus-all comparison is able to confirm motifs significantly associated with binding of CEBPa, FOXA1 or HNF4A in whole mouse liver previously identified by MMARGE’s pairwise comparisons. To make sure that the all-versus-all comparison is sensitive, we shuffled the strain order and repeated the analysis (Figure 6B). To assess how much the results are influenced when very similar strains are shuffled, AJ and C57 were switched, but CAST and SPRET were kept at the same position. The further assess robustness of the results, the more diverse strains were shuffled with the more similar strains. Furthermore, we used completely different mouse genomes (NOD/ShiLtJ (NOD), DBA/2J (DBA), PWK/PhJ (PWK) and WSB/EiJ (WSB)). The color bar in Figure 6b shows the number of differences between the strains. When two very similar strains were changed (AJ with C57) the results are almost the same and the data sets are clustered together. However, as soon as more different strains are switched, the results changed dramatically. Motifs that are significant in all comparisons (e.g. NF1) should be counted as false positive results. This analysis shows that changing very similar data sets with each other does not affect the results, probably because most of the informative loci are found between these two strains and the two more diverse strains.
Figure 6.

Results of all-versus-all analysis. (A) Summary heat map of multiple testing corrected p-values of all-versus-all analysis of CEBP, FOXA1, and HNF4A ChIP-seq data sets from whole liver in AJ, C57, CAST, and SPRET. The analysis confirms the results from the pairwise analysis performed in Figure 5G. The same motifs are highly significance with slight variations independent of considering motif score (MS) or motif existence (ME). (B) Summary heat map of multiple testing corrected p-value of the all-versus-all analysis for CEBP, FOXA1, and HNF4A ChIP-seq data sets with the original order of the strains and shuffled order of the strains to assess sensitivity of MMARGE. The color of the boxes correlates to the number of mutations (from 0 – white to 50 million – brown). When very similar strains are switched (AJ and C57) the MMARGE results are clustered together. As soon as more diverse strains are switched or different strains are used, the results cluster as outliers to the original data and almost all motifs lose significance. (C) UCSC genome browser shot visualizing three human PU.1 datasets. The allele-specific loss of a PRDM1 motif close to an ETS motif causes allele-specific loss of PU.1 binding. (D) Summary heat map of multiple testing corrected p-values of transcription factor motifs significantly associated with PU.1 binding in human lymphoblastoid cell. Many TF motifs found to be significantly associated with PU.1 binding are either known to play important roles in B cell development and maintenance or cancer. By increasing the stringency of peaks included in the analysis (and decreasing the number of observations) the number of significant motifs decreases. Only PRDM1 is found as significant when using a filter of 16 reads.
All-versus-all analysis of heterozygous human data
To show that MMARGE is also able to analyze data from several human individuals with a low number of mutations, 34 PU.1 ChIP-seq datasets from Waszak et al. (25) were analyzed (listed in Supplementary Table S1). The VCF files were downloaded from the 1000 Genomes Project (31) and the individual MMARGE files and genomes were generated. A bowtie2 (28) index was created for each genome (two indices per genotype—one for the complete genome containing mutations on allele 1 and one for mutations on allele 2) and the ChIP-seq reads were mapped against both indices of the corresponding genotype. Only data sets with an overall mappability of 80% were considered in the downstream analysis (22 individuals). Peaks were called on all perfectly aligned reads and all peaks were merged and annotated allele-specific (320,146 peaks). To see how noise influences the results, MMARGE was applied to an unfiltered peak file, as well as a peak file only containing reliable peaks with at least eight reads in at least on individual (16, respectively). The dataset used in this analysis was based on lymphoblastoid cell lines, human B cell lines infected with an Eppstein-Barr virus to immortalize them.
Because the dataset is based on B cells it is not expected that any macrophage specific LDTFs are significant, instead B cell specific LDTFs (like PRDM1 also known as BLIMP-1, E2A etc.) would be expected to show a significant association with PU.1 binding (64). Figure 6C shows a UCSC genome browser session for one locus in three different individuals where one SNP that causes a loss of a PRDM1 motif close to an ETS factor motif is associated with loss of binding of PU.1. Applying the mutation approach systematically to all loci in all individuals and then summarizing the motifs, MMARGE identified the B cell LDTF PRDM1 as highly significant, as well as a motif belonging to the IRF family of transcription factors known to play a role in B cells (Figure 6D) and an ETS motif, important for PU.1 binding. DUX4 has been previously associated with acute lymphoblastic leukemia (ALL) (65) which is coherent with the cancer-like cell type used in this experiment. MMARGE was able to identify many other important transcription factors for B cells including NUR77 and a KLF binding motif (associated with B cell development (66,67)). The more stringent the filtering, the less significant motifs could be found. Filtering by eight reads, about half of the significant motifs could be found. But filtering by 16 reads only found PRDM1 as significant. This highlights the importance of a good quality data set, because a lot of difference is found in lower bound peaks rather than the top peaks. Overall, MMARGE was able to find significant motifs associated with PU.1 binding in human lymphoblastoid cell lines taking advantage of allele-specific binding in many individuals.
Memory usage and runtime analysis
MMARGE can run on any system, however, it requires a substantial amount of memory. All memory and runtime tests are performed on the data presented in this study. The two most memory demanding parts of MMARGE are shifting files from strains coordinates to the reference, as well as to run the pairwise and all-versus-all mutation analysis. For testing a server system with 56 cores (1 GHz each) and 750GB random-access memory (RAM) was used running CentOS and a GPFS file system. Runtime was measured with Linux's time command and memory usage was monitored using the perl module Memory::Usage. During shifting, the memory consumption is mainly dependent on the number of variations found in one individual versus the reference (Supplementary Figure S4A). The size of the file that is shifted does not influence the memory consumption at all. When shifting files for SPRET, the mouse strain with the most variations (42 million), ∼2GB of memory are required. The same is true for the pairwise MMARGE mutation analysis (Supplementary Figure S4B). When analyzing SPRET versus CAST (which in sum have 60 million mutations), the memory consumption is about 3GB. To run the MMARGE all versus all comparison, the memory consumption depends on the number of individuals and therefore number of mutations considered. In the analysis presented in this study, 22 heterozygous human samples were analyzed, which required 9.3GB of RAM.
In comparison to memory, the runtime for shifting is dependent on the size of the file required to shift (Supplementary Figure S4C) and ranges from about 5 min for sam files of 5GB to about 15 minutes to sam files of about 8GB. As shifting is a linear process, threading is not available for it. To analyze the runtime for MMARGE’s pairwise mutation analysis, the motif file provided in the MMARGE package is used. The runtime can vary greatly by using a smaller or larger motif file. As MMARGE offers multi threading, the runtime can be reduced substantially by using more cores (Supplementary Figure S4D). The runtime is dependent on the number of cores used, as well as the number of mutations considered. All pairwise analysis presented in this study took between 10 and 20 min (using 4 cores), or 5 and 15 min (using 8 cores). This makes it possible to run MMARGE on most modern personal computers.
DISCUSSION
We developed a powerful tool to efficiently analyze ChIP-seq and other NGS data to understand the impact of transcription factor motifs on collaborative binding of transcription factors. MMARGE is the first publicly available suite of software tools to integrate natural genetic variation (including InDels) and NGS binding data and provides complementary algorithms to analyze data from different genetic backgrounds in a pairwise manner as well as by utilizing a linear mixed model. It further provides many useful tools to directly look at genetic differences between different genetic backgrounds. By simulating a dataset and also applying MMARGE to real world data, we could show that the algorithm works correctly in identifying motifs significantly associated with the binding of a measured transcription factor.
Here, we applied MMARGE to ChIP-seq data, which requires a well-working antibody for the reference transcription factor. However, MMARGE can also be applied to ATAC-seq data or DNase I hypersensitivity data, which does not require any previous knowledge. In this case, rather than collaborative binding partners for a reference transcription factor, analysis of open chromatin would be expected to recover the dominant collaborative factors needed to establish open chromatin regions. Therefore, MMARGE can potentially be applied to identify key regulatory factors in any cell type as long as parallel datasets from genetically diverse strains or individuals are available.
The algorithm assumes that the binding of the measured factor is only affected by local mutations in transcription factor binding motifs. As a result, sequence changes that influence binding on a global or long-distance scale in trans will not be detected and introduce noise to this. Furthermore, MMARGE only analyzes one motif at a time. More complex relationships between transcription factors (e.g. the requirement for binding of three factors simultaneously) are not considered in the analysis. As in every analysis based on statistical tests, the power of discovery is dependent on the number of observations. A greater number of genetic variations between two individuals provides a better analysis result and will detect more significant motifs. For comparisons with low numbers of genetic variations MMARGE offers a linear mixed model to increase the power of detection by merging all genetic variation between all individuals. This, however, requires substantially more experiments. Furthermore, the software is dependent on a list of position-weight matrices for the detection of TF binding sites. It is known that TF can bind to very weak motifs that cannot be detected by a motif-scanning algorithm but play important roles in regulating gene expression (68). However, MMARGE is dependent on finding motifs based on scanning the DNA for the consensus sequence provided by the PWM. This limits the sensitivity of MMARGE. Improvements in our understanding how to detect motifs in sequence will therefore improve the power of MMARGE. Similar to de-novo motif finding, also MMARGE only detects TF motifs. There are sometimes many similar transcription factors capable of binding the same consensus motif, which MMARGE cannot discriminate. As more TFs and their motifs are characterized, these types of analysis will surely improve.
Genome-wide association studies (GWAS) evaluating common sequence variants associated with diverse phenotypes consistently demonstrate that the majority of variants reside in non-coding regions of the genome (20,69,70). These findings suggest that such variants impose risk by altering promoter and enhancer elements that regulate gene expression. Interpretation of such variants is currently limited because the genomic location of the regulatory elements at which they could potentially exert their effects varies according to cell type. By identifying important motif mutations, MMARGE can provide a new and unique way to analyze transcription factor binding and detect the major collaborative factors involved in the establishment of cell-specific enhancer landscapes. With the advances in sequencing technology and availability of human samples, MMARGE can facilitate the analysis of datasets that provide insights into the relationship between non-coding genetic variation and gene expression in humans.
DATA AVAILABILITY
The MMARGE source code and installation package are freely available on GitHub (https://github.com/vlink/marge/blob/master/MMARGE_v1.0.tar.gz) or Zenodo (10.5281/zenodo.1245209)
The mouse LPM dataset from (21) was downloaded from the GEO database under accession number GSE62826. The data is available at http://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=vlink&hgS_otherUserSessionName=MARGE_LPM_data. The mouse liver data set from (51) was downloaded from ArrayExpress Archive (http://www.ebi.ac.uk/arrayexpress/) under accession number E-MTAB-1414. The data is available at http://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=vlink&hgS_otherUserSessionName=MARGE_Liver_data. The human data set from (25) was downloaded from the ArrayExpress Archive under accession number E-MTAB-3657. The data is accessible at http://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=vlink&hgS_otherUserSessionName=MARGE_human_data.
MMARGE is implemented in Perl and R (49). It has been tested on several UNIX systems, including CentOS and Debian with Perl version 5.20 and higher and R version 3.3 and higher. We provide a script that installs MMARGE and allows download of pre-processed mutation data from the mouse genome project (30) and the genomes from the 1000 Genome Project used in this manuscript. MMARGE requires the Perl core modules POSIX, Getopt::Long, Storable and threads, as well as the modules Set::IntervalTree (http://search.cpan.org/∼benbooth/Set-IntervalTree/), and Statistics-Basic (http://search.cpan.org/∼jettero/Statistics-Basic-1.6611/). It further requires the R packages SeqLogo (https://bioconductor.org/packages/release/bioc/html/seqLogo.html), gridBase (https://CRAN.R-project.org/package=gridBase), lme4 (48), and gplots (https://CRAN.R-project.org/package=gplots). It also requires an installed version of gzip. For the motif mutation analysis MMARGE requires HOMER (1) (http://homer.ucsd.edu/homer/) to be installed and executable. Without a working installation of HOMER, MMARGE’s functionality is limited to only visualization and annotation of the data.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Leslie van Ael for support with figures. We further thank Ty Troutman, Jenhan Tao, and Inge Holtman for running MMARGE and their feedback. We thank Chris Benner, Ty Troutman, Dylan Skola, Jenhan Tao and Zhengyu Ouyang for critically reading the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NIH [CA173903, GM085764, DK091183]; NIH-NHLBI [R00123485 to C.E.R.]. Funding for open access charge: NIH [CA173903, GM085764, DK091183].
Conflict of interest statement. None declared.
REFERENCES
- 1. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K.. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010; 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barish G.D., Yu R.T., Karunasiri M., Ocampo C.B., Dixon J., Benner C., Dent A.L., Tangirala R.K., Evans R.M.. Bcl-6 and NF-kappaB cistromes mediate opposing regulation of the innate immune response. Genes Dev. 2010; 24:2760–2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Carroll J.S., Meyer C.A., Song J., Li W., Geistlinger T.R., Eeckhoute J., Brodsky A.S., Keeton E.K., Fertuck K.C., Hall G.F. et al. . Genome-wide analysis of estrogen receptor binding sites. Nat. Genet. 2006; 38:1289–1297. [DOI] [PubMed] [Google Scholar]
- 4. John S., Sabo P.J., Thurman R.E., Sung M.H., Biddie S.C., Johnson T.A., Hager G.L., Stamatoyannopoulos J.A.. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat. Genet. 2011; 43:264–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lefterova M.I., Steger D.J., Zhuo D., Qatanani M., Mullican S.E., Tuteja G., Manduchi E., Grant G.R., Lazar M.A.. Cell-specific determinants of peroxisome proliferator-activated receptor gamma function in adipocytes and macrophages. Mol. Cell. Biol. 2010; 30:2078–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nielsen R., Pedersen T.A., Hagenbeek D., Moulos P., Siersbaek R., Megens E., Denissov S., Borgesen M., Francoijs K.J., Mandrup S. et al. . Genome-wide profiling of PPARgamma:RXR and RNA polymerase II occupancy reveals temporal activation of distinct metabolic pathways and changes in RXR dimer composition during adipogenesis. Genes Dev. 2008; 22:2953–2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ghisletti S., Barozzi I., Mietton F., Polletti S., De Santa F., Venturini E., Gregory L., Lonie L., Chew A., Wei C.L. et al. . Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity. 2010; 32:317–328. [DOI] [PubMed] [Google Scholar]
- 8. Pennacchio L.A., Ahituv N., Moses A.M., Prabhakar S., Nobrega M.A., Shoukry M., Minovitsky S., Dubchak I., Holt A., Lewis K.D. et al. . In vivo enhancer analysis of human conserved non-coding sequences. Nature. 2006; 444:499–502. [DOI] [PubMed] [Google Scholar]
- 9. Woolfe A., Goodson M., Goode D.K., Snell P., McEwen G.K., Vavouri T., Smith S.F., North P., Callaway H., Kelly K. et al. . Highly conserved non-coding sequences are associated with vertebrate development. PLoS Biol. 2005; 3:e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Heintzman N.D., Hon G.C., Hawkins R.D., Kheradpour P., Stark A., Harp L.F., Ye Z., Lee L.K., Stuart R.K., Ching C.W. et al. . Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009; 459:108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thurman R.E., Rynes E., Humbert R., Vierstra J., Maurano M.T., Haugen E., Sheffield N.C., Stergachis A.B., Wang H., Vernot B. et al. . The accessible chromatin landscape of the human genome. Nature. 2012; 489:75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Visel A., Blow M.J., Li Z., Zhang T., Akiyama J.A., Holt A., Plajzer-Frick I., Shoukry M., Wright C., Chen F. et al. . ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009; 457:854–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Consortium E.P., Bernstein B.E., Birney E., Dunham I., Green E.D., Gunter C., Snyder M.. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bossard P., Zaret K.S.. GATA transcription factors as potentiators of gut endoderm differentiation. Development. 1998; 125:4909–4917. [DOI] [PubMed] [Google Scholar]
- 15. Lee C.S., Friedman J.R., Fulmer J.T., Kaestner K.H.. The initiation of liver development is dependent on Foxa transcription factors. Nature. 2005; 435:944–947. [DOI] [PubMed] [Google Scholar]
- 16. McPherson C.E., Shim E.Y., Friedman D.S., Zaret K.S.. An active tissue-specific enhancer and bound transcription factors existing in a precisely positioned nucleosomal array. Cell. 1993; 75:387–398. [DOI] [PubMed] [Google Scholar]
- 17. Kaikkonen M.U., Spann N.J., Heinz S., Romanoski C.E., Allison K.A., Stender J.D., Chun H.B., Tough D.F., Prinjha R.K., Benner C. et al. . Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Mol. Cell. 2013; 51:310–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Link V.M., Gosselin D., Glass C.K.. Mechanisms underlying the selection and function of macrophage-specific enhancers. Cold Spring Harb. Symp. Quant. Biol. 2015; 80:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Heinz S., Romanoski C.E., Benner C., Allison K.A., Kaikkonen M.U., Orozco L.D., Glass C.K.. Effect of natural genetic variation on enhancer selection and function. Nature. 2013; 503:487–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hindorff L.A., Sethupathy P., Junkins H.A., Ramos E.M., Mehta J.P., Collins F.S., Manolio T.A.. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:9362–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gosselin D., Link V.M., Romanoski C.E., Fonseca G.J., Eichenfield D.Z., Spann N.J., Stender J.D., Chun H.B., Garner H., Geissmann F. et al. . Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell. 2014; 159:1327–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hogan N.T., Whalen M.B., Stolze L.K., Hadeli N.K., Lam M.T., Springstead J.R., Glass C.K., Romanoski C.E.. Transcriptional networks specifying homeostatic and inflammatory programs of gene expression in human aortic endothelial cells. Elife. 2017; 6:e22536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fisher R. The correlation between relatives on the supposition of Mendelian inheritance. Trans. R. Soc. Edinb. 1919; 52:399–433. [Google Scholar]
- 24. Pinheiro J.B., Douglas. Mixed-Effects Models in S and S-PLUS. 2000; Springer. [Google Scholar]
- 25. Waszak S.M., Delaneau O., Gschwind A.R., Kilpinen H., Raghav S.K., Witwicki R.M., Orioli A., Wiederkehr M., Panousis N.I., Yurovsky A. et al. . Population variation and genetic control of modular chromatin architecture in humans. Cell. 2015; 162:1039–1050. [DOI] [PubMed] [Google Scholar]
- 26. Link V.M., Duttke S.H., Chun H.B., Holtman I.R., Westin E., Hoeksema M.A., Abe Y., Skola D., Romanoski C.E., Tao J. et al. . Analysis of genetically diverse macrophages reveals local and domain-wide mechanisms that control transcription factor binding and function. Cell. 2018; 173:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Danecek P., Auton A., Abecasis G., Albers C.A., Banks E., DePristo M.A., Handsaker R.E., Lunter G., Marth G.T., Sherry S.T. et al. . The variant call format and VCFtools. Bioinformatics. 2011; 27:2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Keane T.M., Goodstadt L., Danecek P., White M.A., Wong K., Yalcin B., Heger A., Agam A., Slater G., Goodson M. et al. . Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 2011; 477:289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Genomes Project, C. Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A. et al. . A global reference for human genetic variation. Nature. 2015; 526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Browning S.R., Browning B.L.. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 2007; 81:1084–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Delaneau O., Marchini J., Zagury J.F.. A linear complexity phasing method for thousands of genomes. Nat. Methods. 2011; 9:179–181. [DOI] [PubMed] [Google Scholar]
- 34. Castel S.E., Levy-Moonshine A., Mohammadi P., Banks E., Lappalainen T.. Tools and best practices for data processing in allelic expression analysis. Genome Biol. 2015; 16:195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen J., Rozowsky J., Galeev T.R., Harmanci A., Kitchen R., Bedford J., Abyzov A., Kong Y., Regan L., Gerstein M.. A uniform survey of allele-specific binding and expression over 1000-Genomes-Project individuals. Nat. Commun. 2016; 7:11101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Satya R.V., Zavaljevski N., Reifman J.. A new strategy to reduce allelic bias in RNA-Seq readmapping. Nucleic Acids Res. 2012; 40:e127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stevenson K.R., Coolon J.D., Wittkopp P.J.. Sources of bias in measures of allele-specific expression derived from RNA-sequence data aligned to a single reference genome. BMC Genomics. 2013; 14:536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Degner J.F., Marioni J.C., Pai A.A., Pickrell J.K., Nkadori E., Gilad Y., Pritchard J.K.. Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data. Bioinformatics. 2009; 25:3207–3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. van de Geijn B., McVicker G., Gilad Y., Pritchard J.K.. WASP: allele-specific software for robust molecular quantitative trait locus discovery. Nat. Methods. 2015; 12:1061–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rozowsky J., Abyzov A., Wang J., Alves P., Raha D., Harmanci A., Leng J., Bjornson R., Kong Y., Kitabayashi N. et al. . AlleleSeq: analysis of allele-specific expression and binding in a network framework. Mol. Syst. Biol. 2011; 7:522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D.. The human genome browser at UCSC. Genome Res. 2002; 12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Robinson J.T., Thorvaldsdottir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P.. Integrative genomics viewer. Nat. Biotechnol. 2011; 29:24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Thorvaldsdottir H., Robinson J.T., Mesirov J.P.. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 2013; 14:178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. O’Leary N.A., Wright M.W., Brister J.R., Ciufo S., Haddad D., McVeigh R., Rajput B., Robbertse B., Smith-White B., Ako-Adjei D. et al. . Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016; 44:D733–D745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mathelier A., Fornes O., Arenillas D.J., Chen C.Y., Denay G., Lee J., Shi W., Shyr C., Tan G., Worsley-Hunt R. et al. . JASPAR 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2016; 44:D110–D115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Heinz S., Romanoski C.E., Benner C., Allison K.A., Kaikkonen M.U., Orozco L.D., Glass C.K.. Effect of natural genetic variation on enhancer selection and function. Nature. 2013; 503:487–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Douglas B., Martin M., Ben B., Steve W.. Fitting linear mixed-effects models using lme4. J. Stat. Softw. 2015; 67:1–48. [Google Scholar]
- 49. R Development Core Team R Foundation for Statistical Computing. 2016; Vienna. [Google Scholar]
- 50. Akaike H. Information theory and an extension of the maximum likelihood principle. Proceeding of the Second International Symposium on Information Theory. 1973; 267–281. [Google Scholar]
- 51. Stefflova K., Thybert D., Wilson M.D., Streeter I., Aleksic J., Karagianni P., Brazma A., Adams D.J., Talianidis I., Marioni J.C. et al. . Cooperativity and rapid evolution of cobound transcription factors in closely related mammals. Cell. 2013; 154:530–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dent A.L., Vasanwala F.H., Toney L.M.. Regulation of gene expression by the proto-oncogene BCL-6. Crit Rev Oncol Hematol. 2002; 41:1–9. [DOI] [PubMed] [Google Scholar]
- 53. Gao Z., Ure K., Ables J.L., Lagace D.C., Nave K.A., Goebbels S., Eisch A.J., Hsieh J.. Neurod1 is essential for the survival and maturation of adult-born neurons. Nat. Neurosci. 2009; 12:1090–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kanatsuka A., Tokuyama Y., Nozaki O., Matsui K., Egashira T.. Beta-cell dysfunction in late-onset diabetic subjects carrying homozygous mutation in transcription factors NeuroD1 and Pax4. Metabolism. 2002; 51:1161–1165. [DOI] [PubMed] [Google Scholar]
- 55. Kurebayashi S., Ueda E., Sakaue M., Patel D.D., Medvedev A., Zhang F., Jetten A.M.. Retinoid-related orphan receptor gamma (RORgamma) is essential for lymphoid organogenesis and controls apoptosis during thymopoiesis. Proc. Natl. Acad.. Sci. U.S.A. 2000; 97:10132–10137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sun Z., Unutmaz D., Zou Y.R., Sunshine M.J., Pierani A., Brenner-Morton S., Mebius R.E., Littman D.R.. Requirement for RORgamma in thymocyte survival and lymphoid organ development. Science. 2000; 288:2369–2373. [DOI] [PubMed] [Google Scholar]
- 57. Anguita E., Candel F.J., Chaparro A., Roldan-Etcheverry J.J.. Transcription factor GFI1B in health and disease. Front. Oncol. 2017; 7:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jakobsen J.S., Waage J., Rapin N., Bisgaard H.C., Larsen F.S., Porse B.T.. Temporal mapping of CEBPA and CEBPB binding during liver regeneration reveals dynamic occupancy and specific regulatory codes for homeostatic and cell cycle gene batteries. Genome Res. 2013; 23:592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang N.D., Finegold M.J., Bradley A., Ou C.N., Abdelsayed S.V., Wilde M.D., Taylor L.R., Wilson D.R., Darlington G.J.. Impaired energy homeostasis in C/EBP alpha knockout mice. Science. 1995; 269:1108–1112. [DOI] [PubMed] [Google Scholar]
- 60. Racanelli V., Rehermann B.. The liver as an immunological organ. Hepatology. 2006; 43:S54–S62. [DOI] [PubMed] [Google Scholar]
- 61. Moya M., Benet M., Guzman C., Tolosa L., Garcia-Monzon C., Pareja E., Castell J.V., Jover R.. Foxa1 reduces lipid accumulation in human hepatocytes and is down-regulated in nonalcoholic fatty liver. PLoS One. 2012; 7:e30014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Babeu J.P., Boudreau F.. Hepatocyte nuclear factor 4-alpha involvement in liver and intestinal inflammatory networks. World J. Gastroenterol. 2014; 20:22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wagner M., Zollner G., Trauner M.. Nuclear receptors in liver disease. Hepatology. 2011; 53:1023–1034. [DOI] [PubMed] [Google Scholar]
- 64. Matthias P., Rolink A.G.. Transcriptional networks in developing and mature B cells. Nat. Rev. Immunol. 2005; 5:497–508. [DOI] [PubMed] [Google Scholar]
- 65. Izraeli S. Deciphering “. ”: Novel fusion genes driving B-cell acute lymphoblastic leukemia. B-others. EBioMedicine. 2016; 8:8–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Vu T.T., Gatto D., Turner V., Funnell A.P., Mak K.S., Norton L.J., Kaplan W., Cowley M.J., Agenes F., Kirberg J. et al. . Impaired B cell development in the absence of Kruppel-like factor 3. J. Immunol. 2011; 187:5032–5042. [DOI] [PubMed] [Google Scholar]
- 67. Zikherman J., Parameswaran R., Weiss A.. Endogenous antigen tunes the responsiveness of naive B cells but not T cells. Nature. 2012; 489:160–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Farley E.K., Olson K.M., Zhang W., Brandt A.J., Rokhsar D.S., Levine M.S.. Suboptimization of developmental enhancers. Science. 2015; 350:325–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Degner J.F., Pai A.A., Pique-Regi R., Veyrieras J.B., Gaffney D.J., Pickrell J.K., De Leon S., Michelini K., Lewellen N., Crawford G.E. et al. . DNase I sensitivity QTLs are a major determinant of human expression variation. Nature. 2012; 482:390–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Trynka G., Sandor C., Han B., Xu H., Stranger B.E., Liu X.S., Raychaudhuri S.. Chromatin marks identify critical cell types for fine mapping complex trait variants. Nat. Genet. 2013; 45:124–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The MMARGE source code and installation package are freely available on GitHub (https://github.com/vlink/marge/blob/master/MMARGE_v1.0.tar.gz) or Zenodo (10.5281/zenodo.1245209)
The mouse LPM dataset from (21) was downloaded from the GEO database under accession number GSE62826. The data is available at http://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=vlink&hgS_otherUserSessionName=MARGE_LPM_data. The mouse liver data set from (51) was downloaded from ArrayExpress Archive (http://www.ebi.ac.uk/arrayexpress/) under accession number E-MTAB-1414. The data is available at http://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=vlink&hgS_otherUserSessionName=MARGE_Liver_data. The human data set from (25) was downloaded from the ArrayExpress Archive under accession number E-MTAB-3657. The data is accessible at http://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=vlink&hgS_otherUserSessionName=MARGE_human_data.
MMARGE is implemented in Perl and R (49). It has been tested on several UNIX systems, including CentOS and Debian with Perl version 5.20 and higher and R version 3.3 and higher. We provide a script that installs MMARGE and allows download of pre-processed mutation data from the mouse genome project (30) and the genomes from the 1000 Genome Project used in this manuscript. MMARGE requires the Perl core modules POSIX, Getopt::Long, Storable and threads, as well as the modules Set::IntervalTree (http://search.cpan.org/∼benbooth/Set-IntervalTree/), and Statistics-Basic (http://search.cpan.org/∼jettero/Statistics-Basic-1.6611/). It further requires the R packages SeqLogo (https://bioconductor.org/packages/release/bioc/html/seqLogo.html), gridBase (https://CRAN.R-project.org/package=gridBase), lme4 (48), and gplots (https://CRAN.R-project.org/package=gplots). It also requires an installed version of gzip. For the motif mutation analysis MMARGE requires HOMER (1) (http://homer.ucsd.edu/homer/) to be installed and executable. Without a working installation of HOMER, MMARGE’s functionality is limited to only visualization and annotation of the data.