


Abstract
The two paralogous zinc finger factors CTCF and CTCFL differ in expression such that CTCF is ubiquitously expressed, whereas CTCFL is found during spermatogenesis and in some cancer types in addition to other cell types. Both factors share the highly conserved DNA binding domain and are bound to DNA sequences with an identical consensus. In contrast, both factors differ substantially in the number of bound sites in the genome. Here, we addressed the molecular features for this binding specificity. In contrast to CTCF we found CTCFL highly enriched at ‘open’ chromatin marked by H3K27 acetylation, H3K4 di- and trimethylation, H3K79 dimethylation and H3K9 acetylation plus the histone variant H2A.Z. CTCFL is enriched at transcriptional start sites and regions bound by transcription factors. Consequently, genes deregulated by CTCFL are highly cell specific. In addition to a chromatin-driven choice of binding sites, we determined nucleotide positions critical for DNA binding by CTCFL, but not by CTCF.
INTRODUCTION
In recent years the multifunctional and highly conserved factor CTCF (CCCTC-binding factor) has been identified as a key player in 3D chromatin architecture and gene regulation (1–3). CTCF binds DNA through a combination of 11 zinc-fingers from its central DNA binding domain (4). At its binding sites it can interact with a variety of co-factors, most importantly the cohesin complex to mediate the formation of long-distance DNA interaction and DNA loops (5). Such looping events can then link three-dimensional genomic architecture to a functional output such as the regulation of genes through an enhancer or insulator (6). Utilizing techniques like 3C (chromatin conformation capture) and its genome-wide derivatives such as Hi-C, topologically associated domains (TADs) could be identified and CTCF was found to be enriched in the border areas of such domains (7). Disruption of CTCF binding and binding sites leads to changes in TAD patterns and has effects on proper gene expression programs (8). Taken together, CTCF is one of the central factors in bridging genome architecture to function.
In contrast to the established role of CTCF, the cellular role of the only known CTCF-paralogue, CTCFL, remains to be solved. CTCFL was identified in 2002 (9) and is believed to result from a gene duplication event in the early amniotic evolution (10), with CTCF and CTCFL sharing a highly conserved 11 zinc finger (ZF) DNA binding domain. The N- and C-termini of the two proteins are different, with an amino acid similarity of <20% between mammalian versions (11). First reports described CTCFL expression to be testis specific and mutually exclusive to CTCF. Later, more detailed analysis could show CTCFL to be transiently expressed during spermatogenesis, prior to the onset of meiosis, overlapping with CTCF expression (12). Some functional differences regarding the two proteins have been identified, for instance it seems that only CTCF binds components of the Cohesin complex like Smc1 in mouse (12) or RAD21 in human (13). CTCFL also failed to substitute for a loss of CTCF in CTCF KO experiments (12). Further Knockout experiments of CTCFL showed it to be important in proper testicular development. This is exemplified by the deregulation of important testis-specific genes, such as Gal3st1 and Prss50 (12,14,15). Tissue specificity of CTCFL expression has been questioned (16) by showing a more widespread expression in normal and in cancer cells. Aberrant expression of CTCFL was identified in some cancers (17–19). Research to identify CTCFL as a biomarker for specific cancer types (20) and for therapeutical approaches has been followed up (21).
With the advent of next-generation-sequencing many advances in the field of DNA binding factors have been made. Also for CTCFL, the genome-wide binding patterns have been started to be explored (12,13). Sites of CTCFL binding strongly overlap with CTCF sites and the identified DNA binding motifs of the two proteins are virtually identical (12,13,22). CTCFL seems to preferentially bind to genomic regions of active and open chromatin showing for example an enrichment at transcriptional start sites compared to CTCF (12). CTCFL binding is strongly associated with the presence of active histone modifications like H3K4me3 or H3K27ac (12,13). Most recently, it could be shown that CTCFL binds genomic sites characterized by the presence of two CTCF-motifs in close proximity allowing for simultaneous binding of CTCF and CTCFL (13). In mouse and human, the similarity between the ZF DNA binding domains of the two proteins is ∼70%, on the amino acid level (11), which also might explain some degree of differential binding.
Thus, epigenetic marks and dual binding motifs contribute to binding specificity. However, it has not been analysed, whether epigenetic marks are solely responsible for the binding specificity such that a closed site in a particular tissue is not bound by CTCFL, but will be bound in another tissue with an open chromatin conformation. Here, we find that this is exactly the case. CTCF is binding irrespective of chromatin ‘openness’, whereas CTCFL binding is regulated by epigenetic marks characteristic for open chromatin. In addition, we find that not all CTCF sites can potentially be bound by CTCFL; rather, DNA-sequence specificity restricts CTCFL binding to a sub-set of CTCF sites.
MATERIALS AND METHODS
Cell culture and transfection
Murine NIH3T3 and P19 cells as well as human K562 cells were grown at 37°C with 5% CO2 in Dulbecco's modified Eagle's medium supplemented with 10% (v/v) serum and 1% PenStrep. Differentiation of P19 cells was achieved by supplementing cells grown on adherent dishes with 10 μM retinoic acid. Transfections were performed on adherent cells using jetPEI reagent (Polyplus transfection), which was used in accordance to the manufacturer's instructions. To generate stable clones, cells were transfected with pBI-EGFP-FLAG-mCtcfl and pTA-N, a Tet-off system (Clonetech) turning off the expression of CTCFL in the presence of Doxycycline (2 μg/ml). The transfected cells were selected for puromycin resistance starting 24 h after transfection. The clones were selected in 96-well plates, expanded and characterized by immunoblotting, RT-qPCR and immunofluorescence. CTCFL expression was achieved by growing the cells in medium lacking Doxycycline for 48 h.
ChIP-seq data analysis of K562 ENCODE data
The K562 pre-aligned ChIP-seq data (hg19) were downloaded from ENCODE (23) via the UCSC genome browser portal (Supplementary Table S1) (24). CTCF and CTCFL peaks were called by MACS2 with standard settings (25). Peaks overlapping the ENCODE blacklisted regions for the hg19 regions were removed from the analysis. The set of peaks overlapping between both replicates were used for subsequent analyses. We defined five different categories: all CTCF sites, all CTCFL sites, CTCF/CTCFL co-bound sites as well as CTCF or CTCFL stand-alone sites, respectively. We defined two sites being overlapping in case a minimal overlap of 1 bp between the two peak intervals was observed. All downstream analysis was done in R/BioConductor (26).
The GenomicRanges BioCOnductor function reduce was used in order to merge CTCF and CTCFL peaks to a common set (27). Reads from bam files were imported through Rsamtools functions (Morgan, M., Pagès, H., Obenchain, V. and Hayden, N. (2016) Rsamtools: Binary alignment (BAM), FASTA, variant call (BCF), and tabix file import. R package version 1.24.0.; http://bioconductor.org/packages/release/bioc/html/Rsamtools.html.). We constructed a count matrix containing information about the number of reads per individual peak for all ChIP-seq data sets mentioned above using the countOverlaps function of the GenomicRanges BioC package. Read counts per peak were normalized using the FPKM (fragments per kilobase per million of sequenced reads) method normalizing for the total number of sequencing reads as well as for the size of the given peak interval. In order to control for potential biases introduced by the ChIP-seq technology we decided to subtract the corresponding FPKM value of the input control, respectively. We calculated the normalized read counts per peak N as
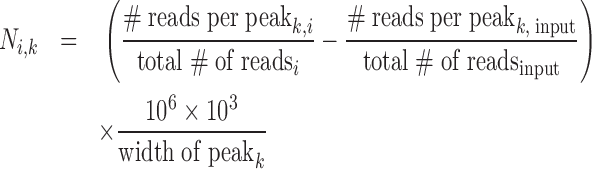 |
with i being the i-th data set and k indicating the peak index ranging from 1 to the total number of peaks under analysis (38 365).
The resulting count matrix was used for generating boxplots in Figure 1 and Supplementary Figure S1. Statistical differences between individual binding categories (CTCFL, CTCF/CTCFL co-bound and CTCF or CTCFL stand-alone sites) and the average CTCF site as reference were calculated by Wilcoxon signed rank test (two-sided alternative: ‘binding in analyzed category is greater than that of CTCF binding’). The corresponding code is available upon request.
Figure 1.

CTCFL binding events positively correlate with active histone modifications and transcription factors but not with components of the cohesin complex in K562 cells. ENCODE ChIPseq data was retrieved (see Materials and methods) and box-plotted over five subgroups (A) of CTCF/CTCFL-binding events. Shown are the FPKM values after subtraction of the corresponding input control values (see supplements) for active histone marks (B) and known transcription factors (C). As a measure of open chromatin we compared the binding sites to DNase-seq experiments (D). Correlation with CTCF interacting components of the cohesin complex was determined (E). Statistical evaluation is listed in Supplementary Table S3.
Comparison to genomic annotations
RefSeq gene annotations for Homo sapiens were downloaded from UCSC homepage (version hg19). Next the genome was partitioned into the following intervals: transcriptional start site (TSS; ±1 kb around RefSeq start sites), TSS upstream (−10 kb to −1 kb), transcriptional end sites (TES; ±1 kb around transcriptional end sites), exons, introns and everything not covered by these classes as intergenic. CTCF and CTCFL peak ranges were intersected with these annotation intervals and the relative association was calculated (as fraction of the complete genome) and compared to the genomic background distribution.
Chromatin immunoprecipitation (ChIP)
ChIP was performed in a one-day protocol as described in (28). Confluent cells growing in 15 cm dishes were fixed with a final concentration of 1% formaldehyde (Calbiochem) for 10 min and quenched for 5 min using 1/7 volume of 1 M Glycine. Cells were washed twice with cold PBS and harvested in 1 ml PBS (+ 1 mM PMSF). After pelleting, cells were taken up in 1 ml IP buffer (150 mM NaCl, 50 mM Tris–HCl (pH 7.5), 5 mM EDTA, NP-40 (0.5% vol/vol), Triton X-100 (1.0% vol/vol)), supplemented with protease inhibitors (Complete Mini, Roche) per 107 cells and incubated for 10 min in ice. The cell solution was then sonified using a Bioruptor (Diagenode) for 15 cycles (30 s ON/OFF) followed by pelleting of cell debris (10 min at 14 000 rpm). The chromatin containing supernatant was transferred to new tubes and diluted 1:10 with IP buffer. 1 ml of this dilution was used in each precipitation. Ten percent were always used in parallel as an input sample. The solution was pre-cleared for 2 h using 30 μl Protein-A/G-Agarose beads (Calbiochem) by rotating at 4°C. After mild centrifugation (5 min; 2000 rpm) the supernatant solution was transferred to new tubes and corresponding antibodies (CTCF N2.2 (29); CTCFL S6 (12)) were added over night at 4°C on a rotating wheel. The antibody/protein/DNA complexes were bound by addition of 30 μl Protein-A/G-Agarose beads for 2 h rotating at 4°C. The beads were then washed 5 times for 5 min using 1 ml IP buffer each. 100 μl 10% Chelex 100 resin (Bio-Rad) was added to the beads, briefly vortexed and boiled for 10 min. An optional Proteinase K digestion was performed. Chelex and beads were spun down and 80 μl supernatant were transferred to a new tube. Beads were re-suspended in 120 μl MilliQ H2O, spun down and the supernatant was pooled with the previous supernatant. This solution was then ready for qPCR and library preparation.
Deep sequencing of ChIP DNA and bioinformatics analysis
Samples were prepared as described for ChIP, but the elution step after DNA purification was performed with H2O instead of elution buffer. If necessary, samples generated with the same antibody were pooled and volume was reduced by evaporation to 30 μl to obtain at least 10 ng of total DNA. Sequencing libraries were prepared from 10 ng of immunoprecipitated DNA with the NEBNext ChIP-Seq Library Prep Reagent (New England Biolabs) according to manufacturer's instructions. Cluster generation was performed using the cBot (Illumina Inc.). Sequencing was done on the HiSeq 2500 (Illumina Inc.) using TruSeq SBS Kit v3 – HS (Illumina) for 50 cycles. Image analysis and base calling were performed using the Illumina pipeline v 1.8 (Illumina Inc.). Raw and processed data have been deposited in the NCBI gene expression omnibus (GEO) under accession number GSE103199.
ChIP-Seq reads were converted to fastq format and aligned to a precompiled hg19 reference index with BOWTIE with -k option set to 1 (30). Sequencing data were controlled for general quality features using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Unambiguously mapped and unique reads were kept for subsequent generation of binding profiles and calling of peaks using MACS v1. (25) at default settings. In case of 3T3 cells uninduced clone 34 served as reference sample. In case of p19 we used corresponding input samples. All downstream analyses were done in R/BioConductor (http://www.bioconductor.org).
Peaks identified by MACS at a Poisson P value < 10−5 and an FDR <5% were used for intersection analysis to determine the overlap in pairwise comparisons. Two peaks were determined to be overlapping in case they had a minimal overlapping interval of 1 bp.
Analysis of CTCF core sequence preferences
The chromosomal locations, hg18 coordinates and CTCF sequences of all CTCF motifs of ranks 1 to 1000 were used as identified (31). We used the liftOver utility of the UCSC genome browser for converting CTCFL and CTCF peak intervals from hg18 to hg19 coordinates (32). The overlap of CTCF and CTCFL peaks with each genomic instance of the top 100 sequences was than calculated and presented as percentages.
Comparison between gene expression changes and CTCFL binding
CTCF/ CTCFL peaks were called as described above. Additional information about the chromatin state of CTCF/CTCFL sites was derived from publicly available H3K27ac data, which is known to mark active cis-regulatory chromatin segments (33):
H3K27ac peaks were called with MACS v1. In order to assign genes to peaks we followed the association rule 1 proposed by the Bejerano lab (36). In this basal plus extension rule we defined basal regulatory regions as the region from −5000 to + 1000 bp around the TSS. We extended this region to maximally 500 kb until it reaches the next neighboring basal domain. Using this set of regions, we assigned each peak to one or multiple genes. Gene expression changes were determined by Affymetrix Gene arrays and corresponding log2-transformed gene expression changes were compared between all genes, genes bound by CTCFL or genes bound by H3K27ac-overlapping CTCFL binding sites. Wilcoxon signed rank test were performed in order to test for statistical differences between groups.
RNA analysis
For microarray analysis RNA was also isolated using RNeasy Mini Kit and Microarrays were performed using either Affymetrix Gene 1.0 ST Platform (NIH3T3 cells) or Affymetrix Gene 2.0 ST platform (P19 cells). In case of p19 cells, raw data was analysed using Affymetrix own software suite (Expression console & Transcriptome analysis console). In case of NIH3T3 cells, CEL files were processed using the Aroma.Affymetrix package with RMA background subtraction and quantile normalization. Deregulated genes were also verified by qPCR.
Electrophoretic mobility shift analysis (EMSA)
Radiolabeled DNA probes were generated by phosphorylation with gamma 32P ATP and subsequently annealed. 100 fMol of probes were incubated with 5 μl of in vitro produced protein (Promega TnT T7 Quick Coupled Transcription/Translation System) per shift. The binding reaction was performed in PBS (pH 7.4, supplemented with 5 mM MgCl2, 1 mM ZnCl2, 1 mM DTT, 0.1% NP-40 and 10% glycerol) for 20 min at room temperature in the presence of 200 ng/μl pdIdC and 25–100 ng/μl salmon sperm DNA. Protein–DNA complexes were analyzed on nondenaturing polyacrylamide gels (5% acrylamide (w/v)) in TAE-buffer. Electrophoresis was performed at 4°C with a field strength of 12 V/cm for 3–4 h.
Oligonucleotides
Sequences of the genomic regions used in the band shift experiments are listed in Supplementary Table S2.
RESULTS
Active chromatin marks correlate with CTCFL binding
Previous publications have addressed the binding specificity of CTCF and CTCFL in respect to clustering of binding sites (13) and to active chromatin marks (12). Here, we wanted to systematically address the binding determinants of CTCF as compared to CTCFL. We utilized the extensive ENCODE database available for K562 cells to identify possible chromatin marks or transcription factors enriched at CTCF or CTCFL binding sites. K562 cells are positive for CTCF as well as for CTCFL. We first analysed the general overlap of binding sites between the two proteins (Figure 1A). There are 38 365 CTCF binding sites compared to only 13 292 for CTCFL with a shared fraction of 9,397 sites. This means that 70% of CTCFL sites are also occupied by CTCF in K562 cells. We then classified the binding sites for the two proteins in 5 different groups: all CTCF sites (CTCF), all CTCFL sites (CTCFL), shared sites (CTCF + CTCFL), only CTCF bound (CTCF only) and only CTCFL bound sites (CTCFL only). To assess the occupancy of histone marks and of DNA binding factors over the selected subgroups of binding sites we retrieved the respective datasets from the ENCODE database (Supplementary Table S1) and box plotted the log2 transformed IP signal/Input for each factor over a 500 bp window around the identified binding sites. For the five histone modifications H3K27 acetylation, H3K4 dimethylation and trimethylation, H3K79 dimethylation and H3K9 acetylation plus the histone variant H2A.Z, which are all associated with an active, open chromatin conformation, we see a strong enrichment at sites with sole CTCFL binding (Figure 1B, blue) and very weak levels at sole CTCF binding sites (Figure 1B, green). Such a distinctive behaviour could not be seen for histone marks not associated with active chromatin (Supplementary Figure S1). When analysing the occupancies of known transcription factors, the same correlation with sole CTCFL binding sites can be seen. The identified factors are CREB1, ETS1, FOS, HDAC2, TAF7, YY1, POL2 and ZBTB7, all of which show the highest levels of binding in the CTCFL-only subgroup (Figure 1C, blue). In order to challenge the correlation of CTCFL binding sites with open chromatin we used ENCODE DNaseI data in comparison to the five CTCF/CTCFL groups (Figure 1D).This again shows a significant correlation of CTCFL-only sites with open chromatin (DNaseI sensitive). In contrast to the above transcription factors, both cohesin factors RAD21 and SMC3 are specifically depleted within the CTCFL-only subgroup (Figure 1E, blue). For both cohesin components an important co-association with CTCF has been shown (5,6,37,38), mediating three-dimensional long-range chromatin contacts. This finding supports previous results showing that the cohesin complex can be specifically recruited by CTCF, rather than by CTCFL (13). Recruitment is mediated by binding of the cohesin component SA2 (39) to the C-terminal domain of CTCF, which differs from the C-terminal domain of CTCFL.
Taken together, we identified a strong positive correlation of active chromatin marks and transcription factors with CTCFL, which is not seen for CTCF. In contrast, cohesin complex components are enriched at CTCF-only sites and depleted from CTCFL-only locations.
Cell-specific binding of CTCFL is driven by chromatin ‘openness’
CTCFL expression is highly restricted to spermatogenesis and to specific cancer types and cell lines (see K562 cells above) and occurs in addition to the ubiquitous expression of CTCF. In order to analyse the binding specificity of both factors, we mimicked the in vivo situation by conditional expression of CTCFL in cell lines in addition to the endogenous expression of CTCF. We generated CTCFL expressing mouse NIH3T3 and P19 cell clones. To investigate possible cell-type specific binding patterns for CTCFL due to differential chromatin composition we generated genome-wide binding maps in the inducible NIH3T3 and P19 clones. First, we confirmed the specificity of the antibodies used. The CTCFL antibody (CTCFL S6 (12)) specifically recognizes murine CTCFL and not human CTCFL (Supplementary Figure S2). In addition to western blots, the most stringent tests for specificity are ChIP experiments. The CTCF antibody shows ChIPseq signals at sites known to be bound by CTCF (29). The CTCFL antibody is specific for CTCFL as ChIPseq peaks are only detectable after CTCFL expression (Supplementary Figures S3 and S4) and a substantial number of sites are specific for CTCFL and devoid of CTCF (Supplementary Figure S4 and see below, Figure 2B). We than studied the general distribution of the factors over genomic features in the two cell clones. We mapped the binding sites to the features and compared these fractions with the genomic percentage of these features (Figure 2A). In the cell clones we find the genomic associations to be fairly similar. CTCFL shows in both cell types a higher enrichment for regions associated with open chromatin, like TSSs (40), as compared to CTCF (Figure 2A). When comparing the cell types in respect to CTCF, the association with TSSs is less frequent in P19 cells with an increase for intergenic binding. Of all identified binding sites only 1,354 are bound by both factors in both cell types, with varying levels of overlap between the datasets (Figure 2B). CTCF sites are generally more conserved between the cell lines with 41% of 3T3-CTCF sites also present in P19 (Figure 2C). In contrast, only 13% of the 3T3-CTCFL sites are shared (Figure 2C). Taken together, for each of the factors CTCF and CTCFL we find a similar distribution in respect to the genomic features in the two cell types. Nevertheless, the binding site choice of CTCFL is highly cell-specific.
Figure 2.

CTCF and CTCFL show similar genome-wide distribution between two independent cell clones, but the binding sites show only a weak overlap. (A) Annotation of the CTCFL binding sites to the associated genomic feature for the ChIPseq results obtained from NIH3T3 & P19 clones. (B) Four-way-Venn diagram showing the overlap of CTCF & CTCFL binding events between the 2 analysed cell clones. (C) Two-way-Venn diagrams showing the single CTCF and CTCFL overlaps between the cell clones.
Cell-specific deregulation of target genes by conditional CTCFL expression
Given the cell specific binding of CTCFL, we wanted to know, whether this is reflected by a cell specific and CTCFL induced change in expression profiles. Here we used the above cell clones with conditional CTCFL expression. We determined the expression profile in the absence and in the presence of CTCFL induction. This we did for the NIH3T3 cell type as well as for the P19 embryonal carcinoma cells. To further test our hypothesis that CTCFL binding is in part determined by chromatin conformation, we differentiated the P19 cell clone. P19 cells are stem cell-like in nature and can easily be differentiated by retinoic acid treatment (41). We used a long-term differentiation protocol (9 days) to ensure changes in gene expression and chromatin composition, followed by CTCFL induction. Thus, we determined the expression pattern for six conditions, NIH3T3, P19 undifferentiated and P19 differentiated in the absence or after induction of CTCFL in each case. Expression profiles were compared and changes in expression for each of the genes and each of the cell types were determined (Figure 3A, Supplementary Table S4). Log2-transformed fold changes were calculated for the contrasts between CTCFL induced versus non-induced cells and sorted by hierarchical clustering (Figure 3A). A striking pattern is evident, indicating that genes induced or repressed by the expression of CTCFL are highly cell and differentiation specific with almost no overlap between the cell types. Quantitative analysis of CTCFL effected genes (Figure 3B) revealed similar magnitudes of target genes being repressed or induced. Furthermore, cell specificity is highly evident, with none of the repressed genes or only three of the induced genes being shared by all of the three cell types. Even within a single cell line, P19, before and after differentiation, the cell specificity of target genes is highly obvious. In order to test for a functional relationship between CTCFL binding, open chromatin and differential gene expression in CTCFL-expressing versus non-expressing cells we compared the gene expression changes of CTCFL-bound or CTCFL/H3K27ac co-bound genes with the gene expression changes of the average gene (Supplementary Figure S5). Especially CTCFL/H3K27ac co-bound genes turned out to become significantly induced (P-values of 2.00e–07 (NIH3T3); 3.36e–20 (p19 undifferentiated); 1.54e–27 (p19 differentiated)) after expression of CTCFL. These expression changes are not dramatic as strong effects are only seen in a short and transient time window (42).
Figure 3.

Cell specificity of CTCFL deregulated genes. (A) Clustered heatmap of genes that are deregulated by CTCFL in either NIH3T3 cells or in P19 undifferentiated or in differentiated cells. Shown are normalized log2-transformed changes of expression intensities (Supplementary Table S4). Hierarchical clustering was performed using average linkage and cosine distance metric. (B) Three-way-Venn diagrams showing the overlap of upregulated or downregulated genes between the three comparisons.
Sequence preference of CTCFL binding
Besides cell specificity of CTCFL target genes, very few genes regulated by CTCFL are shared between cell types (Figure 3 and Supplementary Table S4). One gene to be pointed out is the Adgrg1 gene (or GPR56), which was induced in all three cases. This gene is known to play an important role in spermatogenesis. Mice deficient for GPR56 are impaired in male gonad development and in fertility (43). Furthermore, GPR56 has been shown to be involved in cancer progression (44). We reasoned that in this case, CTCFL binding in three different cell types might be driven by sequence specificity, in addition to chromatin features. Previous identification of the binding consensus could not detect differences between CTCF versus CTCFL (12). To explore a possible sequence specificity for both factors we followed a recently described workflow. In this study, ENCODE datasets were used in order to determine the top 1000 unique 14 bp core sequences bound in each case by CTCF in at least 50 instances in the genome of K562 cells (31). Subsequently, we mapped the CTCFL occupancy to each of these sequences (Figure 4A). A general correlation can be observed with the most frequently CTCF bound sequences also showing the highest percentages of bound CTCFL instances. In addition, single, highly CTCF bound sequences without any CTCFL binding can be detected. To identify possible small DNA sequence differences with an impact on CTCFL occupancy, we grouped the individual core sequences with the highest CTCFL binding into one class and a second class not bound by CTCFL, but highly occupied by CTCF. For this we used DNA sequence alignment and clustering algorithms (45). Comparison of the top 20 sequences of both classes (Supplementary Figure S6) with Clustal Omega (data not shown) identified a single sequence from each of the two classes, respectively, with higher similarity between each other than with any other sequence of the two classes. These two highly similar sequences, CCAGCAGAGGGCGC & TCAGTAGAGGGCGC, show only a two base difference, but at the same time strong differences in CTCFL occupancy (Figure 4 B, crucial bases indicated in red; Supplementary Figure S6). The presence of a T at positions 1 and 5 in the core sequence coincides with a complete lack of CTCFL occupancy. However, when a C is located at these positions, 43% of these sequences are bound by CTCFL in vivo. In contrast to CTCFL, the level of CTCF occupancy is similarly high for both sequences with 98% and 100% of these sequences in the genome showing binding (Figure 4B). These results lead us to propose that these two base changes may have an influence on in vitro binding of CTCFL.
Figure 4.

DNA sequence contributes to differential CTCF and CTCFL binding. ENCODE K562 binding data for CTCF and CTCFL were analysed utilizing the approach by Liu et al. (31). CTCF DNA binding sequences were grouped and sorted by their percentage of instances bound by the CTCF factors in K562 cells. (A) Correlation plot showing the percentage of CTCF and CTCFL bound instances for each identified core motif. (B) Identification of two highly similar sequences showing CTCF occupancy in all instances but with differential CTCFL binding. Sequence differences are marked in red. The number of instances found in the human genome are shown for the CTCF plus CTCFL binding sequence (left) and the CTCFL only binding sequence (right).
This hypothesis was tested by using double-stranded oligonucleotides of a genomic region, 51 base pairs in length, which centres on the identified CCAGCAGAGGGCGC sequence. We chose the binding site HNR (named for the neighbouring gene Hnrnpul1), which is strongly bound by both, CTCF and CTCFL in vivo (Supplementary Figure S7A). We generated the specific mutation TCAGTAGAGGGCGC (HNRmB), which we predicted to be impaired in CTCFL binding. Vice versa a 51-bp genomic region centring on the non CTCFL-bound motif TCAGTAGAGGGCGC, SLC (named for the gene Slc22a18as in which it is located), was chosen. Endogenous chromatin binding demonstrates CTCF occupancy, but no CTCFL binding (Supplementary Figure S7B). We predicted that replacing the ‘T’ at positions 1 and 5 by ‘C’ (CCAGCAGAGGGCGC -SLCpB) should allow for binding by CTCF as well as by CTCFL. Electrophoretic mobility shift assays (EMSA) were carried out with CTCF and CTCFL translated in vitro (Figure 5). As predicted, the oligonucleotides, HNR and SLCpB, were bound by CTCF as well as by CTCFL. Competition with an excess of unlabelled oligonucleotide showed the specificity of binding. Use of the corresponding variants, HNRmB and SLC, resulted in a similar binding of CTCF, as determined from similar competition efficiencies. In contrast, CTCFL did not bind to the two sequences containing the motif TCAGTAGAGGGCGC irrespective of its flanking sequence regions. To further challenge the conclusion that HNR is a binder for CTCF and CTCFL and that CTCFL binding is specifically impaired in the HNRmB mutation or that SLC does not bind CTCFL, but achieves binding upon mutation to SLCpB, we set up a pairwise competition assay for both wildtype and mutant sequences in parallel. This allows for direct comparison of the sequence pairs in binding affinity to CTCFL (Figure 5B). As expected, CTCFL binding to the HNR probe can be efficiently competed by HNR (with only 8% of the signal remaining), whereas competition by the point mutant HNRmB is less efficient. Similarly, CTCFL binding to the SLCpB probe is efficiently competed by this probe (with only 5% of the signal remaining), whereas competition by the wildtype probe SLC is less efficient.
Figure 5.

Specific point mutations determine CTCFL binding specificity. EMSA experiments were performed by incubating in vitro expressed CTCF or CTCFL with 32P-labeled DNA probes. For competition experiments increasing amounts (2.5-, 10- and 40-fold) of un-labeled probes were used. Samples were run on a 5% PAA gel and analysed by autoradiography. Arrows indicate the CTCF and CTCFL specific shifts. Two binding sites were chosen (HNR and SLC). HNR is bound in vivo by both factors, whereas SLC is not bound by CTCFL. This difference is recapitulated by the in vitro binding. (A) Replacing the ‘C’ at positions 1 and 5 by ‘T’ within the HNR site CCAGCAGAGGGCGC created HNRmB. As predicted, this site binds CTCF in vitro, but is unable to bind CTCFL (top right). The reverse experiment replacing the ‘T’ at positions 1 and 5 by ‘C’ within the SLC site TCAGTAGAGGGCGC created SLCpB. This generates a site bound by both factors (bottom left). (B) Pairwise comparison of wildtype and mutant sequence in competition efficiency for CTCFL binding. Remaining shift signals in maximal competition (40-fold) are indicated (Signal). This supports the observation in (A), that HNRmB lost its binding capacity to CTCFL, whereas SLCpB gained binding to CTCFL.
This supports our bioinformatics prediction, derived from in vivo bound sequences, that specifically for CTCF sites devoid of CTCFL binding, specificity is determined by the very sequence and not so much by chromatin conformation.
DISCUSSION
CTCF is a highly conserved and essential factor involved in regulating the crosstalk between distant chromatin regions. A gene duplication event generated a related gene coding for CTCFL (10). The DNA binding domain of both factors is very similar, harbouring eleven, almost identical zinc fingers (9,13). CTCFL expression during spermatogenesis and in some cancer types revealed a problem in understanding mechanistically how this factor in the presence of the ubiquitous factor CTCF is binding to chromatin. Binding site specificity of CTCF in comparison to CTCFL has been studied in several cases. By determining the consensus sequence of all sites bound by CTCF and of all sites bound by CTCFL, almost identical consensus sequences have been generated (12,46). In focussing on the number of consensus sequences within CTCF or CTCFL bound regions, it has been shown, that CTCFL preferentially binds to clustered binding motifs (13). Co-binding by both factors explained the apparent resistance of CTCF competition by CTCFL binding. Clustered binding sites with CTCFL binding have been found to be enriched at active promoters in cancer cells (13). Thus, a specific sequence feature enriched in active promoters could solely determine CTCFL binding selectivity. Alternatively, or in addition, binding site selection might be mediated by chromatin modification marks characteristic for open and active chromatin. Here we find that CTCFL binding sites are highly enriched for H3K27 acetylation, H3K4 di- and trimethylation, H3K79 dimethylation and H3K9 acetylation plus the histone variant H2A.Z. In case such modifications might contribute to binding site selectivity one would expect, that different cell types with different repertoires of active genes, and therefore of active chromatin regions, should respond to CTCFL expression in a cell type specific manner. Here, we find that CTCFL target genes in mouse fibroblasts, embryonal carcinoma cells and neuronal cells are almost not overlapping. This argues for cell specific chromatin marks to contribute to CTCFL function. Therefore we predicted that both, sequence features as well as chromatin marks have a combined effect on CTCFL selectivity.
One example for sequence driven selection is the GPR56 (Adgrg1) gene, which is activated by CTCFL in all three cell types analysed. CTCFL facilitated induction of GPR56 might explain its activity in spermatogenesis (43) as well as in cancer progression (44).
Our bioinformatics analysis identified nucleotide sequences always bound by CTCF, but never by CTCFL. This is another argument for a sequence driven binding selectivity between CTCF and CTCFL. In order to test binding specificity in vitro we chose CTCF and CTCFL bound sequences of the HNR and the SLC locus. Besides locus specific flanking sequences the nucleotide C to T change at consensus positions 3 and 7 resulted in loss of CTCFL binding, but not in CTCF binding. This clearly demonstrated a sequence driven selectivity in binding of the two CTCF factors.
Zinc finger mapping to the consensus sequence revealed fingers 4 to 7 to be involved in recognition of the consensus core sequence (47,48). Fingers 1 to 3 and 8 to 11 are proposed to contact other regions, downstream and upstream flanking the consensus (47). According to the structure of zinc fingers, the amino acids of the alpha helical region are numbered, with positions −1, 2, 3, and 6 being in contact with nucleotides of the binding sequence (49). Within these critical amino acids of fingers 4 to 7, only finger 6 shows an amino acid difference between CTCFL and CTCF (9,46). The critical amino acids are Q-1, G2, T3 and M6 for CTCF and Q-1, G2, T3 and I6 for CTCFL (Figure 6, inset). Thus position 6 of the alpha helix is methionine in case of CTCF and isoleucine in CTCFL. The consensus nucleotides are aligned with the contacting zinc fingers (Figure 6), indicating the potential vicinity between the nucleotides 3 and 7 of the consensus and amino acid M6/I6 of the zinc finger 6. This is in precise agreement with predictions based on empirical calculations of pairwise amino acid–nucleotide interaction energies, showing that the C/T at consensus position 7 is contacted by CTCF zinc finger 6 (50). Furthermore, the recent establishment of the structure of CTCF zinc fingers bound to DNA (51), is in agreement with the alignment shown in Figure 6. We propose that the single amino acid difference between CTCF and CTCFL in zinc finger 6 contributes to the binding specificity within the core consensus.
Figure 6.

CTCF/CTCFL specific zinc finger recognition. The 11 zinc fingers are indicated by their coordinated zinc ion (Zn) and by four amino acids at the alpha-helical positions −1, 2, 3 and 6 as counted from the N-terminal to the C-terminal direction. These amino acids are in contact with nucleotides of the binding sequence (49). The critical finger 6 is magnified in the inset. Single black letters indicate identical amino acids for CTCF and CTCFL. Double red letters indicate the amino acid found in CTCF (first letter) and in CTCFL (second letter). The central fingers 4 to 7 are in contact with the consensus (bottom (47,48)). DNA upstream oft the consensus is bound by fingers 8 to 11 and strongly bent (52). The DNA sequence shown in black letters is bound by CTCF and by CTCFL, whereas the sequence variant indicated by two red T nucleotides is bound by CTCF, but never by CTCFL (see Figure 4).
In summary, we conclude that the binding specificity of the paralogous factors CTCF and CTCFL is determined by three mechanisms. (a) DNA sequence driven specificity has been described for double versus single binding sites (13) in that CTCFL is preferentially bound at double sites. This might be explained by a difference between both factors in binding strength. CTCFL, potentially binding weakly, might require the cooperative binding function of another CTCF molecule bound nearby. In contrast to this prediction, in vitro we do not find a difference in binding affinity. Both factors, when competed with an excess of binding DNA, are competed with comparable DNA amounts (Figure 5). (b) Here, we find that subtle differences in the amino acid sequence of the zinc fingers determine site-specific selectivity. (c) The chromatin driven specificity of binding demonstrated here as well, can be easily attributed to the protein domains outside of the zinc finger region, as these domains are quite different and therefore might differentially interact with chromatin or with chromatin modifying enzymes.
DATA AVAILABILITY
Bioconductor project (http://www.bioconductor.org).
Rsamtools: Binary alignment (BAM), FASTA, variant call (BCF), and tabix file import (http://bioconductor.org/packages/release/bioc/html/Rsamtools.html). Uploaded sequence tracks: (https://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=MarekB&hgS_otherUserSessionName=mm9_Bergmaier_NAR). Raw and processed data have been deposited in the NCBI gene expression omnibus (GEO) under accession number GSE103199.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Joerg Leers for experimental instructions and Leni Schäfer-Pfeiffer for excellent technical assistance.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Deutsche Forschungsgemeinschaft [TRR81; Re 433/23]. Funding for open access charge: DFG [TRR81].
Conflict of interest statement. None declared.
REFERENCES
- 1. Ali T., Renkawitz R., Bartkuhn M.. Insulators and domains of gene expression. Curr. Opin. Genet. Dev. 2016; 37:17–26. [DOI] [PubMed] [Google Scholar]
- 2. Handoko L., Xu H., Li G., Ngan C.Y., Chew E., Schnapp M., Lee C.W., Ye C., Ping J.L., Mulawadi F. et al. . CTCF-mediated functional chromatin interactome in pluripotent cells. Nat. Genet. 2011; 43:630–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Phillips J.E., Corces V.G.. CTCF: master weaver of the genome. Cell. 2009; 137:1194–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ohlsson R., Renkawitz R., Lobanenkov V.. CTCF is a uniquely versatile transcription regulator linked to epigenetics and disease. Trends Genet.: TIG. 2001; 17:520–527. [DOI] [PubMed] [Google Scholar]
- 5. Parelho V., Hadjur S., Spivakov M., Leleu M., Sauer S., Gregson H.C., Jarmuz A., Canzonetta C., Webster Z., Nesterova T. et al. . Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell. 2008; 132:422–433. [DOI] [PubMed] [Google Scholar]
- 6. Wendt K.S., Yoshida K., Itoh T., Bando M., Koch B., Schirghuber E., Tsutsumi S., Nagae G., Ishihara K., Mishiro T. et al. . Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature. 2008; 451:796–801. [DOI] [PubMed] [Google Scholar]
- 7. Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B.. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012; 485:376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lupianez D.G., Kraft K., Heinrich V., Krawitz P., Brancati F., Klopocki E., Horn D., Kayserili H., Opitz J.M., Laxova R. et al. . Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 2015; 161:1012–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Loukinov D.I., Pugacheva E., Vatolin S., Pack S.D., Moon H., Chernukhin I., Mannan P., Larsson E., Kanduri C., Vostrov A.A. et al. . BORIS, a novel male germ-line-specific protein associated with epigenetic reprogramming events, shares the same 11-zinc-finger domain with CTCF, the insulator protein involved in reading imprinting marks in the soma. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:6806–6811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hore T.A., Deakin J.E., Marshall Graves J.A.. The evolution of epigenetic regulators CTCF and BORIS/CTCFL in amniotes. PLoS Genet. 2008; 4:e1000169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tiffen J.C., Bailey C.G., Marshall A.D., Metierre C., Feng Y., Wang Q., Watson S.L., Holst J., Rasko J.E.. The cancer-testis antigen BORIS phenocopies the tumor suppressor CTCF in normal and neoplastic cells. Int. J. Cancer. 2013; 133:1603–1613. [DOI] [PubMed] [Google Scholar]
- 12. Sleutels F., Soochit W., Bartkuhn M., Heath H., Dienstbach S., Bergmaier P., Franke V., Rosa-Garrido M., van de Nobelen S., Caesar L. et al. . The male germ cell gene regulator CTCFL is functionally different from CTCF and binds CTCF-like consensus sites in a nucleosome composition-dependent manner. Epigenet. Chromatin. 2012; 5:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pugacheva E.M., Rivero-Hinojosa S., Espinoza C.A., Méndez-Catalá C.F., Kang S., Suzuki T., Kosaka-Suzuki N., Robinson S., Nagarajan V., Ye Z. et al. . Comparative analyses of CTCF and BORIS occupancies uncover two distinct classes of CTCF binding genomic regions. Genome Biol. 2015; 16:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kosaka-Suzuki N., Suzuki T., Pugacheva E.M., Vostrov A.A., Morse H.C., Loukinov D., Lobanenkov V.. Transcription factor BORIS (Brother of the Regulator of Imprinted Sites) directly induces expression of a cancer-testis antigen, TSP50, through regulated binding of BORIS to the promoter. J. Biol. Chem. 2011; 286:27378–27388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Suzuki T., Kosaka-Suzuki N., Pack S., Shin D.-M., Yoon J., Abdullaev Z., Pugacheva E., Morse H.C., Loukinov D., Lobanenkov V.. Expression of a testis-specific form of Gal3st1 (CST), a gene essential for spermatogenesis, is regulated by the CTCF paralogous gene BORIS. Mol. Cell. Biol. 2010; 30:2473–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jones T.A., Ogunkolade B.W., Szary J., Aarum J., Mumin M.A., Patel S., Pieri C.A., Sheer D.. Widespread expression of BORIS/CTCFL in normal and cancer cells. PLoS One. 2011; 6:e22399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alberti L., Losi L., Leyvraz S., Benhattar J.. Different effects of BORIS/CTCFL on stemness gene expression, sphere formation and cell survival in epithelial cancer stem cells. PLoS One. 2015; 10:e0132977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Martin-Kleiner I. BORIS in human cancers – a review. Eur. J. Cancer. 2012; 48:929–935. [DOI] [PubMed] [Google Scholar]
- 19. Zampieri M., Ciccarone F., Palermo R., Cialfi S., Passananti C., Chiaretti S., Nocchia D., Talora C., Screpanti I., Caiafa P.. The epigenetic factor BORIS/CTCFL regulates the NOTCH3 gene expression in cancer cells. Biochim. Biophys. Acta. 2014; 1839:813–825. [DOI] [PubMed] [Google Scholar]
- 20. Okabayashi K., Fujita T., Miyazaki J., Okada T., Iwata T., Hirao N., Noji S., Tsukamoto N., Goshima N., Hasegawa H. et al. . Cancer-testis antigen BORIS is a novel prognostic marker for patients with esophageal cancer. Cancer Sci. 2012; 103:1617–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Asano T., Hirohashi Y., Torigoe T., Mariya T., Horibe R., Kuroda T., Tabuchi Y., Saijo H., Yasuda K., Mizuuchi M. et al. . Brother of the regulator of the imprinted site (BORIS) variant subfamily 6 is involved in cervical cancer stemness and can be a target of immunotherapy. Oncotarget. 2016; 7:11223–11237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang J., Zhuang J., Iyer S., Lin X., Whitfield T.W., Greven M.C., Pierce B.G., Dong X., Kundaje A., Cheng Y. et al. . Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012; 22:1798–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Consortium E.P. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D.. The human genome browser at UCSC. Genome Res. 2002; 12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W. et al. . Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gentleman R.C., Carey V.J., Bates D.M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J. et al. . Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004; 5:R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lawrence M., Huber W., Pages H., Aboyoun P., Carlson M., Gentleman R., Morgan M.T., Carey V.J.. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 2013; 9:e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nelson J.D., Denisenko O., Bomsztyk K.. Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat. Protoc. 2006; 1:179–185. [DOI] [PubMed] [Google Scholar]
- 29. Weth O., Paprotka C., Günther K., Schulte A., Baierl M., Leers J., Galjart N., Renkawitz R.. CTCF induces histone variant incorporation, erases the H3K27me3 histone mark and opens chromatin. Nucleic Acids Res. 2014; 42:11941–11951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Langmead B. Aligning short sequencing reads with Bowtie. Curr. Protoc. Bioinformatics. 2010; doi:10.1002/0471250953.bi1107s32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu M., Maurano M.T., Wang H., Qi H., Song C.-Z., Navas P.A., Emery D.W., Stamatoyannopoulos J.A., Stamatoyannopoulos G.. Genomic discovery of potent chromatin insulators for human gene therapy. Nat. Biotechnol. 2015; 33:198–203. [DOI] [PubMed] [Google Scholar]
- 32. Hinrichs A.S., Karolchik D., Baertsch R., Barber G.P., Bejerano G., Clawson H., Diekhans M., Furey T.S., Harte R.A., Hsu F. et al. . The UCSC Genome Browser Database: update 2006. Nucleic Acids Res. 2006; 34:D590–D598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Creyghton M.P., Cheng A.W., Welstead G.G., Kooistra T., Carey B.W., Steine E.J., Hanna J., Lodato M.A., Frampton G.M., Sharp P.A. et al. . Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mikkelsen T.S., Xu Z., Zhang X., Wang L., Gimble J.M., Lander E.S., Rosen E.D.. Comparative epigenomic analysis of murine and human adipogenesis. Cell. 2010; 143:156–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Serandour A.A., Avner S., Oger F., Bizot M., Percevault F., Lucchetti-Miganeh C., Palierne G., Gheeraert C., Barloy-Hubler F., Peron C.L. et al. . Dynamic hydroxymethylation of deoxyribonucleic acid marks differentiation-associated enhancers. Nucleic Acids Res. 2012; 40:8255–8265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McLean C.Y., Bristor D., Hiller M., Clarke S.L., Schaar B.T., Lowe C.B., Wenger A.M., Bejerano G.. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010; 28:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rubio E.D., Reiss D.J., Welcsh P.L., Disteche C.M., Filippova G.N., Baliga N.S., Aebersold R., Ranish J.A., Krumm A.. CTCF physically links cohesin to chromatin. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:8309–8314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zuin J., Dixon J.R., van der Reijden M.I., Ye Z., Kolovos P., Brouwer R.W., van de Corput M.P., van de Werken H.J., Knoch T.A., van I.W.F. et al. . Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:996–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xiao T., Wallace J., Felsenfeld G.. Specific sites in the C terminus of CTCF interact with the SA2 subunit of the cohesin complex and are required for cohesin-dependent insulation activity. Mol. Cell. Biol. 2011; 31:2174–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Boyle A.P., Davis S., Shulha H.P., Meltzer P., Margulies E.H., Weng Z., Furey T.S., Crawford G.E.. High-resolution mapping and characterization of open chromatin across the genome. Cell. 2008; 132:311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jones-Villeneuve E.M., Rudnicki M.A., Harris J.F., McBurney M.W.. Retinoic acid-induced neural differentiation of embryonal carcinoma cells. Mol. Cell. Biol. 1983; 3:2271–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nora E.P., Goloborodko A., Valton A.L., Gibcus J.H., Uebersohn A., Abdennur N., Dekker J., Mirny L.A., Bruneau B.G.. Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell. 2017; 169:930–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen G., Yang L., Begum S., Xu L.. GPR56 is essential for testis development and male fertility in mice. Dev. Dyn. 2010; 239:3358–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yang L., Xu L.. GPR56 in cancer progression: current status and future perspective. Future Oncol. 2012; 8:431–440. [DOI] [PubMed] [Google Scholar]
- 45. Sievers F., Higgins D.G.. Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol. Biol. 2014; 1079:105–116. [DOI] [PubMed] [Google Scholar]
- 46. Pugacheva E.M., Teplyakov E., Wu Q., Li J., Chen C., Meng C., Liu J., Robinson S., Loukinov D., Boukaba A. et al. . The cancer-associated CTCFL/BORIS protein targets multiple classes of genomic repeats, with a distinct binding and functional preference for humanoid-specific SVA transposable elements. Epigenet. Chromatin. 2016; 9:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nakahashi H., Kwon K.R., Resch W., Vian L., Dose M., Stavreva D., Hakim O., Pruett N., Nelson S., Yamane A. et al. . A genome-wide map of CTCF multivalency redefines the CTCF code. Cell Rep. 2013; 3:1678–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Burcin M., Arnold R., Lutz M., Kaiser B., Runge D., Lottspeich F., Filippova G.N., Lobanenkov V.V., Renkawitz R.. Negative protein 1, which is required for function of the chicken lysozyme gene silencer in conjunction with hormone receptors, is identical to the multivalent zinc finger repressor CTCF. Mol. Cell. Biol. 1997; 17:1281–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Elrod-Erickson M., Benson T.E., Pabo C.O.. High-resolution structures of variant Zif268-DNA complexes: implications for understanding zinc finger-DNA recognition. Structure. 1998; 6:451–464. [DOI] [PubMed] [Google Scholar]
- 50. Persikov A.V., Singh M.. De novo prediction of DNA-binding specificities for Cys2His2 zinc finger proteins. Nucleic Acids Res. 2014; 42:97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hashimoto H., Wang D., Horton J.R., Zhang X., Corces V.G., Cheng X.. Structural basis for the versatile and methylation-dependent binding of CTCF to DNA. Mol. Cell. 2017; 66:711–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Arnold R., Burcin M., Kaiser B., Muller M., Renkawitz R.. DNA bending by the silencer protein NeP1 is modulated by TR and RXR. Nucleic Acids Res. 1996; 24:2640–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Bioconductor project (http://www.bioconductor.org).
Rsamtools: Binary alignment (BAM), FASTA, variant call (BCF), and tabix file import (http://bioconductor.org/packages/release/bioc/html/Rsamtools.html). Uploaded sequence tracks: (https://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=MarekB&hgS_otherUserSessionName=mm9_Bergmaier_NAR). Raw and processed data have been deposited in the NCBI gene expression omnibus (GEO) under accession number GSE103199.