

Abstract
Simultaneously modeling hybridization and the multispecies coalescent is becoming increasingly common, and inference of species networks in this context is now implemented in several software packages. This article addresses some of the conceptual issues and decisions to be made in this modeling, including whether or not to use branch lengths and issues with model identifiability. This article is based on a talk given at a Spotlight Session at Evolution 2017 meeting in Portland, Oregon. This session included several talks about modeling hybridization and gene flow in the presence of incomplete lineage sorting. Other talks given at this meeting are also included in this special issue of Systematic Biology.
Keywords: Displayed trees, gene flow, likelihood, phylogenetic networks, trait evolution
A subset of the phylogenetic literature deals with networks, in which evolution is represented graphically by networks rather than trees. Network representations of evolution can in turn be either rooted or unrooted (Fig. 1). A fundamental distinction here should be made between implicit networks which are drawn in order to depict signals in the data that are not tree-like, such as used by popular programs like NeighborNet (Bryant and Moulton 2002) and SplitsTree (Huson 1998), but are not intended to model the cause of data not being tree-like, versus approaches that explicitly model violations of tree-like evolution due to biological processes such as hybridization or horizontal transfer. NeighborNet and SplitsTree both draw unrooted networks, and are intended to represent whether distances or frequencies of splits, respectively, can be fit onto a tree in a mathematical sense, regardless of the biological mechanism (Huson et al. 2010; Huson and Scornavacca 2011; Morrison 2011; Bapteste et al. 2013). The nontree-like signal could be due to misestimation of trees, model misspecification, and other causes as well as actual hybridization or horizontal transfer. Unrooted networks can also be drawn that explicitly model hybridization, such as in the software SNaQ (Solís-Lemus et al. 2017) (Fig. 1). This article focuses on explicit phylogenetic networks, for which reticulation nodes represent hybridization events rather than conflicts in the input trees.
Figure 1.

Rooted (a) and unrooted (b) explicit networks. The internal nodes are numbered to help show that (b) is the unrooted version of (a). In rooted networks, a typical assumption is that hybridization nodes (i.e., node 2) have two incoming hybridization edges and one outgoing edge, while non-hybridization tree nodes have one incoming edge and two outgoing edges. Hybridization events result in cycles in the undirected graph, such as that made by nodes 2, 3, 5, and 4 in (a) and nodes 2, 3, and 4 in (b). The network in (b) is obtained from (a) by suppressing node 5 (the root node in (a)) and treating the path from nodes 3 to 4 as a single edge. In both graphs hybridization edges are shown as gray directed edges going into a node. In a), all edges are interpreted as directed away from the root and toward the tips of the network, while in b), only the two hybridization edges are directed. Such a network is called semidirected (Solís-Lemus and Ané 2016).
Much of the literature on phylogenetic networks uses combinatorial approaches (Choy et al. 2005; Huson et al. 2010; Huson and Scornavacca 2011; Bapteste et al. 2013). Many of these involve minimization problems, such as finding the network with the minimum number of hybridization events to explain two conflicting trees (Bordewich and Semple 2007; van Iersel and Linz 2013). However, a relatively new trend is using probabilistic approaches for modeling hybridization that lend themselves to maximum likelihood inference (Meng and Kubatko 2009; Kubatko 2009; Yu et al. 2012; Solís-Lemus and Ané 2016; Wen et al. 2016). Many of these methods are motivated by simultaneously modeling hybridization and incomplete lineage sorting, two biological processes that can lead gene trees to conflict with one another, a phenomenon called gene tree incongruence. One reason for thinking about modeling hybridization and incomplete lineage sorting simultaneously is that if two species or populations are able to hybridize, then it is reasonable to think that they are closely enough related that incomplete lineage sorting is likely to be a prominent cause of gene tree incongruence.
A recent set of spotlight talks at Evolution 2017 in Portland, Oregon focused on issues of modeling hybridization and gene flow. Papers in this issue include Burbrink and Gehara (2018), which finds evidence for ancient hybridization in New World kingsnakes using recent software which models hybridization and coalescence simultaneously. Blischak et al. (2018) introduce software for hybridization detection using invariants in the site pattern probabilities, a technique which has also lead to some success in establishing model identifiability. Long and Kubatko (2018) examine postspeciation gene flow between sister taxa, which can lead to anomalous gene trees (AGTs, gene trees more probable than the gene tree with the same topology as the species tree), and examines robustness of SVDquartets (Chifman and Kubatko 2014) compared to other species tree methods in this setting. Morales and Carstens (2018) also examine postspeciation gene flow in an empirical example for Myotis bats. Bastide et al. (2018) introduce methods for analyzing trait evolution on networks as opposed to trees, which promises to greatly expand methodology for trait evolution modeling.
This article talks about some of the conceptual issues that arise in making modeling decisions when trying to understand hybridization and incomplete lineage sorting simultaneously and discusses some of the issues raised at that meeting.
To outline the article, some of the modeling issues include the following:
Modeling multiple sources of incongruence (e.g., hybridization, incomplete lineage sorting (ILS), recombination, etc.)
Using horizontal versus nonhorizontal hybridization edges
Identifiability and distinguishability
The role of displayed trees
Branch lengths in the network
Branch lengths in the gene trees
Rooted versus unrooted networks
Modeling trait evolution on networks
Distances between networks
Gene Tree Incongruence
Following Rosenberg (2002), I will distinguish between gene tree incongruence, meaning gene trees at different loci having different topologies, versus gene tree discordance, meaning that the gene tree and species tree have different topologies. There are many sources of gene tree incongruence. Conceptually, it is helpful to distinguish between biological sources of incongruence versus statistical sources of incongruence. Examples are summarized in Table 1.
Table 1.
Causes of incongruence
| Source of incongruence | Category | Notes |
|---|---|---|
| Incomplete lineage sorting | Biological | Affects entire genome |
| Hybridization | Biological | Affects entire genome |
| Postspeciation gene flow | Biological | Overlaps with hybridization but is often modeled for gradual speciation as opposed to instantaneous mergers of previously isolated populations |
| Horizontal gene transfer | Biological | Affects segments of a genome |
| Recombination | Biological | Can cause incongruence within genes |
| Ancient population structure | Biological | Can mimic hybridization in terms of gene tree probabilities |
| Low mutation rate | Biological | Affects data quality (low information in data) |
| High mutation rate | Biological | Affects data quality (noisy sequences) |
| Short alignments | Statistical | Affects data quantity (and quality, similar to low mutation rates) |
| Incorrect substitution model | Statistical | Can cause branch length errors and long branch attraction |
| Incorrect biological modeling | Statistical | For example assuming gene trees at linked loci are independent |
| Sequence misalignment/errors | Statistical | Data quality problem |
| Incorrect species assignment | Statistical | Model misspecification |
It should be stressed that these biological processes can make true gene trees conflict independently of how those gene trees are estimated. By “true gene trees,” I mean the actual pattern of ancestor–descendant relationships for the genetic locus in question. Conceptually, it makes sense to talk about the true gene tree even if there is no mutation. In this case, there might not be evidence available to recover the true gene tree, but that is conceptually a separate issue.
Short alignments, especially when the mutation rate is low, can cause a lack of informative sites, effectively making the sample size (i.e., the sequence length) too small. On the other hand, a mutation rate that is too high leads to saturated sequences, in which there is too much noise to recover a clear signal. An incorrect substitution model can also lead to incorrect estimation of the number of multiple mutations at a site, which can lead to incorrectly estimated branch lengths or topologies. The case of incorrect assignment of species to sequences would at least cause discordance of gene trees with the species tree even if it did not cause gene trees to be incongruent with each other. All of these statistical and data quality considerations can lead to poorly estimated gene trees, which might disagree with each other even if the true underlying gene trees are identical.
There are several papers that discuss distinguishing hybridization from ILS (e.g., Holland et al. 2008; Joly et al. 2009; Choleva et al. 2014). This is unfortunate terminology because both processes often co-occur and are not necessarily competing explanations for gene tree incongruence. High levels of ILS can even be beneficial for inferring hybridization (Zhu and Degnan 2017). A reason for this is that when lineages fail to coalesce, they can simultaneously trace multiple paths through a network topology, thus giving information about how often lineages tend to come from one ancestor rather than another.
As mentioned earlier, if species are closely enough related to hybridize, they might also have high levels of ILS precisely because they are closely related. Generally, we expect coalescence effects to always be occurring to some extent because the time to coalescence between two gene lineages predates the time of speciation. If consecutive speciation events are far enough apart, then the extra time to coalescence will almost never persist far enough in the past for gene trees to be discordant with the species tree. In this case, the multispecies coalescent model (MSC) predicts a very low level of incongruence, but this is still compatible with the MSC. It makes sense to think of the MSC as a null hypothesis, and other biological processes, such as recombination, population structure, gene flow, etc. to occur in addition.
Consequently, instead of thinking of distinguishing ILS from other models, I think of the following as possible models (or hypotheses) we might be interested in:
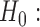 MSC (null model)
MSC (null model)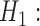 MSC
MSC 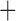 population structure
population structure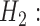 MSC
MSC 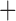 hybridization
hybridization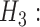 MSC
MSC 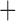 recombination
recombination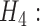 MSC
MSC  population structure
population structure  hybridization etc.
hybridization etc.
In other words, we can think of the MSC as a null model, and more complicated models might invoke additional biological processes. In some cases, it might be difficult to distinguish some of these models. For example, if there are three species, 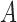 ,
, 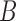 , and
, and 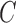 , and the species tree is
, and the species tree is 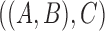 , then the MSC model by itself predicts that the two discordant gene trees
, then the MSC model by itself predicts that the two discordant gene trees 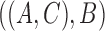 and
and 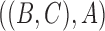 have equal probability (Nei 1987). If the proportions of these two discordant trees differ significantly from each other, then this is evidence against the null hypothesis of the MSC, and this is sometimes used as evidence against the MSC as adequately describing the data (Degnan and Rosenberg 2009; Ané 2010; Chung and Ané 2011; Song et al. 2012). However, the
have equal probability (Nei 1987). If the proportions of these two discordant trees differ significantly from each other, then this is evidence against the null hypothesis of the MSC, and this is sometimes used as evidence against the MSC as adequately describing the data (Degnan and Rosenberg 2009; Ané 2010; Chung and Ané 2011; Song et al. 2012). However, the 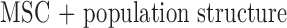 and
and 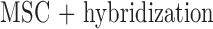 models can both predict asymmetries in the two discordant topologies (Slatkin and Pollack 2008; Meng and Kubatko 2009; DeGiorgio and Rosenberg 2016). Consequently, distinguishing these two models can be quite difficult.
models can both predict asymmetries in the two discordant topologies (Slatkin and Pollack 2008; Meng and Kubatko 2009; DeGiorgio and Rosenberg 2016). Consequently, distinguishing these two models can be quite difficult.
Although population structure can lead to some similar patterns in the data as hybridization, we will restrict most of our attention to hybridization. The study of the coalescent in the presence of population structure has been studied for a long time from a population genetic perspective and is often called the structured coalescent (Takahata 1988; Hein et al. 2005). A recent approach applying the structured coalescent in a phylogeographic context is Müller et al. (2017). Theunert and Slatkin (2017) discuss distinguishing recent admixture from ancient population structure as applied to human evolution in modeling admixture of Denisovans and ancestors of Melanesians. This can be done using site patterns, such as the ABBA-BABA test (Durand et al. 2011) or using linkage disequilibrium, in which lack of independence of nearby genetic loci is evidence of recent admixture, and independence increases over time due to recombination.
A lot of progress has been made in understanding the MSC by itself as a null model. Research for the MSC combined with hybridization is more recent, and it is quite challenging to study models that allow two biological processes simultaneously. It might be more realistic to allow all sources of gene tree incongruence to occur simultaneously, but this seams infeasible from a modeling point of view. Adding multiple processes greatly increases the number of parameters in the models and reduces their mathematical tractability. For example, we should expect recombination to be occurring—but properly modeling recombination leads to ancestral recombination graphs, which are themselves reticulating (Hein et al. 2005; Gusfield 2014). To model recombination with coalescence and hybridization would lead to networks within networks—a much more complicated problem than modeling gene trees within species networks. Simulation studies of recombination in the context of the MSC have also not shown very large effects (Lanier and Knowles 2012). Understanding two processes at a time seems to be a worthwhile goal.
Using Horizontal Versus Nonhorizontal Hybridization Edges
A fundamental decision at the modeling stage is whether hybridization edges are allowed to have a time component or are represented as horizontal edges (with a length of 0 to link populations that are contemporary with each other). If edges are not horizontal, then the lengths of the hybridization edges introduce new parameters into the problem. The two parents of a hybridization node might occur at different times, meaning that there might be two new branch lengths introduced for every hybridization node.
One reason for including nonhorizontal edges is that even if lineages could not coalesce on hybridization edges (i.e., branches leading into a hybridization node), the existence of unsampled or extinct lineages could lead to the desirability of having nonhorizontal edges (Fig. 2). Such lineages are sometimes called “ghost” lineages because although they exist and effect probabilities of gene trees, they are not seen due to being unsampled.
Figure 2.

Networks with “ghost lineages” and horizontal hybridization edges and corresponding networks with nonhorizontal edges. All networks have one hybridization event. In a) and c) dotted lines indicate evolving species that are not sampled either due to extinction or incomplete sampling. In a) and c), all hybridization edges (edges that lead into hybridization nodes) are drawn horizontally. However, the existence of unsampled species 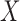 in a), and unsampled species
in a), and unsampled species 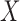 and
and 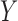 in c) means that there was a time more ancient than the hybridization event when lineages from
in c) means that there was a time more ancient than the hybridization event when lineages from 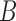 and
and 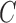 might have coalesced but could not have coalesced with lineages from
might have coalesced but could not have coalesced with lineages from 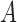 or
or  . The probabilities of coalescence events, gene trees, and sequence evolution in network b) are equivalent to those in network a), and similarly d) is equivalent in this sense to c).
. The probabilities of coalescence events, gene trees, and sequence evolution in network b) are equivalent to those in network a), and similarly d) is equivalent in this sense to c).
Conceptually, I argue that is not a problem to have populations evolve for some time before they subsequently merge, leading to nonhorizontal hybridization edges even without extinction or incomplete sampling. From a modeling point of view, however, it increases the number of parameters in the problem and the likelihood calculations become more complicated, but current software, such as phylonet (Than et al. 2008), ms (Hudson 2002), hybrid-Lambda (Zhu et al. 2015), PhyloNetworks (Solís-Lemus et al. 2017), hybrid-coal (Zhu and Degnan 2017), and *BEAST (Zhang et al. 2018) can handle networks with this structure. The prior for networks used by Zhang et al. (2018) is called a birth-hybridization prior in which a network is evolved forward in time, and the waiting time until the next event (either speciation or hybridization) is an exponential random variable, leading to nonhorizontal edges even in the absence of extinction or lack of sampling. From a forward-in-time perspective, whether an evolving population will turn out to correspond to a speciation edge or a hybridization edge depends on what happens in the future—whether the next event for that population is a merger or a divergence. This consideration makes it biologically awkward to treat hybridization edges as different from tree edges.
To envision how nonhorizontal edges could occur, imagine an irregularly shaped lake with a population of fish (Fig. 3). Over time, water levels in the lake change, so that the shallower, narrow areas can become dry, leading to separating the lake into separate, smaller lakes. This would lead to genetic isolation and the inability of genes in isolated regions to coalesce. In Fig. 3, if we imagine the sequence of water levels, we would expect lineages sampled from regions 1 and 2 in the present (the bottom of the figure) to sometimes coalesce fairly recently in the most recent case where regions 1 and are connected. However, going back in time, the three regions are separated again. Going back in time, imagine that regions 2 and 3 become connected while region 1 remains isolated. In this case, genes from regions 2 and 3 can coalesce. Such a sequence of habitat fragmentation and mergers could lead to patterns of genetic isolation that could create opportunities to coalesce reflected in a hybridization network with nonhorizontal hybridization edges, even if all species (or subspecies) were sampled in the present and there was no extinction.
Figure 3.
An example illustrating a possible biological interpretation for nonhorizontal edges without assuming extinction or incomplete sampling. a) Gray regions represent an irregularly shaped lake (or habitat) that becomes more or less fragmented over time due to changing water levels (for example). The network b) represents the history of genetic isolation that might be expected from such a sequence of geographic isolation. c) The network as a sequence of populations (boxes) with discrete generations, and gene tree 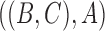 obtained by both lineages from
obtained by both lineages from 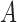 and
and 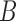 going to the right within the network as we trace their ancestry from the present to the past.
going to the right within the network as we trace their ancestry from the present to the past.
Identifiability and Distinguishability
A goal in probabilistic modeling is to have a class of models that is identifiable. This means that each model in the class leads to a distinct set of probabilities (or probability densities) for possible data. Thus, given enough data to determine the relevant probabilities, it is possible to determine which model in the class lead to the observed data. If two models lead to the exact same probability distribution for the data, then no amount of data can distinguish those two models, and the class of models as a whole is not identifiable. Here, there are several possibilities for what to count as data. For example, one could use rooted gene trees, unrooted gene trees, or DNA sequences or alignments. For the most part, I will restrict the discussion to identifiability given gene trees without considering error in gene trees that are estimated from DNA sequences. Ideally, for methods that use gene tree topologies (either rooted or unrooted), we would like to show that networks are still identifiable from imperfectly estimated gene trees. In the context of estimating species trees (as opposed to networks), it also true that it is easier to show that methods have desirable theoretical properties from known gene trees than from gene trees estimated with error (Roch and Warnow 2015).
Showing that a class of models is identifiable is often difficult, and in phylogenetics, identifiability results are often established long after a class of models is used (e.g., Allman and Rhodes 2003). A more modest goal is to understand when two models are or are not distinguishable, rather than understanding identifiability for the whole class of models of interest. Tools from algebraic geometry, particularly phylogenetic invariants, have often been used to establish identifiability of gene trees for different classes of substitution models (Allman and Rhodes 2003). Algebraic properties of site pattern probabilities also underlies the software SVDquartets and the software HyDe introduced in this issue for hybridization detection (Blischak et al. 2018).
For phylogenetic networks, as well as for phylogenetic trees, there are two aspects of identifiability: 1) the network topology and 2) given the topology, the parameters in the network such as branch lengths and inheritance probabilities. For some small examples with small numbers of taxa, the number of possible gene tree topologies can be fewer than the number of parameters to be estimated. This is a problem in terms of estimability from frequencies of gene tree topologies. In particular, we can think of each distinct gene tree probability as a function of the branch lengths and inheritance probabilities in the network, leading to a system of equations. If there are more parameters than equations in the system, then there will not be a unique solution to the system. This is typically only a problem for very small networks, such as with three or four taxa and one lineage sampled per species, and arises for the “bad diamonds” in Solís-Lemus and Ané (2016). Generally, identifying the network topology seems to be the greater interest, and I will focus on that first.
In the literature of combinatorial approaches, phylogenetic networks are often considered indistinguishable if they display the same trees (Fig. 4). A network is said to display a particular tree if removing some subset of hybridization edges leads to that particular tree remaining. The network in Fig. 2b displays the trees 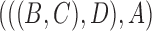 and
and 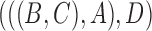 . The network in Fig. 2d only displays the tree topology
. The network in Fig. 2d only displays the tree topology 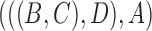 but has a different biological meaning from a tree. If branch lengths are taken into account, Fig. 2d displays two trees with different sets of branch lengths but the same topology. In some of the network literature, networks are often thought of as attempts to represent a collection of input trees by a network that displays those trees with a minimum number of hybridization events, often called the hybridization number. Attempts to determine the hybridization number have received considerable attention, particularly in the case of two input trees (Bordewich and Semple 2007; van Iersel and Linz 2013; van Iersel et al. 2014, 2016a, 2017).
but has a different biological meaning from a tree. If branch lengths are taken into account, Fig. 2d displays two trees with different sets of branch lengths but the same topology. In some of the network literature, networks are often thought of as attempts to represent a collection of input trees by a network that displays those trees with a minimum number of hybridization events, often called the hybridization number. Attempts to determine the hybridization number have received considerable attention, particularly in the case of two input trees (Bordewich and Semple 2007; van Iersel and Linz 2013; van Iersel et al. 2014, 2016a, 2017).
Figure 4.

Example of two networks (top row) that are distinct but each display the same three trees (bottom row). For example, removing the edges with lengths 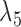 and
and 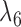 from
from 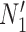 and
and 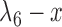 and
and 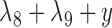 in
in 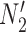 both result in displayed tree
both result in displayed tree 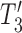 . The networks are distinguishable under the NMSC using gene tree topologies if there is one (or more) lineages sampled per species. The example is reprinted from Zhu and Degnan (2017) and is a modification of a figure from Pardi and Scornavacca (2015). The example from Pardi and Scornavacca (2015) can be obtained by removing taxon
. The networks are distinguishable under the NMSC using gene tree topologies if there is one (or more) lineages sampled per species. The example is reprinted from Zhu and Degnan (2017) and is a modification of a figure from Pardi and Scornavacca (2015). The example from Pardi and Scornavacca (2015) can be obtained by removing taxon 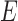 .
.
If incongruence in the gene trees can only be due to hybridization events, then the idea that networks must display different trees to be distinguishable makes sense. As will be explained below, however, when there is incomplete lineage sorting, gene lineages in the network do not always follow paths of displayed trees. An extreme case is given by Zhu et al. (2016), in which it is shown that the most probable gene tree topology can disagree with the topologies of all displayed trees. This result is a network analog of the result that the most probable gene tree is not necessarily concordant with the species tree, a phenomenon referred to as AGTs (Degnan and Rosenberg 2006). Another example is shown in Solís-Lemus et al. (2016), in which gene flow between nonsister taxa results in anomalous unrooted gene trees (AUGTs) (Degnan 2013). The Solís-Lemus et al. (2016) example occurs even in the case of four taxon unrooted gene trees, for which AUGTs are not possible in the traditional multispecies coalescent (which assumes no hybridization and no gene flow between distinct species) (Degnan and Rosenberg 2006; Larget et al. 2010; Allman et al. 2011. In this issue, Long and Kubatko (2018) show that continuous gene flow between sister taxa can also result in rooted AGTs, even in the case of three taxa.
In empirical studies, there is often a very large set of conflicting gene trees, in some cases, with every gene tree having a unique topology—for example, Salichos and Rokas (2013) give an example with 23 taxa and all 1070 gene trees being unique. Just as a single species tree can give rise to a large number of incongruent gene trees, we should expect that large numbers of incongruent gene trees can be compatible with a small number of hybridization events, because incongruence can be due to ILS as well as hybridization.
There are two senses of identifiability that are important in discussions of inferring trees and networks. One is that, we want to be able to identify the tree or network topology that fits the data. In particular, invariant methods often identify patterns in the data that hold for a particular topology regardless of the branch lengths or other parameters (Allman and Rhodes 2003). A second sense of identifiability is that given a particular topology, we wish to be able to infer the branch lengths and other real-valued parameters associated with the model, such as inheritance probabilities. This can often be done by solving a system of equations relating probabilities of gene trees to the parameters in the network. Often, however, methods of inferring species trees or networks only determine the topology and not the branch lengths.
So far, results regarding identifiability for species network topologies under the network multispecies coalescent (NMSC) have focused on using quartets in the unrooted gene trees to infer features of unrooted versions of the species network (Solís-Lemus and Ané 2016; Baños 2017). The results show that for level-1 networks, for which cycles do not overlap, cycles of length four can be detected, with a fifth taxon being necessary to detect which node is the hybridization node, and the hybrid nodes of cycles of length five can also be detected. In this setting, detecting hybridization nodes can allow for detecting the direction of some edges in the network, and consequently the inferred networks are called semidirected. For examples, see Solís-Lemus and Ané (2016).
Having two or more taxa descended from a hybrid node is particularly helpful for identifying hybridization events, even for networks that are more complicated than level-1. For example, there is a cycle of four nodes with branch lengths 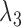 ,
, 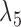 ,
, 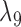 , and
, and 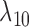 for network
for network 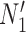 in Fig. 4. The hybridization node is where the branches with lengths
in Fig. 4. The hybridization node is where the branches with lengths 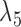 and
and 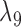 meet, from which lineages from species
meet, from which lineages from species 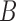 and
and 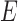 can both potentially be present. Networks in which only one lineage is sampled that is descended from a hybrid node can have identifiability problems. For the examples in Fig. 4, if there is only one lineage sampled from species
can both potentially be present. Networks in which only one lineage is sampled that is descended from a hybrid node can have identifiability problems. For the examples in Fig. 4, if there is only one lineage sampled from species 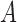 ,
, 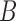 ,
, 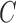 , and
, and 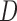 , and no lineage is sampled from
, and no lineage is sampled from 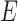 , then the networks
, then the networks 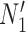 and
and 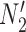 cannot be distinguished using gene trees, even with known branch lengths (Pardi and Scornavacca 2015; Zhu and Degnan 2017). However, if both
cannot be distinguished using gene trees, even with known branch lengths (Pardi and Scornavacca 2015; Zhu and Degnan 2017). However, if both 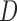 and
and 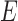 are sampled, then the two networks are distinguishable, meaning that they give different probability distributions on the gene trees and even the gene tree topologies, even though these networks are not level-1 due to the overlapping cycles (Zhu and Degnan 2017).
are sampled, then the two networks are distinguishable, meaning that they give different probability distributions on the gene trees and even the gene tree topologies, even though these networks are not level-1 due to the overlapping cycles (Zhu and Degnan 2017).
However, a few insights regarding identifiability have emerged. One is that the methods for determining probabilities of gene tree topologies in Yu et al. (2012) and Zhu and Degnan (2017) do not make an explicit use of displayed trees. The algorithms work exactly the same way whether or not a gene tree happens to be displayed by the network, and the algorithm does not always decompose the species network into only its displayed trees. This has results for simulation as well. An early paper linking hybridization networks and ILS (and allowing for both simultaneously) (Holland et al. 2008) simulated gene trees under species networks by first finding a tree displayed by the network, and then simulating the gene trees under the MSC on that displayed species tree. However, if two or more lineages are sampled from descendants of a hybrid node (either two or more lineages from the same species or from distinct species), then the two lineages might trace different paths through the network. The gene tree should be thought of as coalescing directly in the network in this case, not in a tree displayed by the network, and this will lead to a different distribution of gene trees. Consequently, limiting gene trees to those that can evolve on a species tree displayed by the network can lead to an incorrect distribution of gene trees. Simulating directly from the network is the approach taken in the program hybrid-Lambda (Zhu et al. 2015).
The idea that the gene tree evolves on a species tree displayed by the network is reasonable provided there is only one lineage sampled from a descendant of a hybrid node, and this is the approach taken in Meng and Kubatko (2009) and Kubatko (2009). In these papers, the trees displayed by the network are called parental trees. The more general situation in which there can be several species descended from hybrid nodes does not make explicit use of displayed trees.
Future work on identifiability should keep in mind cases where gene trees do not evolve on trees displayed by the networks. Model identifiability will also have to describe the space of phylogenetic networks. For example, are multiple edges between two nodes allowed? Possibilities for making claims of identifiability are to limit the complexity of the network in some way, for example, to limit the number of hybridization events, or to limit results to level-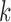 networks to some small values of
networks to some small values of 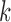 , where the level of a network describes the number of reticulations in each biconnected component (Choy et al. 2005).
, where the level of a network describes the number of reticulations in each biconnected component (Choy et al. 2005).
To give a hint of the algebraic argument that has been used to show identifiability of rooted species trees from unrooted gene trees (Allman et al. 2011), consider the three possible models in Fig. 5. Here, our interest is only in distinguishing whether the data come from network (a) (which is a tree), (b) (also and tree), or (c). Probabilities of rooted gene tree topologies are shown in Table 2 (for space reasons, network (d) is not included, and network (c) is only shown for 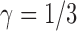 ). Network (a) has five algebraically distinct probabilities, network (b) has seven algebraically distinct probabilities, and networks (c) and (d) each have nine algebraically distinct probabilities (Table 2, Yu et al. 2011). By algebraically distinct, we mean that the polynomials representing the probabilities are distinct. For certain special choices of branch lengths, it might be possible to make some of the probabilities numerically not distinct, but such choices of branch lengths would technically have probability 0 for trees or networks generated by birth–death processes. Representing probabilities as polynomials allows the use of methods from algebraic geometry and algebraic statistics to investigate identifiability issues and to perform statistical inference (Drton et al. 2009; Allman et al. 2011; Chifman and Kubatko 2015). Looking at the number of distinct gene tree probabilities is informative about the network topologies. Networks (c) and (d) have the same number of distinct gene tree probabilities, so a worry might be whether we can distinguish them. However, if the topology is known to be of the form of network (d), then there are nine distinct gene tree probabilities and six parameters, leading to an overdetermined system of equations. Network (c) is a special case of Network (d) where some of the branch lengths are equal to 0. Network (d) can also be distinguished from the others using unrooted trees as data (Solís-Lemus et al. 2016).
). Network (a) has five algebraically distinct probabilities, network (b) has seven algebraically distinct probabilities, and networks (c) and (d) each have nine algebraically distinct probabilities (Table 2, Yu et al. 2011). By algebraically distinct, we mean that the polynomials representing the probabilities are distinct. For certain special choices of branch lengths, it might be possible to make some of the probabilities numerically not distinct, but such choices of branch lengths would technically have probability 0 for trees or networks generated by birth–death processes. Representing probabilities as polynomials allows the use of methods from algebraic geometry and algebraic statistics to investigate identifiability issues and to perform statistical inference (Drton et al. 2009; Allman et al. 2011; Chifman and Kubatko 2015). Looking at the number of distinct gene tree probabilities is informative about the network topologies. Networks (c) and (d) have the same number of distinct gene tree probabilities, so a worry might be whether we can distinguish them. However, if the topology is known to be of the form of network (d), then there are nine distinct gene tree probabilities and six parameters, leading to an overdetermined system of equations. Network (c) is a special case of Network (d) where some of the branch lengths are equal to 0. Network (d) can also be distinguished from the others using unrooted trees as data (Solís-Lemus et al. 2016).
Figure 5.

Four networks with branch lengths. Networks a)–b) are trees with two branch length parameters in coalescent units. Network c) has two branch length parameters and an inheritance probability parameter 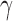 which determines the probability of going left u for each lineage at the hybrid node. Here the hybridization edges are horizontal, so it assumed that the lineages for
which determines the probability of going left u for each lineage at the hybrid node. Here the hybridization edges are horizontal, so it assumed that the lineages for 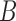 and
and 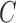 either coalesce more recently than the hybridization event, or they don’t, in which case each independently enters the population ancestral to
either coalesce more recently than the hybridization event, or they don’t, in which case each independently enters the population ancestral to 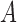 or
or 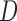 before (going backwards in time) coalescence is possible. In d), branch
before (going backwards in time) coalescence is possible. In d), branch 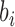 has length
has length 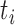 , and the hybridization edges (with lengths
, and the hybridization edges (with lengths 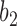 and
and 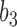 ) are independent populations. In d), if both lineages from
) are independent populations. In d), if both lineages from 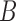 and
and 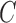 take the same path through the network (say, to the left), then there is the possibility that they could coalesce on an edge more ancient than the hybridization event, but more recent than the species divergence between the population which is ancestral to
take the same path through the network (say, to the left), then there is the possibility that they could coalesce on an edge more ancient than the hybridization event, but more recent than the species divergence between the population which is ancestral to 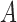 ,
, 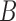 , and
, and 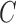 (i.e., the population corresponding to branch
(i.e., the population corresponding to branch 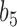 ). This network therefore has more parameters than the network in (c). The number of parameters can be reduced by one if there is the constraint that
). This network therefore has more parameters than the network in (c). The number of parameters can be reduced by one if there is the constraint that 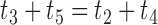 , but this isn’t required by the model.
, but this isn’t required by the model.
Table 2.
Probabilities of gene tree topologies for different species trees and networks shown in Figure 5 with 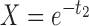 ,
, 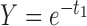 , and where
, and where 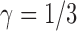 is the probability that lineages go to the left
is the probability that lineages go to the left
| Network/tree from Fig. 5 | |||
|---|---|---|---|
| Gene tree | (a) | (b) | (c) |
1. 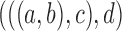
|
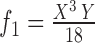
|
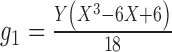
|
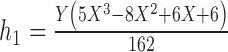
|
2. 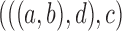
|
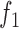
|
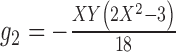
|
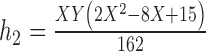
|
3. 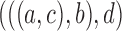
|
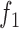
|
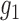
|
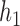
|
4. 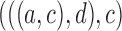
|
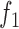
|
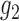
|
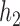
|
5. 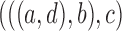
|
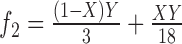
|
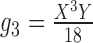
|
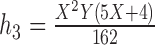
|
6. 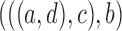
|
|
|
|
7. 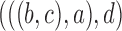
|
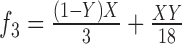
|
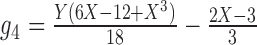
|
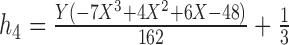
|
8. 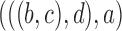
|
|
|
|
9. 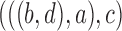
|
|
|
|
10. 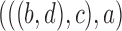
|
|
|
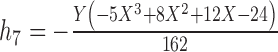
|
11. 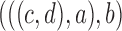
|
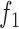
|
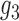
|
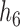
|
12. 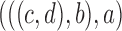
|
|
|
|
13. 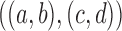
|
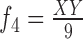
|
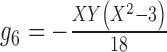
|
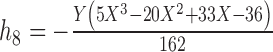
|
14. 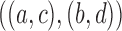
|
|
|
|
15. 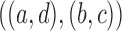
|
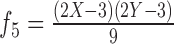
|
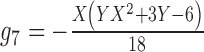
|
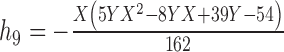
|
A further problem with identifiability that seems more difficult to address is that there might be other causes of gene tree incongruence above and beyond the MSC and hybridization. The previous paragraph gave an idea of how to distinguish the models 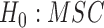 versus
versus 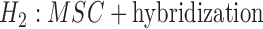 , for example, but did not indicate distinguishing between
, for example, but did not indicate distinguishing between 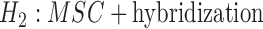 versus
versus 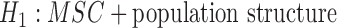 , for example. Both hybridization and population structure can lead to increased complexity in gene tree distributions and can lead to inconsistency in usual species tree reconstruction methods. In particular, Slatkin and Pollack (2008) demonstrate that the most probable topology does not necessarily match the species tree topology, even with three taxa. Consequently, many popular two-stage methods of inferring species trees can be inconsistent when there is ancestral population structure, meaning that they can fail to recover the species tree even with arbitrarily large numbers of loci (DeGiorgio and Rosenberg 2016). Another example where two classes of models might give rise to similar patterns is that population structure might give the appearance of distinct species when the MSC is used for species delimitation without accounting for population structure (Sukumaran and Knowles 2017). Generally, identifiability results require an assumption that the correct class of models is first given (such as only coalescence and hybridization as sources of incongruence), and identifiability is hopefully shown within that assumed class of models.
, for example. Both hybridization and population structure can lead to increased complexity in gene tree distributions and can lead to inconsistency in usual species tree reconstruction methods. In particular, Slatkin and Pollack (2008) demonstrate that the most probable topology does not necessarily match the species tree topology, even with three taxa. Consequently, many popular two-stage methods of inferring species trees can be inconsistent when there is ancestral population structure, meaning that they can fail to recover the species tree even with arbitrarily large numbers of loci (DeGiorgio and Rosenberg 2016). Another example where two classes of models might give rise to similar patterns is that population structure might give the appearance of distinct species when the MSC is used for species delimitation without accounting for population structure (Sukumaran and Knowles 2017). Generally, identifiability results require an assumption that the correct class of models is first given (such as only coalescence and hybridization as sources of incongruence), and identifiability is hopefully shown within that assumed class of models.
A final difficulty to be addressed is that most identifiability results so far have focused on known gene trees, and have not dealt with problems in estimating gene trees. Usually methods that are shown to be statistically consistent on known gene trees are then tested in simulation to observe their robustness to misestimation in the gene trees (e.g., Liu et al. 2009; Huang et al. 2010; Leaché and Rannala 2011; Wu 2012; Chou et al. 2015). It would of course be desirable to have theoretical results on identifiability of models (and consistency of inference methods) from gene trees estimated with errors as large as are typically observed in empirical studies. An attempt in this direction is given by Roch and Warnow (2015), which uses bounds in errors of estimated triplets to bound the error for the whole tree and thereby prove consistency for a triplet-based method from estimated gene trees. One approach for potentially reducing error introduced by using estimated gene trees is to estimate gene tree frequencies in a Bayesian framework and later use these Bayesian estimates of frequencies, where uncertainty in the individual gene trees has been accounted for and integrated out. This approach is used in BUCKy (Ané et al. 2007; Larget et al. 2010) which estimates both gene tree frequencies and species trees. The estimated gene tree frequencies from BUCKy can be used as input to other methods that use gene tree topology frequencies. There is some discussion in the literature of the impact of gene tree accuracy on species tree methods (Huang et al. 2010; DeGiorgio and Degnan 2014; Roch and Warnow 2015). However, what is important is that the estimated distribution of gene trees or their summary statistics (quartets, triples, clusters, etc.), matches the true distribution rather than that the individual estimated gene trees match the true gene trees.
In addition to distinguishing networks from gene trees, tests of hybridization are often done based directly on sequence data. A popular method is called the ABBA-BABA test (Durand et al. 2011), which is used for subsets of four taxa or populations. If the correct tree is 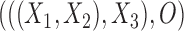 (where
(where 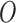 is the outgroup), then a common allele pattern should be BBAA, where taxa
is the outgroup), then a common allele pattern should be BBAA, where taxa 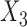 and
and 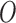 share the same ancestral allele, and
share the same ancestral allele, and 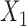 and
and 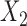 share the same new allele. If
share the same new allele. If 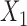 and
and 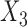 share an allele not shared by
share an allele not shared by 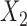 , then this is evidence of admixture between
, then this is evidence of admixture between 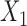 and
and 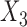 and can result in pattern BABA. Similarly, admixture between
and can result in pattern BABA. Similarly, admixture between 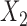 and
and 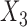 could lead to pattern ABBA. If there is no admixture, then frequencies of patterns ABBA and BABA should be similar. Comparing frequencies of the discordant patterns (called the
could lead to pattern ABBA. If there is no admixture, then frequencies of patterns ABBA and BABA should be similar. Comparing frequencies of the discordant patterns (called the 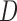 -statistic) can give evidence for whether there was a violation of tree-like evolution of the taxa due to ancient hybridization or population structure. Although the ABBA-BABA test was originally given for four populations, Pease and Hahn (2015) extend the idea to a five-taxon setting (a balanced four-taxon rooted tree plus a fifth taxon as an outgroup) to help identify the direction of gene flow using a statistic called
-statistic) can give evidence for whether there was a violation of tree-like evolution of the taxa due to ancient hybridization or population structure. Although the ABBA-BABA test was originally given for four populations, Pease and Hahn (2015) extend the idea to a five-taxon setting (a balanced four-taxon rooted tree plus a fifth taxon as an outgroup) to help identify the direction of gene flow using a statistic called 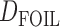 . The results are consistent with those of Solís-Lemus and Ané (2016) in that adding a fifth taxon improves identifiability by allowing both the detection of the hybrid node and the direction of gene flow when the cycle has enough nodes. With four taxa, they can detect cycles with four nodes, but not the direction of gene flow. Their work suggests that it will be useful to examine identifiability of networks from sequence data in addition to gene tree topologies. Pease and Hahn (2015) also point out that the ABBA-BABA test and
. The results are consistent with those of Solís-Lemus and Ané (2016) in that adding a fifth taxon improves identifiability by allowing both the detection of the hybrid node and the direction of gene flow when the cycle has enough nodes. With four taxa, they can detect cycles with four nodes, but not the direction of gene flow. Their work suggests that it will be useful to examine identifiability of networks from sequence data in addition to gene tree topologies. Pease and Hahn (2015) also point out that the ABBA-BABA test and 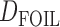 can be misled by unsampled “ghost” lineages. Recent admixture can also be detected by using linkage disequilibrium, where nonindependence of nearby genetic loci is evidence of recent admixture, with the degree of nonindependence declining over time due to recombination (Corander and Marttinen 2006).
can be misled by unsampled “ghost” lineages. Recent admixture can also be detected by using linkage disequilibrium, where nonindependence of nearby genetic loci is evidence of recent admixture, with the degree of nonindependence declining over time due to recombination (Corander and Marttinen 2006).
Branch Lengths in the Networks and Gene Trees
Species networks can be considered either topologically or with branch lengths. Branch lengths are particularly useful for probabilistic models. Together with inheritance probabilities, parameters that determine the probabilities that a lineage is inherited from each of its parental populations, these real-valued parameters allow probabilistic modeling of gene trees within species networks.
A difficult question is whether branch lengths in the gene trees should be used. Several of the current algorithms for inferring species networks from gene trees only use the topological information, ignoring potential information in the branch lengths. Intuitively, it would seem to be preferable to use the information in the branch lengths as well. In some cases, two networks might be indistinguishable using only gene tree topologies yet distinguishable using gene trees with branch lengths. As an extreme case, if there is a species network with only two species, 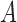 and
and 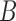 , then there is only one gene tree topology,
, then there is only one gene tree topology, 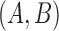 . Yet the species divergence history for these two species might have undergone periods of genetic isolation followed by population mergers before the final cessation of gene flow. This would lead to networks where a species diverged into two populations, the two populations subsequently merged, and this process might have been repeated several times. This could occur for example in cases of episodic glaciation or sea level changes, where there are alternating periods of gene flow and genetic isolation. In these cases, coalescence times could potentially be multimodal, or at least have higher variance than would be predicted under a model where gene flow stopped only once (DeGiorgio et al. 2011). The distribution of branch lengths in the gene trees would then give information regarding times when the species had split and then merged before the final divergence. Information in coalescence times is especially used for inferring complex demographic histories and changes in ancestral population sizes such as in hidden Markov models (HMMs) (Dutheil et al. 2009; Schiffels and Durbin 2014).
. Yet the species divergence history for these two species might have undergone periods of genetic isolation followed by population mergers before the final cessation of gene flow. This would lead to networks where a species diverged into two populations, the two populations subsequently merged, and this process might have been repeated several times. This could occur for example in cases of episodic glaciation or sea level changes, where there are alternating periods of gene flow and genetic isolation. In these cases, coalescence times could potentially be multimodal, or at least have higher variance than would be predicted under a model where gene flow stopped only once (DeGiorgio et al. 2011). The distribution of branch lengths in the gene trees would then give information regarding times when the species had split and then merged before the final divergence. Information in coalescence times is especially used for inferring complex demographic histories and changes in ancestral population sizes such as in hidden Markov models (HMMs) (Dutheil et al. 2009; Schiffels and Durbin 2014).
The use of branch lengths from gene trees has a parallel in species tree inference, where methods that use gene trees with branch lengths, such as STEAC (Liu et al. 2009) and STEM (Kubatko et al. 2009), have so far been outperformed in simulation by methods that use only estimated topologies, such as NJ (Liu and Yu 2011) and ASTRAL (Mirarab et al. 2014). Reasons for the underperformance of STEM have been explored in the case that species are closely related and recently separated, leading to underestimation of divergence times (DeGiorgio and Degnan 2014). The performance of STEAC relative to the related method STAR (which can be interpreted as STEAC with all internal branch lengths replaced with the value
(Liu and Yu 2011) and ASTRAL (Mirarab et al. 2014). Reasons for the underperformance of STEM have been explored in the case that species are closely related and recently separated, leading to underestimation of divergence times (DeGiorgio and Degnan 2014). The performance of STEAC relative to the related method STAR (which can be interpreted as STEAC with all internal branch lengths replaced with the value 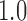 ) is less well understood. In simulations, STEAC had very similar performance as STAR under idealized conditions, such as there being a molecular clock, but did not perform as well when there were molecular clock violations (Liu et al. 2009).
) is less well understood. In simulations, STEAC had very similar performance as STAR under idealized conditions, such as there being a molecular clock, but did not perform as well when there were molecular clock violations (Liu et al. 2009).
Methods for inferring species networks from gene trees under the NMSC can also be divided into those that use topologies only (Meng and Kubatko 2009; Yu et al. 2011, 2012; Solís-Lemus and Ané 2016) and those that use gene trees with branch lengths (Kubatko 2009; Yu et al. 2014; Wen et al. 2016). Using branch lengths in gene trees to infer networks might suffer from the same problems encountered when inferring species trees, namely that when gene trees are estimated using maximum likelihood, divergences can be underestimated (due to identical or nearly identical DNA sequences at just one locus), making the maximum likelihood estimate of the distances between distinct taxa to be very low or zero (DeGiorgio and Degnan 2014). This problem can be ameliorated by some extent by using Bayesian estimates of gene trees. The prior for branch lengths means that even identical sequences will result in a nonzero branch length separating distinct taxa in the Bayesian estimates of the gene trees (DeGiorgio and Degnan 2014; Wen et al. 2016).
Another complication with methods that employ branch lengths is that accurate estimation of branch lengths requires more assumptions than using topologies. For example, branch lengths on different gene trees have to calibrated to adjust for possibly different mutation rates or mutation models at the different loci (Kubatko et al. 2009; Rhodes 2017), whereas no such calibration is needed when using topologies. In Bayesian frameworks especially, allowing locus-specific and species-specific mutation rates can be accommodated, for example by assuming that mutation rates come from a distribution (i.e., a random effects model) without requiring estimation of a separate mutation rate parameter for each locus or by using a relaxed clock (Ogilvie et al. 2017). It is possible that for shallower species trees and networks, getting the substitution model exactly correct is less essential, especially since multiple mutations at a single site are less common and the molecular clock is more likely to be approximately correct (Burgess and Yang 2008). However, shallower trees can have very low variation in sequences, which leads to the underestimation problem described above, in which topology-based methods can outperform branch-length based summary methods even under the molecular clock and simple substitution models (DeGiorgio and Degnan 2014).
Bayesian (and likelihood) approaches that use branch lengths or sequence data have the advantage that they can potentially infer all of the parameters of the NMSC, including times of speciation in generations and ancestral population sizes (Rannala and Yang 2003; Wen and Nakhleh 2017; Zhang et al. 2018). Methods that infer species network branch lengths from gene tree topologies only are limited to inferring branch lengths in coalescent units, in which the generation time and population size parameters are confounded.
Rooted Versus Unrooted Networks
In addition to deciding whether or not to use branch lengths to infer networks, one must also decide whether to infer a rooted or an unrooted network. In the brief history of methods to infer species trees from gene trees under the MSC, there has been a shift from methods that use rooted gene trees (roughly 2006–2010) to methods that use unrooted gene trees (roughly 2010 to the present). Earlier rooted methods include minimizing deep coalesce (Maddison and Knowles 2006), STAR (Liu et al. 2009), STEM (Kubatko et al. 2009), MP-EST (Liu et al. 2010), whereas later unrooted methods include NJ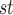 (Liu and Yu 2011) and ASTRAL (Mirarab et al. 2014). Although these last two methods are among the best-performing overall, it is not clear to what extent this is due to the gene trees being unrooted since these methods also use different criteria to estimate the species trees. The division between earlier, rooted methods and later, unrooted methods is also only approximate, with BUCKy (Ané et al. 2007) being used early with unrooted trees and STELLS (Wu 2012) a later addition to the rooted gene tree methods.
(Liu and Yu 2011) and ASTRAL (Mirarab et al. 2014). Although these last two methods are among the best-performing overall, it is not clear to what extent this is due to the gene trees being unrooted since these methods also use different criteria to estimate the species trees. The division between earlier, rooted methods and later, unrooted methods is also only approximate, with BUCKy (Ané et al. 2007) being used early with unrooted trees and STELLS (Wu 2012) a later addition to the rooted gene tree methods.
Methods for estimating networks in the NMSC started developing just a few years later, but again started with rooted methods (Meng and Kubatko 2009; Kubatko 2009; Yu et al. 2011, 2012; Yu and Nakhleh 2015b), but the more recent method SNaQ uses unrooted trees (Solís-Lemus and Ané 2016). An interesting question is whether the same shift from rooted to unrooted methods will continue for inferring networks with newer methods focusing on unrooted gene trees.
Apart from the trend, a question is whether it is actually better to use unrooted gene trees as input than rooted gene trees. One reason that the use of unrooted gene trees is natural is that fast likelihood programs such as RAxML (e.g., Stamatakis 2006) output unrooted trees. Keeping this in mind, two possible pipelines for inferring species trees (or networks) are:
Infer unrooted gene trees
Root gene trees using an outgroup
Input rooted gene trees to a rooted species tree (or network) method
versus
Infer unrooted gene trees
Input unrooted gene trees to an unrooted species tree (or network) method
Root the species tree (or network) using an outgroup
An important advantage for the second approach is that an outgroup only has to be used once, which, in addition to slightly reducing the computation time, reduces the possibility that gene trees are incorrectly rooted. In particular, an outgroup at the species level is not necessarily an outgroup at the level of an individual locus. Coalescence can fail to occur between some lineages in the interval between the root of the ingroup taxa and the root of the tree with the outgroup taxa. Consequently, if the outgroup is too close to the ingroup taxa, several gene trees might be incorrectly rooted, which could reduce the accuracy of methods for inferring rooted species trees or networks (Gatesy et al. 2007; Simmons and Gatesy 2015), even if the outgroup is genuinely an outgroup at the species level. This could be avoided by choosing an outgroup that was sufficiently far away, but this could introduce other errors due to saturated sequences (too much mutation on the branch leading to the outgroup) and can lead to the inclusion of an outgroup making the tree of the ingroup taxa less accurate than analyzing the ingroup taxa by themselves (Holland et al. 2003).
A second advantage is that for some groups of organisms, an appropriate outgroup might be hard to find (Boykin et al. 2010), or it might not be known in advance whether a candidate outgroup is too close or too far to the ingroup taxa to avoid causing problems. In addition to the two pipelines above, Allman et al. (2011) showed that it is theoretically possible to infer a rooted species tree directly from unrooted gene tree topologies without using an outgroup. Although this method has been tested using approximate Bayesian computation (Alanzi and Degnan 2017), it is expected that using an outgroup (if available) to ultimately root the tree (or network) will typically be easier than inferring the root directly from the unrooted trees. SNaQ (Solís-Lemus and Ané 2016) is able to infer some rooted information (direction of some hybridization edges) in networks from unrooted trees. An open question is whether complete, rooted networks can be inferred from unrooted trees under some conditions.
Modeling Trait Evolution on Networks
Models of trait evolution allow general traits, rather than just DNA or protein sequences, to evolve along a tree. Traits can be discrete or continuous. Continuous traits are often modeled using Brownian motion or an Ornstein–Uhlenbeck (OU) model (Felsenstein 2004). The idea is that there is a mean trait value for a population which changes over time, either drifting, such as from a Brownian motion, which assumes that traits vary according to a normal distribution with variance proportional to time. Alternatively, in the OU model the distance of a trait value to some optimal value influences the variance and direction of the trait evolution.
A new idea, presented at the Evolution meeting, is to model trait evolution on a network instead of a tree. This more general approach might be more realistic in cases where there has been hybridization between populations. One approach is to treat mean values of traits in a hybrid population as weighted averages of the parental populations plus an additional amount allowing for a shift just after a hybridization event, thus allowing for transgressive evolution, in which the hybrid population can have more extreme values than either parental population (Bastide et al. 2018).
A conceptual issue here is whether trait evolution should be modeled on a tree or network at the species versus gene levels. For the network models considered in this article, gene trees are embedded in species networks, but the gene trees themselves are still tree-like. This is in contrast to other network structures such as networks depicting horizontal gene transfer events or ancestral recombination graphs, in which the correct graph structure is not tree-like at the level of the gene. These new network approaches to trait evolution open the door to thinking about traits evolving on combinations of trees that are due to a network structure.
Distances Between Networks
Another area that needs more work is distances between networks. A number of distances have been based on the Robinson–Foulds (RF) (Robinson and Foulds 1981) and rooted triple distances (Critchlow et al. 1996), which were designed for trees, and generalized to networks (Cardona et al. 2009a,b; Nahkleh 2010). For the RF distance on trees, the set of clusters in each tree is listed. For example, for the trees in Fig. 5a,b, the clusters for tree (a) are 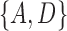 and
and 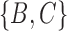 , while for tree (b), the clusters are
, while for tree (b), the clusters are 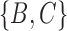 and
and 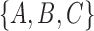 . Each tree has one cluster that is not in the other tree, leading to a total of two clusters not shared between the two trees. This leads to a RF distance of 2. The approach can be generalized to networks in different ways. For example, one could list all the clusters associated with each of the displayed trees associated with a network. For example, in networks (c) and (d) from Fig. 5, the clusters associated with the network are
. Each tree has one cluster that is not in the other tree, leading to a total of two clusters not shared between the two trees. This leads to a RF distance of 2. The approach can be generalized to networks in different ways. For example, one could list all the clusters associated with each of the displayed trees associated with a network. For example, in networks (c) and (d) from Fig. 5, the clusters associated with the network are 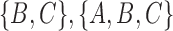 , and
, and 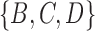 . Clusters associated with the displayed trees are called softwired clusters and distances based on these are called softwired distances (Huson et al. 2010). Alternatively, the hardwired cluster distance is based on listing clusters associated with the tree edges in the network. This leads to one cluster for each tree edge. For example, in networks (c) and (d) from Fig. 5, the hardwired clusters are
. Clusters associated with the displayed trees are called softwired clusters and distances based on these are called softwired distances (Huson et al. 2010). Alternatively, the hardwired cluster distance is based on listing clusters associated with the tree edges in the network. This leads to one cluster for each tree edge. For example, in networks (c) and (d) from Fig. 5, the hardwired clusters are 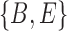 ,
, 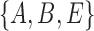 ,
, 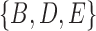 , and
, and 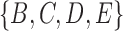 . The hardwired cluster distance has been used to measure error in reconstructed versus true networks in simulations (Yu and Nakhleh 2015a).
. The hardwired cluster distance has been used to measure error in reconstructed versus true networks in simulations (Yu and Nakhleh 2015a).
A similar approach is to use rooted triples instead of clusters. Here each rooted triple associated with a tree or network is listed, and the number of rooted triples not shared can be treated as the distance. For example, tree (a) in Fig. 1 has triples 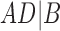 ,
, 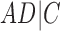 ,
, 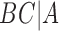 , and
, and 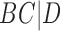 , while tree (b) has triples
, while tree (b) has triples 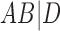 ,
, 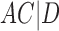 ,
, 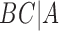 , and
, and 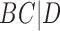 , leading to a total of four triples that are not shared. Triples from the displayed trees in a network can be used to describe distances between networks as well.
, leading to a total of four triples that are not shared. Triples from the displayed trees in a network can be used to describe distances between networks as well.
Although there are many proposed distances between networks, current approaches seem to implicitly assume that networks with identical displayed trees should have a distance of 0 between them (e.g., Cardona et al. 2009a). Under the NMSC, this is sometimes but not always appropriate because networks displaying the same trees can be distinguished in many cases. Typically, distances proposed for networks are proven to satisfy properties of metrics (in particular, the distance between two networks is 0 if and only if the networks are isomorophic) only for restricted classes of networks such as level-1 networks (with no overlapping cycles) or tree-child networks (for which every internal node is the parent of extant taxon or a tree node) (Cardona et al. 2009c). Typical dissimilarity measures return 0 for the networks in Fig. 4 even though these networks induce different distributions on the gene trees and can be distinguished. Ideally, a metric on networks for the NMSC is desired where the distance between networks is not 0 when the networks are distinguishable.
Discussion
This is an exciting time for the application of phylogenetic networks and the modeling of multiple biological mechanisms that simultaneously contribute to gene tree heterogeneity. The spotlight session at Evolution in 2017 focused on a few directions in which this topic is growing, and there is still much room for work in this area. Traditional phylogenetic tree inference has had several decades to mature with the development of parsimony and distance methods in the 1960s and 1970s (see Felsenstein (2004) for an historical sketch), application of maximum likelihood starting in the 1980s (Felsenstein 1981), and Bayesian methods in the 1990s (Rannala and Yang 1996). In that time, a lot of work was done on refining algorithms, thinking of properties of tree space and how to search that space effectively with moves such as nearest neighbor interchange, subtree prune and regraft, and tree bisection and rearrangement. Birth–death models were also developed as priors for the space of species trees (Rannala and Yang 1996).
Work on phylogenetic networks is much less mature, with the space of phylogenetic networks that we wish to work with still not very clearly defined, and there is a need for understanding moves in network space and networks priors in order to do Bayesian inference on networks. Recent progress in these directions include generalizing the nearest neighbor interchange and subtree prune and regraft algorithms for networks (Huber et al. 2016a, 2016b; Gambette et al. 2017). Moves for networks include flipping the direction of a reticulation edge, adding a reticulation between randomly selected edges, and deleting a randomly selected edge (Gambette et al. 2017). Network priors are needed for Bayesian inference of species networks. This is not as simple as the birth–death prior typically used for trees partly because the number of possible networks is much larger, and also because the number of parameters depends on the number of reticulations. One approach is to condition on the number of hybridization events (Jones et al. 2013), while a more ambitious approach is to search a space with an uncertain number of hybridization events (Wen et al. 2016). A third approach is to expand birth–death models to birth–death-hybridization models, where there is a rate at which pairs of lineages can hybridize in addition to rates for lineages to speciate or go extinct (Zhang et al. 2018).
Acknowledgments
The author thanks Cécile Ané and two anonymous reviewers for very helpful comments.
Funding
This work was supported by National Institutes of Health [Grant R01 GM117590].
References
- Alanzi A.R., Degnan J.H.. 2017. Inferring rooted species trees from unrooted gene trees using approximate Bayesian computation. Mol. Phylogenet. Evol. 116:13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allman E.S., Rhodes J.A.. 2003. Phylogenetic invariants for the general Markov model of sequence mutation. Math. Biosci. 186:113–144. [DOI] [PubMed] [Google Scholar]
- Allman E.S., Degnan J.H., Rhodes J.A.. 2011. Identifying the rooted species tree from the distribution of unrooted gene trees under the coalescent. J. Math. Biol. 62:833–862. [DOI] [PubMed] [Google Scholar]
- Ané C. 2010. Reconstructing concordance trees and testing the coalescent model from genome-wide data sets In: Knowles L.L., Kubatko L.S., editors. Estimating species trees: theoretical and practical aspects. Hoboken, NJ: Wiley-Blackwell; p. 35–52. [Google Scholar]
- Ané C., Larget B., Baum D.A., Smith S.D., Rokas A.. 2007. Bayesian estimation of concordance factors. Mol. Biol. Evol. 24:412–426. [DOI] [PubMed] [Google Scholar]
- Baños H. 2017. Identifying species network features from gene tree quartets under the coalescent model. arXiv:1711.10545v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bapteste E., van Iersel L., Janke A., Kelchner S., Kelk S., McInerney J.O., Morrison D.A., Nakhleh L., Steel M., Stougie L., Whitfield J.. 2013. Networks: expanding evolutionary thinking. Trends Genet. 29:439–441. [DOI] [PubMed] [Google Scholar]
- Bastide P., Solís-Lemus C., Kriebel R., Sparks W., Ané C.. 2018. Phylogenetic comparative methods on phylogenetic networks with reticulations. Syst. Biol. 67:800–820. [DOI] [PubMed] [Google Scholar]
- Blischak P., Chifman J., Wolfe A., Kubatko L.S.. 2018. Hyde: a python package for genome-scale hybrid detection. Syst. Biol. 67:821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordewich M., Semple C.. 2007. Computing the hybridization number of two phylogenetic trees is fixed-parameter tractable. IEEE/ACM Trans. Comput. Biol. Bioinform. 4:458–466. [DOI] [PubMed] [Google Scholar]
- Boykin L.M., Kubatko L.S., Lowrey T.K.. 2010. Comparison of methods for rooting phylogenetic trees: a case study using Orcuttieae (Poaceae: Chloridoideae). Mol. Phylogenet. Evol. 54:687–700. [DOI] [PubMed] [Google Scholar]
- Bryant D., Moulton V.. 2002. NeighborNet: an agglomerative method for the construction of planar phylogenetic networks In: Proceedings of 2nd Int’l Workshop Algorithms in Bioinformatics (WABI02), Vol. 2452. Lecture Notes in Computer Science. Berlin, Germany: Springer; p. 375–391. [Google Scholar]
- Burbrink F., Gehara M.. 2018. The biogeography of deep time phylogenetic reticulation. Syst. Biol. 67:743–755. [DOI] [PubMed] [Google Scholar]
- Burgess R., Yang Z.. 2008. Estimation of hominoid ancestral population sizes under bayesian coalescent models incorporating mutation rate variation and sequencing errors. Mol. Biol. Evol. 25:1979–1994. [DOI] [PubMed] [Google Scholar]
- Cardona G., Llabrés M., Rosselló F., Valiente G.. 2009a. Metrics for phylogenetic networks I: Generalizations of the Robinson–Foulds metric. IEEE/ACM Trans. Comput. Biol. Bioinform. 6:46–61. [DOI] [PubMed] [Google Scholar]
- Cardona G., Llabrés M., Rosselló F., Valiente G.. 2009b. Metrics for phylogenetic networks II: Nodal and triplets metrics. IEEE/ACM Trans. Comput. Biol. Bioinform. 6:454–469. [DOI] [PubMed] [Google Scholar]
- Cardona G., Llabrés M., Rosselló F., Valiente G.. 2009c. On Nakhleh’s metric for reduced phylogenetic networks. IEEE/ACM Trans. Comput. Biol. Bioinform. (TCBB) 6:629–638. [DOI] [PubMed] [Google Scholar]
- Chifman J., Kubatko L.. 2014. Quartet inference from SNP data under the coalescent model. Bioinformatics 30:3317–3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chifman J., Kubatko L.. 2015. Identifiability of the unrooted species tree topology under the coalescent model with time-reversible substitution processes, site-specific rate variation, and invariable sites. J. Theor. Biol. 374:35–47. [DOI] [PubMed] [Google Scholar]
- Choleva L., Musilova Z., Kohoutova-Sediva A., Paces J., Rab P., Janko K.. 2014. Distinguishing between incomplete lineage sorting and genomic introgressions: complete fixation of allospecific mitochondrial DNA in a sexually reproducing fish (cobitis; teleostei), despite clonal reproduction of hybrids. PLOS One 9:e80641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou J., Gupta A., Yaduvanshi S., Davidson R., Nute M., Mirarab S., Warnow T.. 2015. A comparative study of SVDquartets and other coalescent-based species tree estimation methods. BMC Genomics 16:S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy C., Jansson J., Sadakane K., Sung W.-K.. 2005. Computing the maximum agreement of phylogenetic networks. Theor. Comput. Sci. 335:93–107. [Google Scholar]
- Chung Y., Ané C.. 2011. Comparing two Bayesian methods for gene tree/species tree reconstruction: simulations with incomplete lineage sorting and horizontal gene transfer. Syst. Biol. 60:261–275. [DOI] [PubMed] [Google Scholar]
- Corander J., Marttinen P.. 2006. Bayesian identification of admixture events using multilocus molecular markers. Mol. Ecol. 15:2833–2843. [DOI] [PubMed] [Google Scholar]
- Critchlow D.E., Pearl D.K., Qian C.. 1996. The triples distance for rooted bifurcating phylogenetic trees. Syst. Biol. 45:323–334. [Google Scholar]
- DeGiorgio M., Degnan J.H.. 2014. Robustness to divergence time underestimation when inferring species trees from estimated gene trees. Syst. Biol. 63:66–82. [DOI] [PubMed] [Google Scholar]
- DeGiorgio M., Rosenberg N.A.. 2016. Consistency and inconsistency of consensus methods for inferring species trees from gene trees in the presence of ancestral population structure. Theor. Popul. Biol. 110:12–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGiorgio M., Degnan J.H., Rosenberg N.A.. 2011. Coalescence-time distributions in a serial founder model of human evolutionary history. Genetics 189:579–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degnan J.H. 2013. Anomalous unrooted gene trees. Syst. Biol. 62:574–590. [DOI] [PubMed] [Google Scholar]
- Degnan J.H., Rosenberg N.A.. 2006. Discordance of species trees with their most likely gene trees. PLoS Genetics 2:e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degnan J.H., Rosenberg N.A.. 2009. Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol. Evol. 24:332–340. [DOI] [PubMed] [Google Scholar]
- Drton M., Sturmfels B., Sullivant S.. 2009. Lectures on Algebraic Statistics, Vol. 39, Oberwolfach Seminars. Basel Boston Berlin: Birkhäuser Verlag. [Google Scholar]
- Durand E.Y., Patterson N., Reich D., Slatkin M.. 2011. Testing for ancient admixture between closely related populations. Mol. Biol. Evol. 28:2239–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutheil J.Y., Ganapathy G., Hobolth A., Mailund T., Uyenoyama M.K., Schierup M.H.. 2009. Ancestral population genomics: the coalescent hidden Markov model approach. Genetics 183:259–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. 1981. Evolutionary trees from gene frequencies and quantitative characters: finding maximum likelihood estimates. Evolution 35:1229–1242. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. 2004. Inferring phylogenies. Sunderland, MA: Sinauer. [Google Scholar]
- Gambette P., van Iersel L., Jones M., Lafond M., Pardi F., Scornavacca C.. 2017. Rearrangement moves on rooted phylogenetic networks. PLoS Comput. Biol. 13:e1005611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatesy J., DeSalle R., Wahlberg N.. 2007. How many genes should a systematist sample? Conflicting insights from a phylogenomic matrix characterized by replicated incongruence. Syst. Biol. 56:355–363. [DOI] [PubMed] [Google Scholar]
- Gusfield D. 2014. ReCombinatorics: the algorithmics of ancestral recombination graphs and explicit phylogenetic networks. Cambridge, MA: MIT Press. [Google Scholar]
- Hein J., Schierup M.H., Wiuf C.. 2005. Gene genealogies, variation and evolution: a primer in coalescent theory. Oxford, UK: Oxford University Press. [Google Scholar]
- Holland B., Penny D., Hendy M.. 2003. Outgroup misplacement and phylogenetic inaccuracy under a molecular clock—a simulation study. Syst. Biol. 52:229–238. [DOI] [PubMed] [Google Scholar]
- Holland B.R., Benthin S., Lockhart P.J., Moulton V., Huber K.T.. 2008. Using supernetworks to distinguish hybridization from lineage-sorting. BMC Evolut. Biol. 8:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H., He Q., Kubatko L.S., Knowles L.L.. 2010. Sources of error inherent in species-tree estimation: impact of mutational and coalescent effects on accuracy and implications for choosing among different methods. Syst. Biol. 59:573–583. [DOI] [PubMed] [Google Scholar]
- Huber K.T., Linz S., Moulton V., Wu T.. 2016a. Spaces of phylogenetic networks from generalized nearest-neighbor interchange operations. J. Math. Biol. 72:699–725. [DOI] [PubMed] [Google Scholar]
- Huber K.T., Moulton V., Wu T.. 2016b. Transforming phylogenetic networks: moving beyond tree space. J. Theor. Biol. 404:30–39. [DOI] [PubMed] [Google Scholar]
- Hudson R.R. 2002. Generating samples under a Wright–Fisher neutral model of genetic variation. Bioinformatics 18:337–338. [DOI] [PubMed] [Google Scholar]
- Huson D. 1998. SplitsTree: a program for analyzing and visualizing evolutionary data. Bioinformatics 14:68–73. [DOI] [PubMed] [Google Scholar]
- Huson D., Rupp R., Scornavacca C.. 2010. Phylogenetic networks: concepts, algorithms and applications. New York: Cambridge University Press. [Google Scholar]
- Huson D.H., Scornavacca C.. 2011. A survey of combinatorial methods for phylogenetic networks. Genome Biol. Evol. 3:23–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joly S., McLenachan P.A., Lockhart P.J.. 2009. A statistical approach for distinguishing hybridization and incomplete lineage sorting. Am. Nat. 174:E54–E70. [DOI] [PubMed] [Google Scholar]
- Jones G., Sagitov S., Oxelman B.. 2013. Statistical inference of allopolyploid species networks in the presence of incomplete lineage sorting. Syst. Biol. 62:467–478. [DOI] [PubMed] [Google Scholar]
- Kubatko L., Carstens B., Knowles L.. 2009. STEM: species tree estimation using maximum likelihood for gene trees under coalescence. Bioinformatics 25:971–973. [DOI] [PubMed] [Google Scholar]
- Kubatko L.S. 2009. Identifying hybridization events in the presence of coalescence via model selection. Syst. Biol. 58:478–488. [DOI] [PubMed] [Google Scholar]
- Lanier H., Knowles L.. 2012. Is recombination a problem for species-tree analyses? Syst. Biol. 61:691–701. [DOI] [PubMed] [Google Scholar]
- Larget B.R., Kotha S.K., Dewey C.N., Ané C.. 2010. Bucky: gene tree/species tree reconciliation with Bayesian concordance analysis. Bioinformatics 26:2910–2911. [DOI] [PubMed] [Google Scholar]
- Leaché A.D., Rannala B.. 2011. The accuracy of species tree estimation under simulation: a comparison of methods. Syst. Biol. 60:126–137. [DOI] [PubMed] [Google Scholar]
- Liu L., Yu L.. 2011. Estimating species trees from unrooted gene trees. Syst. Biol. 60:661–667. [DOI] [PubMed] [Google Scholar]
- Liu L., Yu L., Edwards S.V.. 2010. A maximum pseudo-likelihood approach for estimating species trees under the coalescent model. BMC Evol. Biol. 10:302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Yu L., Pearl D.K., Edwards S.V.. 2009. Estimating species phylogenies using coalescence times among sequences. Syst. Biol. 58:468–477. [DOI] [PubMed] [Google Scholar]
- Long C., Kubatko L.S.. 2018. The effect of gene flow on coalescent-based species-tree inference. Syst. Biol. 67:770–785. [DOI] [PubMed] [Google Scholar]
- Maddison W.P., Knowles L.L.. 2006. Inferring phylogeny despite incomplete lineage sorting. Syst. Biol. 55:21–30. [DOI] [PubMed] [Google Scholar]
- Meng C., Kubatko L.S.. 2009. Detecting hybrid speciation in the presence of incomplete lineage sorting using gene tree incongruence: a model. Theor. Popul. Biol. 75:35–45. [DOI] [PubMed] [Google Scholar]
- Mirarab S., Reaz R., Bayzid M.S., Zimmermann T., Swenson M.S., Warnow T.. 2014. ASTRAL: genome-scale coalescent-based species tree estimation. Bioinformatics 30:i541–i548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales A., Carstens B.. 2018. Evidence that Myotis lucifugus ‘subspecies’ are five non-sister species, despite gene flow. Syst. Biol. 67:756–769. [DOI] [PubMed] [Google Scholar]
- Morrison D.A. 2011. Introduction to phylogenetic networks. Uppsala, Sweden: RJR Productions. [Google Scholar]
- Müller N.F., Rasmussen D.A., Stadler T.. 2017. The structured coalescent and its approximations. Mol. Biol. Evol. 34:2970–2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahkleh L. 2010. A metric on the space of reduced phylogenetic trees. IEEE/ACM Trans. Comput. Biol. Bioinform. 7:218–222. [DOI] [PubMed] [Google Scholar]
- Nei M. 1987. Molecular evolutionary genetics. Columbia University Press. [Google Scholar]
- Ogilvie H.A., Bouckaert R.R., Drummond A.J.. 2017. StarBEAST2 brings faster species tree inference and accurate estimates of substitution rates. Mol. Biol. Evol. 34:2101–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardi F., Scornavacca C.. 2015. Reconstructible phylogenetic networks: do not distinguish the indistinguishable. PLoS Comput. Biol. e1004135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pease J.B., Hahn M.W.. 2015. Detection and polarization of introgression in a five-taxon phylogeny. Syst. Biol. 64:651–662. [DOI] [PubMed] [Google Scholar]
- Rannala B., Yang Z.. 1996. Probability distribution of molecular evolutionary trees: a new method of phylogenetic inference. J. Mol. Evolut. 43:304–311. [DOI] [PubMed] [Google Scholar]
- Rannala B., Yang Z.. 2003. Bayes estimation of species divergence times and ancestral population sizes using DNA sequences from multiple loci. Genetics 164:1645–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes J.A. 2017. Topological metrizations of trees, and new quartet methods of tree inference. arXiv preprint arXiv:1704.02004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D., Foulds L.. 1981. Comparison of phylogenetic trees. Math. Biosci. 53:131–147. [Google Scholar]
- Roch S., Warnow T.. 2015. On the robustness to gene tree estimation error (or lack thereof) of coalescent-based species tree methods. Syst. Biol. 64:663–676. [DOI] [PubMed] [Google Scholar]
- Rosenberg N.A. 2002. The probability of topological concordance of gene trees and species trees. Theor. Pop. Biol. 61:225–247. [DOI] [PubMed] [Google Scholar]
- Salichos L., Rokas A.. 2013. Inferring ancient divergences requires genes with strong phylogenetic signals. Nature 497:327–331. [DOI] [PubMed] [Google Scholar]
- Schiffels S., Durbin R.. 2014. Inferring human population size and separation history from multiple genome sequences. Nat. Genet. 46:919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons M.P., Gatesy J.. 2015. Coalescence vs. concatenation: sophisticated analyses vs. first principles applied to rooting the angiosperms. Mol. Phylogenet. Evol. 91:98–122. [DOI] [PubMed] [Google Scholar]
- Slatkin M., Pollack J.L.. 2008. Subdivision in an ancestral species creates asymmetry in gene trees. Mol. Biol. Evol. 25:2241–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solís-Lemus C., Ané C.. 2016. Inferring phylogenetic networks with maximum pseudolikelihood under incomplete lineage sorting. PLoS Genet. 12:e1005896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solís-Lemus C., Bastide P., Ané C.. 2017. Phylonetworks: a package for phylogenetic networks. Mol. Biol. Evol. 34:3292–3298. [DOI] [PubMed] [Google Scholar]
- Solís-Lemus C., Yang M., Ané C.. 2016. Inconsistency of species tree methods under gene flow. Syst. Biol. 65:843–851. [DOI] [PubMed] [Google Scholar]
- Song S., Liu L., Edwards S.V., Wu S.. 2012. Resolving conflict in eutherian mammal phylogeny using phylogenomics and the multispecies coalescent model. Proc. Natl. Acad. Sci. USA 109: 14942–14947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2006. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690. [DOI] [PubMed] [Google Scholar]
- Sukumaran J., Knowles L.L.. 2017. Multispecies coalescent delimits structure, not species. Proc. Natl. Acad. Sci. USA 114: 1607–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahata N. 1988. The coalescent in two partially isolated diffusion populations. Genet. Res. 52:213–222. [DOI] [PubMed] [Google Scholar]
- Than C., Ruths D., Nakhleh L.. 2008. PhyloNet: a software package for analyzing and reconstructing reticulate evolutionary relationships. BMC Bioinformatics 9:322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theunert C., Slatkin M.. 2017. Distinguishing recent admixture from ancestral population structure. Genome Biol. Evol. 9:427–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Iersel L., Linz S.. 2013. A quadratic kernel for computing the hybridization number of multiple trees. Inform. Process. Lett. 113:318–323. [Google Scholar]
- van Iersel L., Kelk S., Lekić N., Scornavacca C.. 2014. A practical approximation algorithm for solving massive instances of hybridization number for binary and nonbinary trees. BMC Bioinformatics 15:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Iersel L., Kelk S., Lekić N., Whidden C., Zeh N.. 2016a. Hybridization number on three rooted binary trees is EPT. SIAM J. Discrete Math. 30:1607–1631. [Google Scholar]
- van Iersel L., Kelk S., Stamoulis G., Stougie L., Boes O.. 2017. On unrooted and root-uncertain variants of several well-known phylogenetic network problems. Algorithmica. 10.1007/s00453-017-0366-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen D., Nakhleh L.. 2017. Co-estimating reticulate phylogenies and gene trees from multi-locus sequence data. Syst. Biol. doi: 10.193/sysbio/syx085. [DOI] [PubMed] [Google Scholar]
- Wen D., Yu Y., Nakhleh L.. 2016. Bayesian inference of reticulate phylogenies under the multispecies network coalescent. PLoS Genet. 12:e1006006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y. 2012. Coalescent-based species tree inference from gene tree topologies under incomplete lineage sorting by maximum likelihood. Evolution 66:763–775. [DOI] [PubMed] [Google Scholar]
- Yu Y., Nakhleh L.. 2015a. A distance-based method for inferring phylogenetic networks in the presence of incomplete lineage sorting In: International Symposium on Bioinformatics Research and Applications. Berlin, Germany: Springer; p. 378–389. [Google Scholar]
- Yu Y., Nakhleh L.. 2015b. A maximum pseudo-likelihood approach for phylogenetic networks. BMC Genomics 16(Suppl 10):S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y., Degnan J.H., Nakhleh L.. 2012. The probability of a gene tree topology within a phylogenetic network with applications to hybridization detection. PLoS Genet. 8:e1002660–e1002660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y., Dong J., Liu K.J., Nakhleh L.. 2014. Maximum likelihood inference of reticulate evolutionary histories. Proc. Natl. Acad. Sci. USA 111:16448–16453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y., Than C., Degnan J.H., Nakhleh L.. 2011. Coalescent histories on phylogenetic networks and detection of hybridization despite incomplete lineage sorting. Syst. Biol. 60:138–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C., Ogilvie H.A., Drummond A.J., Stadler T.. 2018. Bayesian inference of species networks from multilocus sequence data. Mol. Biol. Evol. 35:504–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Yu Y., Nakhleh L.. 2016. In the light of deep coalescence: revisiting trees within networks. BMC Bioinformatics 17:415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S., Degnan J.H.. 2017. Displayed trees do not determine distinguishability under the network multispecies coalescent. Syst. Biol. 66:283–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S., Degnan J.H., Goldstien S.J., Eldon B.. 2015. Hybrid-lambda: simulation of multiple merger and kingman gene genealogies in species networks and species trees. BMC Bioinformatics 16:292. [DOI] [PMC free article] [PubMed] [Google Scholar]