


Abstract
Muscle-specific transcription factor MyoD orchestrates the myogenic gene expression program by binding to short DNA motifs called E-boxes within myogenic cis-regulatory elements (CREs). Genome-wide analyses of MyoD cistrome by chromatin immnunoprecipitation sequencing shows that MyoD-bound CREs contain multiple E-boxes of various sequences. However, how E-box numbers, sequences and their spatial arrangement within CREs collectively regulate the binding affinity and transcriptional activity of MyoD remain largely unknown. Here, by an integrative analysis of MyoD cistrome combined with genome-wide analysis of key regulatory histones and gene expression data we show that the affinity landscape of MyoD is driven by multiple E-boxes, and that the overall binding affinity—and associated nucleosome positioning and epigenetic features of the CREs—crucially depend on the variant sequences and positioning of the E-boxes within the CREs. By comparative genomic analysis of single nucleotide polymorphism (SNPs) across publicly available data from 17 strains of laboratory mice, we show that variant sequences within the MyoD-bound motifs, but not their genome-wide counterparts, are under selection. At last, we show that the quantitative regulatory effect of MyoD binding on the nearby genes can, in part, be predicted by the motif composition of the CREs to which it binds. Taken together, our data suggest that motif numbers, sequences and their spatial arrangement within the myogenic CREs are important determinants of the cis-regulatory code of myogenic CREs.
INTRODUCTION
MyoD is a member of the basic helix-loop-helix transcriptional regulators and the principal driver of the myogenic differentiation program (1–3). Sequence-specific recognition of MyoD to DNA is dependent on the core hexanucleotide (CANNTG) sequence (3), termed the E-box motif. However, despite the ubiquitous genomic distribution of E-boxes, multiple regulatory mechanisms ensure that MyoD is selectively tethered to specific chromatin regions. These regulatory mechanisms include chromatin accessibility (4,5), occurrence of relatively denser clustering of MyoD binding motifs within the putative myogenic cis-regulatory elements (CREs) (6,7) and various cooperative interactions between MyoD and other trans and cis factors to regulate binding specificity and affinity (6,8–10). Recent genome-wide analysis of MyoD binding pattern in myogenic cells suggests that on average it binds to relatively large chromatin regions (average MyoD chromatin immnunoprecipitation sequencing (ChIP-seq) peak width of 400 bp) encompassing multiple unique or recurrent E-box motifs (6). Binding of MyoD over such relatively large chromatin regions raises the question of how variables such as motif sequences, numbers and their spatial arrangement within CREs create a context dependent environment to regulate chromatin state and direct MyoD activity. More specifically, how the interactions among cis-based variables within the myogenic CREs regulate the affinity landscape of MyoD and the dynamic range of target gene expression remains largely unknown.
Genome-wide binding data of transcription factor occupancy together with high-resolution mapping of various regulatory histone marks have provided an extensive catalog of CREs in mice and human (11,12). Moreover, the availability of whole genome sequence data of mouse (13) and human (14) allows comparative genomic analysis of the CRE sequences, to compare sequence composition and evolutionary conservation of homologous CREs across the species. These datasets, together with quantitative analysis of TF binding sites and analysis of regulatory histone marks, provide important input to interrogate the dynamic interplay between transcription factors and their CREs in regulation of chromatin state and control of gene expression.
To gain further insights into the mechanistic basis of how MyoD’s interaction with the CREs implements the myogenic cis-regulatory code, we decided to perform a multi-pronged analysis of MyoD binding. We based our study on our own ChIP-seq reporting MyoD binding in differentiating primary mouse muscle cells (15), as well as two independently obtained MyoD ChIP-seq datasets in differentiating C2C12 myogenic cells (7,16). In addition, we determined the relationship between MyoD occupancy and nucleosome positioning, using genome wide histone H3 ChIP-seq as a proxy measure, enhancer activity status using the H3K4me1 histone mark and the associated promoter status using the H3K4me3 and H3K27me3 marks. To study the evolutionary conservation of binding sites, we used single nucleotide polymorphism (SNP) data from a study of 17 mouse strains. At last, we related MyoD binding, motifs in CREs and chromatin state to changes in gene expression during differentiation, as assayed by RNA-seq. From these analyses, we have determined that number, sequence and spatial arrangement of E-box motifs within myogenic CREs in combination regulate MyoD binding to DNA and determine the differential gene expression output. Importantly, we have derived an index which we term ‘motif score’ to quantify total MyoD binding to CREs and to predict differential gene expression output—solely dependent on the numbers, sequences and the spatial arrangement of E-boxes within MyoD-bound CREs. Our comparative genomic analysis on the rates of SNPs in E-boxes suggests that the motifs contributing most strongly to our motif score are also those that are under the strongest conserving selection, whereas other motif variants and other E-boxes genome wide show less or no evidence for selection. Together, our data suggests that sequence variation, numbers and the spatial arrangement of E-box motifs within myogenic CREs together regulate the binding specificity and affinity landscape of MyoD cistrome.
MATERIALS AND METHODS
Mice and animal care
Care of animals was in accordance with institutional guidelines as regulated by the Canadian Council of Animal Care (CCAC). All protocols are first approved by Animal Research Ethics Board at the University of Ottawa and these protocols are reviewed on an annual basis. Animals were euthanized by CO2 inhalation in a chamber specially designed for such use. This procedure is in accordance with the standard operating procedures of the University Animal Facility as recommended by the CCAC.
Cell culture
Primary muscle progenitor cells were isolated from the hind limbs of 4–6 weeks old wild-type mice by Fluorescent Activated Cell Sorting as described previously (15). The cells were maintained on collagen coated culture dishes in growth media (Ham’s F10 supplemented with 20% Fetal Bovine Serum (FBS), 2.5 ng/ml bFGF, 1% penicillin/streptomycin) in a 37°C incubator with 5% CO2. Fully confluent cultures of primary myoblasts were differentiated by switching to differentiation media (Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% horse serum) for 48 h.
Antibodies
The following antibodies were used in this study: anti-H3K4me3 (Abcam 8580), anti-H3K27me3 (Millipore 07–449), anti-H3K4me1 (Cell Signaling Technologies 5326), anti-Histone H3 (Millipore 05–499).
ChIP-seq
Chromatin Tandem Affinity Purification for MyoD was carried out as described previously (15,17). In addition, we performed ChIP-seq for H3K4me3, H3K27me3, H3K4me1 and total histone H3 as follow: briefly, 8 × 107 cells were cross linked with 1% formaldehyde in 1 × phosphate-buffered saline buffer at room temperature. Chromatin was sheered by sonication to an average fragment length of 200 bp. ChIP was performed on 20 mg of cell lysate using 20 μg of antibody at the ratio of 1:1000 antibody to antigen. ChIP library construction was performed from 10 ng of ChIP DNA using standard Illumina ChIP library preparation protocol and as described previously (15,17). Sequencing was performed on GAIIX Genome Analyzer. To achieve a sufficient coverage for histone marks these samples were run in duplicates on two lanes of the flow cell as technical replicates. Sequenced reads from the two lanes we subsequently pooled together to achieve higher depth and better genome coverage.
ChIP-seq peak calling
ChIP-seq reads from the control and the experiments were filtered for polymerase chain reaction-induced duplications and mapped to the mm9 (NCBI37) mouse genome assembly by ELAND. These parameters were used to be consistent with previously the published MyoD dataset (6). The filtering resulted in removal of all but one sequence read with identical 5′ end position. Reads from the control and experiments were fed to MACS version 1.37 (18), after empirically estimating the mean fragment length of the mapped sequenced reads by mappability sensitive cross-correlation, MaSC (19), to identify genomic loci enriched for transcription factor occupancy (peaks).
Motif analysis
Motif analysis was performed on the full length of the DNA sequence under peaks, unless specified otherwise. The full DNA sequence of mm9 was obtained using UCSC Genome Browser tools (http://genome.ucsc.edu). Sequences were submitted to MEMECHIP (http://meme.nbcr.net/meme/cgi-bin/meme-chip.cgi) software using all vertebrate motif database to identify highly enriched consensus motifs.
Fragment length estimation
We applied MaSC analysis to estimate mean fragment length in our single-read sequencing data as described previously (19). The use of MaSC significantly improves the accuracy of the location of the enriched genomic regions and improves the peak calling process (19).
RNA-seq analysis
Total RNA was isolated from proliferating primary myoblasts or primary myotubes which were obtained by feeding >90% confluence primary myoblasts with differentiation medium (DMEM supplemented with 5% horse serum) for 48 h, as described previously (6). The quality and quantity of RNA was assessed by the Agilent Bio analyzer. A total of 2 μg RNA was used as input for mRNA-seq analysis. Cluster generation was carried out following Illumina TruSeq SR Cluster Kit v5 and Illumina Cluster Station was used for cluster generation (15). Sequencing was done using TruSeq SBS Kit v5 – GA (36-cycle) on Illumina GAIIX Genome Analyzers. Reads from RNA-seq samples were mapped to the mm9/NCBI37 mouse genome assembly and to splice sites predicted from UCSC splicing models (refFlat.txt.gz) (20) using the eland_rna analysis option of the Illumina GERALD pipeline v1.7 (Illumina) using default parameters. The Illumina CASAVA pipeline (v1.7) was used to aggregate the mapped reads and to quantify the transcripts present in the original sample, using the readBases method. The edgeR Bioconductor package (21) was used to compare expression between pairs of samples using the negative binomial exact test. Fold change and P-values were corrected by multiple testing using the Bonferroni method.
Microarray gene expression analysis
Affymetrix MoGene-1_0-st-v1 was also used for expression analysis of primary myoblasts and myotubes (48 h in differentiation media). Six microarrays were used in this analysis and are available as GenBank Accession (GEO GSE24811). Transcript Cluster Identifiers (TCID) were normalized with robust multi-array averaging (22) using the Bioconductor R package (23). The results were log2 transformed and were analyzed using significance analysis of microarrays. TCIDs mapping to zero or more than one gene (ENSEMBL v67) were excluded from the analysis.
Circos plot for visualization of genome-wide binding and expression
To visualize data on a genome wide scale we generated a concentric circos plot with nine tracks representing MyoD ChIP-seq, genome wide regulatory histone data (H3K4me1, H3K4me3 and H3K27me3), and differential gene expression output superimposed on motif sequences within the CREs. We first identified all peaks in our MyoD dataset for which a gene was located within +/- 5kb of the peak, resulting in retention of 1800 out of 10 756 peaks. Next, we sorted the 1800 peak set in order of decreasing MyoD peak tag density (mean number of sequenced reads within the MyoD peaks divided by the peak length) and binned data into 100 percentiles. We then computed density for each track as follow: starting from the outermost circle; track 1, MyoD peak reads density (number of sequenced reads within MyoD peaks divided by peak length); track 2, H3K4me1 read density within MyoD peaks; H3K4me3 read density within ±500 bp windows overlapping the TSSs of the MyoD peak-associated genes; track 4, H3K27me3 read density within ±500 bp windows overlapping the TSSs of MyoD peak associated genes; track 5, total histone H3 reads within ±500 bp windows overlapping the TSSs of MyoD peak associated genes; track 6, number of 100% G/C center dinucleotide E-boxes in peaks; track 7, number of 50% G/C center dinucleotide E-boxes; track 8, number of 0% G/C center dinucleotide E-boxes; track 9 absolute expression value (RNA-seq) of the associated gene to MyoD peak. Each track was plotted by calculating mean value for each bin forming the final set of 100-element vectors. The tracks are grouped so that each group has a single scale. The tag numbers are normalized to a total of 10 million reads per dataset.
The color map (as shown) ranges from dark red (high values) to dark blue (low values), passing through orange, yellow and green in sequence. For group one (tracks 1–5), the colors map to values ranging from a scale of 2 to 110 (blue to red), based on the range of values for the MyoD peaks track. Accordingly, the H3K4me1 track maps from dark blue to a shade of green (scale of 0 to ∼40), the H3K4me3 track maps from a shade of green to light yellow (scale of ∼40 to ∼90), the H3K27me3 track maps from lighter shades of blue down to dark blue (scale of ∼9 down to ∼0. At last, the total histone H3 reads stays relatively uniform with minor oscillation between a scale of ∼5 and ∼7. Due to the normalization of ChIP-seq datasets to 10 million reads and binning and subsequent averaging, the values for these ‘tag-number’ tracks end up being real numbers instead of integers. For group two (tracks 6–8) representing E-box numbers and sequences, the full set of colors are remapped to a range of values from ∼0 to ∼6, corresponding to the overall range for the E-boxes tracks. The 100% G/C E-box track ranges from scale of 1 to 6, mapping to most of the color spectrum with a stronger patch of red at the top, the 50% G/C E-box track varies from scale of 0.5 to 4 with a weaker red patch at the top, and the 0% G/C E-box track stays below 1 with a much weaker trend mapping mainly to the blue shades. At last, the gene expression track values (group three with a lone member) range from about 0.2 to 2, and the colors are remapped to this range, independent of the other tracks.
Associating peaks to genes
To minimize the chance of false associations, we used a conservative approach to associate peaks to genes, for each gene, we first considered if any peak(s) overlapped a 5 kbp window centered the TSS of the gene, and associated only the closest peak to the gene. (There were no ties.) Among these associations, if any peak were associated with two different genes, we retain only the gene to whose TSS it was closest. In this way, every peak is associated to at most one gene and vice versa, and the associations are the closest among possible choices.
SNP analysis
Mouse genomic sequences from 17 strains were used in this analysis (13). Using C57BL/6 as the reference genome, there are estimated to be 6.4 million SNPs in which there is an alternative allele in at least one of the 17 strains compared to the C57BL/6 reference strain as reported previously (13). We first measured SNPs rates for A, C, G and T across the reference genome. Using an expression match that counts both unique and overlapping CANNTG, we determined a total of 14 206 895 E-boxes in the reference genome. Next, we determined the rates of SNPs for each of the hexanucleotides in E-boxes within MyoD peaks and those outside of the peaks.
Luciferase assay
Luciferase constructs harboring various configurations of E-box motifs were synthesized and sub cloned into pGL4.23 [luc2/minP] vector. DEL represent deleted E-box; GC represents CAGCTG motif; AT represents CAATTG motif and CA represents CACATG motif. The E-box containing sequence is a multimerization (3×) of a MyoD target described previously (15). Dual luciferase assay was performed by co-transfection of mouse MyoD- and E47-expressing plasmids in Cos7 cells using Promega Dual luciferase assay kit. Luciferase values were normalized to renilla (Luciferase/Renilla) and were plotted relative to a construct with mutated E-box sequences (DEL-DEL-DEL).
RESULTS
This study was conducted to gain mechanistic insight into how the spatial arrangement, numbers and the sequences of E-boxes within myogenic CREs regulate the affinity landscape of MyoD and the expression of associated genes. We first analyzed genome wide occupancy of MyoD in the differentiating primary (6) and C2C12 myogenic cell line-derived myotubes (7,16). To quantify the similarity between the datasets we first measured the overlap in their peaks. Specifically, for each dataset we counted the number of peaks that occurs in the other two datasets (Supplementary Table S1). As an out-group, we added a fourth ChIP-seq dataset for Pax7 (17), an unrelated transcription factor. From this analysis we observed that there is substantial overlap in MyoD binding among datasets (Supplementary Figure S1 and Table S1). To analyze the relationship between MyoD occupancy and the epigenetic state of the CREs we performed ChIP-seq for H3K4me1, H3K4me3 and H3K27me3 in differentiating primary myotubes. H3K4me1 marks active enhancer elements, while H3K4me3 and H3K27me3 mark promoters and transcription start sites (TSS) in an opposing fashion. Next, as a proxy measurement for nucleosome occupancy we performed ChIP-seq of pan histone H3 in primary myotubes. To analyze the relationship between MyoD occupancy, the status of chromatin at the myogenic CREs and gene expression output, we performed Affymetrix gene expression microarray and RNA-seq analyses of differentiating primary muscle cells. At last, to examine the levels of selective constraint between MyoD binding motifs within the myogenic CREs and their genomic counterparts we performed genome wide comparative analysis of SNPs in the genomic sequences of 17 strains of laboratory mice (13).
Distribution of MyoD binding motifs in myogenic CREs
Analysis of MyoD binding data from three independent datasets (6,7,16) revealed significant overlap in binding pattern despite differences in cell types and the ChIP assay protocols used (Supplementary Figure S1 and Table S1). As expected, MyoD peaks were highly enriched for the canonical MyoD binding motif, the E-box sequence CANNTG, compared to their random genomic distribution in all three datasets (Figure 1A–C and Supplementary Figure S2). The presence of the degenerate center dinucleotide NN in the E-box motif results in 16 possible sequence configurations, comprising 10 unique motif classes based on sequence reverse complementarity: dinucleotide GC, CG, CC or GG, AC or GT, etc. Analysis of the E-box sequences in MyoD peaks confirmed the preference of MyoD binding to E-boxes with GC-rich center dinucleotide (GC, CC, CG, GG) during muscle cell differentiation (Figure 1C and D). This includes the CACGTG E-box which, although present with low absolute frequency, is nevertheless two-fold enriched in MyoD peaks over its genome-wide frequency. However, many peaks lack the preferred CA[G/C]TG E-box motif (Figure 1D), implying that the ‘lower GC content’ E-box sequences also have an important role in driving MyoD binding. Moreover, with an average of 3.89 E-box motifs per peak, and with some peaks having 10 E-boxes or more (Figure 1E and Supplementary Figure S2d), it appears likely that binding in many regions may be driven by multiple E-boxes with various sequences.
Figure 1.

E-box preferences of MyoD peaks (A) E-boxes are significantly enriched in MyoD peaks at ∼1.5-fold over genomic levels. (B) Consensus logo of all E-boxes in MyoD peaks shows the hexa-nucleotide sequence with a variable dinucleotide and two flanking invariable dinucleotide. (C) Frequencies of E-boxes with different center dinucleotide in MyoD peaks versus the genome as a whole show very strong enrichment for the GC dinucleotide, moderate enrichment for the other 100% GC dinucleotide and depletion of 0% GC dinucleotide. (D) A heat map showing the presence of at least one E-box (blue) of each type in peaks reveals that the majority of peaks contain at least one GC-rich E-box, but a substantial minority rely on non-GC-rich E-boxes (or have no E-box at all). (E) Overall E-box numbers also vary widely across peaks, raising the question of how E-box types and numbers are jointly utilized to regulate binding affinity. (F) In an analysis of 17 laboratory mouse genomes, the overall rate of SNPs to A, C, G and T nucleotides, and the observed rates of SNPs to E-boxes in MyoD peaks. (G) Rates of SNPs to the center dinucleotide of E-boxes in MyoD peaks or genome wide show preservation of GC-rich E-boxes but not AT-rich E-boxes.
Variant sequences in the MyoD-bound motifs are under selection
The hexanucleotide E-box sequence, CANNTG contains a central variable dinucleotide that is termed ‘degenerate’ and two flanking peripheral dinucleotides that are invariable and are required for recognition and binding of MyoD to DNA (3). To analyze the functional relevance of the variable dinucleotide within the E-box motif, we performed comparative genomic analysis of the rates of SNPs across 17 mouse strains for which genome sequence data is publicly available (13). We hypothesized that SNP substitution rates within the variant sequences in MyoD-bound CREs would be significantly lower than SNPs rates genome wide, and that functionally important E-box variants would be the most conserved. To test this hypothesis, we first calculated SNP rates for A, T, G and C nucleotides across the 17 mouse genomes using the C57 strain as reference (Figure 1F). Next, we calculated SNPs rates for the nucleotides in E-boxes within the MyoD peaks (Figure 1F and Supplementary Figure S2e). Notably, the rates of SNPs at all the invariant E-box positions, namely the initial CA and the closing TG, are substantially lower than those for the corresponding nucleotides genome wide. In the center dinucleotide, SNPs to C and G nucleotides occurred at lower than genomic rates, but SNPs to A and T nucleotides occurred at rates comparable to those genome wide in all three datasets (Figure 1F and Supplementary Figure S2e). We also analyzed SNPs occurring in the center dinucleotide of E-boxes in MyoD peaks versus all 14.2 million E-boxes genome wide (Figure 1G and Supplementary Figure S2f). For the GC, CG and GG/CC center dinucleotide, we found that rates of SNPs were substantially lower than the genomic counterparts, although the SNP rate for the CG dinucleotide is largest overall, both in MyoD peaks and genome wide. In contrast, the AT and TA center dinucleotide had SNPs at greater than genomic rates. Collectively, these results suggest that certain E-boxes in MyoD peaks are under conserving selection; particularly the GC-rich center dinucleotide is subjected to the highest rate of selection (Figure 1F and G; Supplementary Figure S2e and f).
Quantitative analysis of MyoD binding to the myogenic CREs
The frequencies of different E-box motif classes within the MyoD peaks are dramatically different than their genomic distribution (Figure 1A and C; Supplementary Figure S2a and b) and the E-boxes are differentially conserved (Figure 1F and G), suggesting that the degenerate sequences in the E-box motif are functionally relevant. To investigate this question further, we first sought to determine the quantitative notion of MyoD binding affinity with greatest functional relevance. Some previous studies have used peak height (maximum height or read pileup within the peak) (24) or read density (number of reads in the peak divided by its width, or equivalently the average height of the pileup) (25). Peak height makes most sense for transcription factors that bind single sites within CREs, but with varying affinities due to sequence specificity or other factors. Read density similarly emphasizes binding intensity. MyoD binds different E-boxes with different affinities, but is also expected to bind multiple E-boxes within the same peak. Therefore, we adopted the total MyoD reads in a peak as a proxy for total in vivo binding affinity to that locus (26). Indeed, we found that total MyoD reads in peaks are highly correlated both to the widths of peaks, which reflect multiplicity of binding sites (Figure 2A and Supplementary Figure S3a) and to the read densities within the peaks, which reflect binding intensity (Figure 2B and Supplementary Figure S3b).
Figure 2.

Relationship between motif sequence and enrichment of MyoD on target sites. (A) The numbers of MyoD reads in peaks, a proxy for total binding affinity to a region, are strongly correlated to the widths of peaks. (B) MyoD reads are also correlated to read density. (C) Peak reads, height and read density, as well as total reads in a 1 kb-radius window around the peak summit, correlate moderately to nearby gene expression change. The reads also correlate strongly to the epigenetic status of the peak. (D) Peaks with greater numbers of reads include, on average, greater numbers of E-boxes of all kinds, with the increase strongest for E-boxes with GC-rich center dinucleotide. (E) However, when we look at fixed-width regions centered on peak summits, only GC-rich E-boxes show an increasing trend and GC-poor E-boxes decline with increasing total read count. (F) Analysis of GC 100% and GC50% E-boxes in a 200-bp window around the summit again shows the dominant effect of 100% GC E-boxes, but provides some evidence for increased binding resulting from GC50% E-boxes as well.
To further validate this choice, we performed correlation analysis to test whether variables such as total MyoD reads, read density or peak height were related to important functional correlates of regulatory binding, including histone H3 reads in peaks, H3K4me1 reads and the expression changes between growing and differentiating muscle cells of peak-associated genes (Figure 2C and Supplementary Figure S3c). Of the three variables, total MyoD reads correlates most strongly to the histone H3 and H3K4me1 reads (Figure 2C), and this further justifies our choice of using total MyoD reads in a peak as the most relevant measure of MyoD binding. Correlations to gene expression are modest, albeit positive; we return to this issue later.
To examine the relationship between sequenced reads in MyoD peaks and motif sequence, we performed correlation analysis between the numbers of E-boxes and the numbers of reads in peaks by grouping E-box motifs based on the GC content of the center dinucleotide of the E-box into three possible classes GC 0% (CAAATG, CAATTG, CATATG, CATTTG), GC 50% (CAACTG, CAAGTG, CACATG, CACTTG, CAGATG, CAGTTG, CATCTG, CATGTG) and GC 100% (CACCTG, CACGTG, CAGCTG, CAGGTG). Figure 2D shows that when we divide the peaks into percentiles by their total number of MyoD reads, the total number of E-boxes of each type increases roughly linearly with the number of reads. The increase is most dramatic for the GC 100% E-boxes (i.e. GC, GG/CC, CG), of which there are less than one, on average, in the lowest affinity peaks and over five, on average, in the highest affinity peaks.
Since wider peaks tend to have more reads (Figure 2A), one cannot a priori conclude that increasing E-box counts causes greater MyoD binding. For this reason, we also looked at the relationship between E-box counts within ±1 kb of peak summits to peaks reads. This region is large enough to include nearly all MyoD peaks (Supplementary Figure S3), but is still within the range of plausibility for CREs. Figure 2D shows that in these windows, GC 50% E-box numbers are largely independent of peak reads, and GC 0% E-box counts decline with increasing enrichment of MyoD (Figure 2E and Supplementary Figure S3e). On the other hand, it is the GC 100% E-box count that increases, and therefore those motifs are the strongest candidates for driving MyoD binding in differentiating muscle cells. This does not imply that the GC 50% or GC 0% E-boxes are unimportant. As pointed out above and in Figure 1D, some peaks have no GC 100% E-boxes. Further, E-boxes with lower GC content may have subtle, positive contributions to binding (as discussed below). The dominant quantitative relationship, however, is between the number of GC 100% E-boxes in a regulatory region and the amount of MyoD binding.
To determine the relationship between E-box sequences, peak GC content and the dinucleotide composition of CREs on the enrichment of MyoD we performed linear regression analysis to de-complex the effect of the above variable on the overall enrichment of MyoD. We found that although peak GC content has a non-trivial affect on overall MyoD enrichment, the E-box GC content is the main driver of MyoD enrichment to CREs (Supplementary Figure S4). Notably, the effects of the E-box GC content on the enrichment of MyoD to CREs are independent of the peak GC content (Supplementary Figure S4) or the dinucleotide composition of the peaks in general (Supplementary Figure S5).
To further investigate the relationship between E-box sequences and enrichment of MyoD on CREs, we counted the number of GC 100% and GC 50% E-boxes within 200 bp windows of the peak summits. In these regions, where binding is most intense, over 99.95% of peaks have five E-boxes or fewer. Therefore, we can enumerate different combinations of E-box counts. Figure 2F presents a heat map showing the relationship between the average peak reads and the number of GC 100% and GC 50% E-boxes, with blue representing the combinations with the smallest average number of reads and yellow the largest (see also Supplementary Figure S6). The plot reiterates the dominant effect of the number of GC 100% E-boxes on peak reads. Black ‘greater than’ signs pointing upward, which are the predominant case, indicate combinations where one greater GC 100% E-box results in higher average read count, whereas gray downward signs represent the occasional exception. Similarly, black rightward greater than signs indicate combinations where addition of one more GC 50% E-box results in higher average read count. Across all three datasets, comparing the average MyoD reads in a set of peaks with the reads in peaks with one additional GC 100% E-box near the summit, in 101 out of 120 comparisons, the extra GC 100% E-box is associated with higher average reads (P < 0.0001 by proportion test against the null hypothesis of equal chance). Comparing peak sets with one additional GC 50% E-box, we found greater average MyoD reads in 71 out of 120 comparisons, a more modest but still statistically significant effect (P = 0.0446). Collectively, this analysis reveals that the total MyoD peak reads are the most relevant quantification of peak intensity with respect to regulation, and that GC 100% E-boxes are most strongly associated with MyoD binding, with evidence for weaker influence by GC 50% E-boxes.
Functional consequence of spatial organization of motifs within MyoD-bound CREs
CREs contain recurrent and unique motifs for various transcription factors. However, how the multiplicity of motifs and their spatial arrangements within CREs contribute to binding affinity and gene expression output remains largely unknown. To analyze the functional consequences of spatial placement of E-box motifs within the MyoD-bound myogenic CREs on the enrichment of MyoD on targets, we performed a spatial analysis of E-boxes within peaks and the flanking regions not bound by MyoD (Figure 3A and Supplementary Figure S7). This shows a significant enrichment of GC-rich motifs at the peak summit in all three MyoD ChIP-seq datasets (Figure 3A and Supplementary Figure S7). While GC-poor motifs showed no significant preference to the peak summit location (Figure 3A and Supplementary Figure S7). To eliminate the possibility that variation in peak length or the size of the CREs may contribute to the above observation we performed similar analysis on the 25% widest and the 25% narrowest MyoD peaks (Figure 3B and Supplementary Figure S7). By counting E-boxes within ±1 kb of peak summits and by dividing the region into 21 equal-sized bins, we observed that irrespective of the width of the peak or the size of the putative CREs, GC-rich motifs are significantly more enriched at the MyoD peak summit, while GC-poor motifs showed no spatial preference within the CREs or the flanking regions (Figure 3B and Supplementary Figure S8).
Figure 3.

Spatial distribution of E-boxes in the vicinities of peaks, and their contribution to total peak reads. (A) The relative frequency of E-boxes (divided into three groups based on the GC content of their center dinucleotide) in peaks and flanking regions, each of which is divided into seven equal-sized bins for MyoD ChIP-seq replicates. (B) E-box frequencies in 21 equal-sized bins spanning 2 kb centered on the peak summits, for the 10% narrowest peaks and 10% widest peaks. (C) E-box frequencies per bin, in the same 21 bins across all peaks, along with average MyoD reads divided by 10 (so that scale is comparable). (D) For each of the three E-box categories, the maximum likelihood regression coefficients and 95% confidence intervals for a linear model predicting MyoD reads in each of the 21 bins as a function of the E-box counts in the same bins.
The most significant difference between narrow and wide peaks is that wide peaks show enrichment for GC100% E-boxes over a wider domain surrounding the peak summit. This observation suggests that MyoD may actively binds multiple GC100% E-boxes within those CREs, and that the observed peak width, which we have already determined to be correlated to the total peak reads, is controlled in part by the spatial distribution of the E-boxes within the CREs. In other words, the strategy for having a CRE with a lot of MyoD binding is to have multiple GC-rich E-boxes spread over that region, increasing the likelihood of strong MyoD binding to that regulatory domain. However, whether MyoD binds these E-boxes strictly simultaneously or not, cannot be determined from the ChIP-seq data.
On the other hand, the enrichment of the GC-rich E-boxes near the peak summits mimics the enrichment of the MyoD reads themselves (Figure 3C; Supplementary Figures S7 and 8). This observation suggests the hypothesis that although GC-rich E-boxes are enriched toward the summits of peaks, each E-box may contribute roughly equally to the binding affinity, and hence the same number of MyoD reads. However, when we regress the number of MyoD reads in each of the 21 bins on the numbers of E-boxes, we find that the GC100% E-boxes near the summit contribute more reads than do the peripheral counterparts (Figure 3D and Supplementary Figure S7). A similar pattern holds true for the GC50% E-boxes, although the effect is much weaker. This observation suggests that MyoD binding is stronger at central (i.e. summit) of the peaks, suggesting that motif locations within the CREs have a strong effect on binding independent of the motif sequence. Below, we delve into one possible explanation based on chromatin state.
The relationship between tag density and summit ‘motif switching’
The observation that GC-rich E-boxes contribute more to the summit of the peaks and to the overall enrichment of MyoD on target CREs prompted us to focus our attention on the E-boxes closest to the summit of the peaks, herein called the summit E-box, and to analyze the relationship between the summit motifs, other motifs (peripheral) within CREs and overall peaks reads. We found that on average, a peak whose summit E-boxes have higher GC content tends to have more total reads (Figure 4A and Supplementary Figure S6). However, the numbers of the GC100% E-boxes within ±200 bp window of the summit seem to relate linearly to the MyoD read counts, independent of the summit E-box type (Figure 4B and Supplementary Figure S6). In other words, in the regions of most concentrated binding, we see no compelling evidence for synergistic binding, which would appear as either a super linear increase of binding with the number of E-boxes or clear evidence for the interaction of the summit E-boxes with the peripheral counterparts.
Figure 4.

The role of the summit E-box in binding affinity. (A) MyoD reads per peak grouped depending on the GC-content of the E-box nearest the peak summit. (B) Mean MyoD reads within peaks as a function of number of E-boxes, separating by the type of the summit E-box. (C and D) Heatmaps showing MyoD reads as a function of numbers of 100% GC and 50% GC E-boxes within 200 bp of the peak summit, and separating by the type of the summit E-box. (E–G) The relationship between tag density (mean sequenced reads) and single summit E-box motif and enrichment of MyoD reads. Peak summit was determined by MACS and summit E-box is the closest motif to the summit.
Joint analysis of the numbers of GC100% and GC50% E-boxes, conditioned on the summit E-box type (Figure 4C and D) shows effects like that seen in Figure 2E, suggesting that MyoD binding is predominantly controlled by numbers of GC100% E-boxes; however, each additional GC50% E-box tends to also increase binding modestly. Nevertheless, because peaks with more GC100% E-boxes tend to have stronger binding, the stronger peaks are more likely to have a GC100% E-box near the summit (Figure 4E–G and Supplementary Figure S6e–g). For example, the peaks with largest MyoD signal are twice as likely to have a GC100% summit E-box compared with peaks with weakest MyoD signal. Thus, a peak characteristic strongly related to overall MyoD binding is that as we look at peaks with increasing MyoD signal, the summit E-box tends to switch from being AT-rich (GC-poor) to being GC-rich (Figure 4 and Supplementary Figure S6).
Variation within MyoD binding motifs regulates nucleosome occupancy and the chromatin state at myogenic CREs
The binding dynamics of transcription factors are affected by competition with nucleosomes (27,28). To analyze the effect of motif-dependent variation in MyoD binding on the state of chromatin at the myogenic CREs, we performed ChIP-seq of histone H3 mono-methyl lysine 4 (H3K4me1), histone H3 lysine 4 tri-methyl (H3K4me3), histone H3 lysine 27 tri-methyl (H3K27me3) and total histone H3 in primary myotubes. The combination of these three regulatory histones demarcates global gene promoters (H3K4me3 and H3K27me3) and distal enhancers (H3K4me1). ChIP-seq analysis of H3K4me1 shows that in myoblasts and myotubes a combined number of 123 477 genomic regions are enriched for this regulatory histone mark (Supplementary Figure S10). While 54 560 peaks are common between myoblasts and myotubes, 48 995 peaks are specific to myotubes. Gene ontology analysis of genes associated with H3K4me1 peaks shows significant enrichment of biological processes associated with muscle differentiation (Supplementary Figure S11). To examine the regulatory effect of motif variation on histone H3 occupancy at the motif locations, we analyzed histone H3 occupancy centered on three classes (GC 0%, GC 50% and GC 100% center dinucleotide) of E-boxes within MyoD-bound CREs. Importantly, we found that GC-rich E-box motifs had the highest depletion of histone H3, while GC-poor motif had the lowest depletion (Figure 5A and Supplementary Figure S9) (for GC 100% E-boxes, pileup height at E-box is significant at P < 10−49 and for GC 50% E-boxes P = 0.0081). Conversely, GC-rich E-boxes were most enriched for the H3K4me1 mark, whereas GC-poor E-boxes demonstrated the least enrichment (Figure 5B and Supplementary Figure S9) (pileup height at ± 200 bp for GC 100% E-boxes versus GC 50% E-boxes are statistically significant at P < 10−36 by Mann–Whitney U-test; GC 100% versus GC 0% P < 10−24; GC 50% versus GC 0% P = 0.0028). Collectively, this analysis suggests that variant sequences within the E-box motifs play an important role in regulation of chromatin at the CREs.
Figure 5.

Dynamic interplay between MyoD and nucleosomes regulates MyoD binding and differential gene expression output. (A) Pileup analysis of the distribution of total Histone H3 (pan histone H3 ChIP-seq reads) centered on motifs with 0, 50 and 100% GC content center dinucleotide. (B) A similar pileup analysis of H3K4me1 reads. (C) Average differential gene expression for MyoD target genes associated with peaks depending on the total MyoD reads in the peaks, grouped by deciles. (D) Similar differential expression, but where peaks are ranked by the E-box motif score, which combines E-box types, numbers and positions into an overall weighted prediction of MyoD affinity.
The combined effects of E-box numbers, sequences and positioning on nearby gene expression. To analyze the effect of variation in E-box sequences within CREs on gene expression, we first performed steady state gene expression analysis using microarrays and RNA-seq on primary muscle cells at the progenitor stage and after 48 h of differentiation (see ‘Materials and Methods’ section). Using peak to gene association by proximity, we associated MyoD peaks to genes whose TSS are within 5 kb, and for which no other peak or gene is closer. Importantly, the MyoD peak read counts are themselves statistically associated to changes in gene expression (Figure 5C; Pearson correlations 0.298 and 0.231 for Affymetrix and RNA-seq estimates of gene expression respectively, with P < 10−30 for both).
Because we previously found a relationship between numbers and variant sequences of E-boxes in MyoD peaks with reads in those peaks, and because the MyoD reads are related to gene expression, we hypothesized that it would be possible to predict gene expression based on the E-box composition under the peaks. Specifically, we used the regression coefficients depicted in Figure 3D to weigh the E-boxes within 1 kb of the summits of MyoD peaks, generating what we term E-box motif scores for each peak, a prediction of the total MyoD binding activity due to the E-box content of the peak. We then associated those with the expression fold changes of nearby genes (TSS within 5 kb). Figure 5D shows a clear, increasing relationship between E-box motif scores of peaks and the log fold change of nearby genes, as assayed either by Affymetrix chip or RNA-seq (one-way ANOVA P-values < 10−6 for each).
Next, we analyzed genome-wide correlation between the E-box motif sequences and the enrichment of MyoD, the enrichment of H3K4me1 on the myogenic CREs, the enrichment of H3K4me3 and H3K27me3 on the associated TSSs and differential gene expression (RNA-seq). Our data visualized in a circos plot (Figure 6) show a positive correlation between the enrichment of MyoD signal with that of H3K4me1 and the GC content of the associated E-boxes for differentially upregulated genes. Importantly, we saw a positive relationship between MyoD and H3K4me1 enrichment with the enrichment of the H3K4me3 on their associated TSS (see ‘Materials and Methods’ section). At last, we found a genome-wide pattern of positive correlation between differential expression of genes upregulated during differentiation with the enrichment of MyoD, H3K4me1 and the high affinity E-box motifs (Figure 6). Analysis of conservation scores between E-boxes within MyoD peaks versus their genomic counterparts shows a high degree of conservation for MyoD-bound E-boxes (Supplementary Figure S13).
Figure 6.

Genome-wide relationship between MyoD binding affinity, nucleosome occupancy and gene expression. Circos plot showing the relationship between sequence variation in MyoD binding motif and the affinity landscape of MyoD binding to DNA and differential expression of target genes. Each track on the concentric circles represents one dataset. The MyoD track (outermost circle) shows ranked peak tag density (PTD) (number of sequenced reads within peak divided by the peak length) for genome-wide MyoD binding sites. The H3K4me1 track shows PTD of histone H3 mono methyl lysine 4 that overlap with MyoD peaks in myotubes. H3K4me3 track shows PTD of histone H3 tri-methyl lysine 4 on the TSS of genes that are associated with MyoD peaks (Supplementary Data). H3K27me3 track shows tag density of histone H3 tri-methyl lysine 27 on the TSS of genes associated with MyoD peaks. H3 track shows PTD of total histone H3 overlapping with MyoD peaks. The three motif tracks show average number of E-box motifs divided into three categories of 0, 50 and 100% GC content of their center dinucleotide. The RNA track (RNA-seq) shows differential expression of MyoD target genes (Supplementary Data). Data is binned into 100 bins and average value within each bin is plotted.
At last, to determine the effect of motif numbers and sequences on gene expression output in vivo we synthesized CREs with various configurations of motif numbers and sequences and performed luciferase assay by co-transfecting mouse MyoD- and E47-expressing plasmids (Figure 7). Notably, the luciferase activity increases as a function of the number E-boxes for both high and low GC content motifs. Consistent with our genome wide analysis, we observed that addition of E-box motifs with GC100% in their variable center dinucleotide contribute more to MyoD activity compared with those with lower GC content. Taken together, these data suggest that variation in E-box numbers and motif sequences and their spatial arrangement within the myogenic CREs regulates nucleosome position, histone marks and nearby gene expression.
Figure 7.

The effect of motif numbers and sequences on gene expression output. Synthetic CREs were generated and sub cloned into pGL4.23 luciferase construct. (A) Schematic showing the numbers and sequences of E-boxes within CREs. DEL represents location of deleted E-box; GC represents CAGCTG; AT represents CAATTG, CA represents CACATG. (B) Relative luciferase activity after normalization to renilla. Dual luciferase assay was performed in Cos7 cells by co transfecting mouse MyoD and E47 expression vectors.
DISCUSSION
The binding of MyoD to CREs encompassing multiple E-box motifs prompted us to investigate the regulatory affects of three cis factors, namely motif sequences, numbers and their spatial orientation within MyoD-bound CREs on the affinity landscape of MyoD and the dynamic range of gene expression output. Functional consequences of transcription factor binding site variation on binding affinity and gene expression have previously been linked to phenotypic variation among individuals (29), demonstrating the important role of transcription factor binding motif sequence in fine-tuning gene expression. In our study, analysis of MyoD binding to myogenic CREs revealed the important contribution of E-box sequence to the affinity landscape of MyoD and differential gene expression output in differentiating myotubes (Figures 2, 3, 5). This observation is consistent with previous in vitro (30,31) and in vivo (6) experiments that demonstrated differential affinity of MyoD to various E-box motifs. We also demonstrate the functional consequences of variation in E-box sequences on nucleosome position (Figure 5A) and on the modification of enhancer chromatin at regions enriched by for the H3K4me1 mark (Figure 5B). Collectively, these analyses revealed that variation in E-box motifs acts as a major determinant of MyoD binding affinity and the status of enhancer chromatin (Figure 5), an effect that is independent of the GC content (Supplementary Figure S4) or the dinucleotide composition of the MyoD peak regions (Supplementary Figure S5).
Furthermore, our analysis of SNP rates within the E-box motifs shows evidence for selection in a position-dependent manner within the motif. For example, the first two (CA) and last two (TG) nucleotides of the E-box receive SNPs at ∼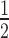 to ¾ the rate for those nucleotides genome wide (Figure 1). Similarly a C in position three or, equivalently, a G in position four receives SNPs at approximately half the genomic rate. Reduction in SNP substitutions (Figure 1) together with a high degree of conservation of E-box motifs within MyoD-bound CREs (Supplementary Figure S13) confirms a functional role for variable sequences within the motifs.
to ¾ the rate for those nucleotides genome wide (Figure 1). Similarly a C in position three or, equivalently, a G in position four receives SNPs at approximately half the genomic rate. Reduction in SNP substitutions (Figure 1) together with a high degree of conservation of E-box motifs within MyoD-bound CREs (Supplementary Figure S13) confirms a functional role for variable sequences within the motifs.
Because MyoD-bound peaks cover an average of 400 bp containing on average four E-boxes per peak (Figure 1 and Supplementary Figure S2), we sought to analyze the possibility of cooperative or synergistic interaction among E-boxes within these CRE domains. Correlation analysis between the number of GC-rich E-boxes in a CRE and the MyoD enrichment showed a nearly linear relationship between the two (Figure 2D and Supplementary Figure S3). We found no evidence for a synergistic or super linear effect, as might be expected if binding of one MyoD molecule to one site increases binding at other sites. However, we did find a positional effect, in which E-boxes closer to the center of a peak contributed more to the overall enrichment of MyoD binding in that region (Figure 3), compared to their peripheral counterparts. The causality can also flow in the opposite direction. In other words, the E-boxes having strongest MyoD binding may determine the peak summit location. Regardless, we observed that peaks with greater overall binding were more likely to have GC rich E-boxes and for the E-box nearest to the peak center to be GC rich (Figure 4). These analyses suggest a model in which motifs with high GC content may tether MyoD to specific locations within the CREs, thereby determine the peak summit location. This mechanism is likely for the majority of MyoD peaks in which the summit motif is GC-rich E-box. However, in a substantial minority of peaks where the summit motif is not GC-rich, tethering of MyoD to a defined region of CREs maybe affected by local structure of enhancer chromatin or other unknown factors.
To study the effect of E-box motifs and their relationship with the epigenetic status of CREs, we examined several related regulatory outcomes. First, we observed a direct correlation between the displacements of histone H3 and sequence variation within MyoD binding motifs on the myogenic CREs. Notably, GC-rich motifs had the highest influence on the displacement of histone H3 on the CREs, which is statistically significant by a simple one-sided sign test (P = 5.9705e-50) or Mann–Whitney U-test (P = 4.4258e-72) (Figure 5A). Similarly, we found strong associations between E-box content of a CRE and the strength of the epigenetic enhancer mark H3K4me1 (Figure 5B). Together, these analyses suggest that numbers and the sequences of E-box motifs within MyoD-bound CREs have a significant functional effect on the enhancer chromatin.
Importantly, we also found that the E-box content of CREs is not limited to predicting local binding and chromatin-related properties. When we associated myogenic CREs to nearby genes, we discovered a quantitative, increasing relationship between MyoD read enrichment on the CRE and expression fold-change between growth and differentiation conditions (differential expression) (Figure 5C). Furthermore, E-box numbers and sequences have quantitative effect on gene expression output in vivo as determined by MyoD/E47 driven luciferase activity (Figure 7). There have been many attempts to explain gene expression as a function of transcription factor binding, including reports of correlations between absolute gene expression and ChIP-seq reads for various factors near the TSS of those genes (32,33). In such studies, one must be mindful of false associations between transcription factors and genes arising because of chromatin accessibility bias in the ChIP-seq signal, which is often large in the vicinity of highly expressed genes (34). Crucially, we have shown that our E-box motif score, which depends purely on the sequence of the CRE, is quantitatively associated with increasing differential gene expression. Given that gene expression is influenced by a plethora of other factors, including the binding of other transcription factors, the epigenetic state of their associated CREs, and post-transcriptional targeting by microRNAs, our identifiable and quantitative association between E-box motifs and nearby differential gene expression is remarkable. Since many transcription factors binding to DNA do not lead to functional consequence on chromatin or on gene expression output (35), it will be interesting to study if CRE sequence-based motif scores can be inferred for other transcription factors.
Collectively, our analysis of MyoD binding suggests that three variables, namely motif sequences, their numbers and their spatial location within the CREs act as major determinants of the myogenic cis-regulatory code. While some transcription factors, such as Pax3/7, Oct4/Pou5f1 and REST/NRSF, bind to relatively more complex DNA motifs, many other transcription factors bind short motifs similar to that of MyOD. Indeed, many transcription factors bind E-boxes specifically. For any such transcription factors, the question arises as to how motif sequence, their number and location within CREs result in binding specificity and affinity. The approach we have described here utilizes MyoD binding to DNA as a specific example, and provides a template for analyzing the binding of other factors to DNA and for understanding how they too balance motif variation, frequency and positioning to achieve regulation.
DATA AVAILABILITY
ChIP-seq data for H3K4me1, H3K4me3, H3K27me3 and Histone H3 in primary myotubes and RNA-seq data for primary myoblasts and myotubes is available through series accession GSE80588. Sample accessions ChIP-seq: GSM2131164 H3K4me1-2DM, GSM2131165 H3K4me3-2DM, GSM2131166 H3K27me3-2DM, GSM2131167 Histone H3 2DM, GSM2131168. Sample accessions RNA-seq: EV-GM, GSM2131169 RNA-seq EV-2DM.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to acknowledge the technical assistance of the Ottawa Bioinformatics Core Facility (University of Ottawa/Ottawa Hospital Research Institute).
Authors’ contribution : Conceived and designed the experiments: V.D.S., T.J.P. and M.A.R. Performed the experiments: V.D.S. and D.N. Analyzed the data: V.D.S., P.R., D.N., G.A.P., C.J.P., H.Y., T.J.P. and M.A.R. Wrote the paper: V.D.S., T.J.P. and M.A.R.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Canadian Institutes for Health Research [MOP-81288 to M.A.R.]; Ontario Ministry of Research and Innovation; National Institutes of Health [R01AR044031]; Canadian Stem Cell Network; Canada Research Chair Program; Canada Research Chair program (to V.D.S.); Natural Sciences and Engineering Research Council of Canada (to VDS); Natural Sciences and Engineering Research Council of Canada (to T.J.P.). Funding for open access charge: Operating grant from the corresponding authors.
Conflict of interest statement. None declared.
REFERENCES
- 1. Davis R.L., Weintraub H., Lassar A.B.. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell. 1987; 51:987–1000. [DOI] [PubMed] [Google Scholar]
- 2. Weintraub H., Genetta T., Kadesch T.. Tissue-specific gene activation by MyoD: determination of specificity by cis-acting repression elements. Genes Dev. 1994; 8:2203–2211. [DOI] [PubMed] [Google Scholar]
- 3. Ma P.C., Rould M.A., Weintraub H., Pabo C.O.. Crystal structure of MyoD bHLH domain-DNA complex: perspectives on DNA recognition and implications for transcriptional activation. Cell. 1994; 77:451–459. [DOI] [PubMed] [Google Scholar]
- 4. Fong A.P., Yao Z., Zhong J.W., Cao Y., Ruzzo W.L., Gentleman R.C., Tapscott S.J.. Genetic and epigenetic determinants of neurogenesis and myogenesis. Dev. Cell. 2012; 22:721–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mousavi K., Zare H., Dell’Orso S., Grontved L., Gutierrez-Cruz G., Derfoul A., Hager G.L., Sartorelli V.. eRNAs promote transcription by establishing chromatin accessibility at defined genomic loci. Mol. Cell. 2013; 51:606–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Soleimani V.D., Yin H., Jahani-Asl A., Ming H., Kockx C.E.M., van IJcken W.F.J., Grosveld F., Rudnicki M.A.. Snail regulates MyoD binding-site occupancy to direct enhancer switching and differentiation-specific transcription in myogenesis. Mol. Cell. 2012; 47:457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cao Y., Yao Z., Sarkar D., Lawrence M., Sanchez G.J., Parker M.H., MacQuarrie K.L., Davison J., Morgan M.T., Ruzzo W.L. et al. Genome-wide MyoD binding in skeletal muscle cells: a potential for broad cellular reprogramming. Dev. Cell. 2010; 18:662–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kophengnavong T., Michnowicz J.E., Blackwell T.K.. Establishment of distinct MyoD, E2A, and twist DNA binding specificities by different basic region-DNA conformations. Mol. Cell. Biol. 2000; 20:261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wilson E.M., Rotwein P.. Control of MyoD function during initiation of muscle differentiation by an autocrine signaling pathway activated by insulin-like growth factor-II. J. Biol. Chem. 2006; 281:29962–29971. [DOI] [PubMed] [Google Scholar]
- 10. Bergstrom D.A., Penn B.H., Strand A., Perry R.L.S., Rudnicki M.A., Tapscott S.J.. Promoter-specific regulation of MyoD binding and signal transduction cooperate to pattern gene expression. Mol. Cell. 2002; 9:587–600. [DOI] [PubMed] [Google Scholar]
- 11. Shen Y., Yue F., McCleary D.F., Ye Z., Edsall L., Kuan S., Wagner U., Dixon J., Lee L., Lobanenkov V.V. et al. A map of the cis-regulatory sequences in the mouse genome. Nature. 2012; 488:116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Andersson R., Gebhard C., Miguel-Escalada I., Hoof I., Bornholdt J., Boyd M., Chen Y., Zhao X., Schmidl C., Suzuki T. et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014; 507:455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Keane T.M., Goodstadt L., Danecek P., White M.A., Wong K., Yalcin B., Heger A., Agam A., Slater G., Goodson M. et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 2011; 477:289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Genomes Project, C. Abecasis G.R., Auton A., Brooks L.D., DePristo M.A., Durbin R.M., Handsaker R.E., Kang H.M., Marth G.T., McVean G.A. et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012; 491:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Soleimani V.D., Yin H., Jahani-Asl A., Ming H., Kockx C.E.M., van IJcken W.F.J., Grosveld F., Rudnicki M.A.. Snail regulates MyoD binding-site occupancy to direct enhancer switching and differentiation-specific transcription in myogenesis. Mol. Cell. 2012; 47:457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mullen A.C., Orlando D.A., Newman J.J., Lovén J., Kumar R.M., Bilodeau S., Reddy J., Guenther M.G., DeKoter R.P., Young R.A.. Master transcription factors determine cell-type-specific responses to TGF-beta signaling. Cell. 2011; 147:565–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Soleimani V.D., Punch V.G., Kawabe Y., Jones A.E., Palidwor G.A., Porter C.J., Cross J.W., Carvajal J.J., Kockx C.E., van IJcken W.F. et al. Transcriptional dominance of pax7 in adult myogenesis is due to high-affinity recognition of homeodomain motifs. Dev. Cell. 2012; 22:1208–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ramachandran P., Palidwor G.A., Porter C.J., Perkins T.J.. MaSC: mappability-sensitive cross-correlation for estimating mean fragment length of single-end short-read sequencing data. Bioinformatics. 2013; 29:444–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fujita P.A., Rhead B., Zweig A.S., Hinrichs A.S., Karolchik D., Cline M.S., Goldman M., Barber G.P., Clawson H., Coelho A. et al. The UCSC Genome Browser database: update 2011. Nucleic Acids Res. 2011; 39:D876–D882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Robinson M.D., McCarthy D.J., Smyth G.K.. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Irizarry R.A., Ooi S.L., Wu Z., Boeke J.D.. Use of mixture models in a microarray-based screening procedure for detecting differentially represented yeast mutants. Stat. Appl. Genet. Mol. Biol. 2003; 2:Article 1. [DOI] [PubMed] [Google Scholar]
- 23. Gentleman R.C., Carey V.J., Bates D.M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J. et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004; 5:R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Won K.J., Ren B., Wang W.. Genome-wide prediction of transcription factor binding sites using an integrated model. Genome Biol. 2010; 11:R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Biddie S.C., John S., Sabo P.J., Thurman R.E., Johnson T.A., Schiltz R.L., Miranda T.B., Sung M.H., Trump S., Lightman S.L. et al. Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol. Cell. 2011; 43:145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rey G., Cesbron F., Rougemont J., Reinke H., Brunner M., Naef F.. Genome-wide and phase-specific DNA-binding rhythms of BMAL1 control circadian output functions in mouse liver. PLoS Biol. 2011; 9:e1000595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li M., Hada A., Sen P., Olufemi L., Hall M.A., Smith B.Y., Forth S., McKnight J.N., Patel A., Bowman G.D. et al. Dynamic regulation of transcription factors by nucleosome remodeling. eLife. 2015; 4:doi:10.7554/eLife.06249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ballare C., Castellano G., Gaveglia L., Althammer S., González-Vallinas J., Eyras E., Le Dily F., Zaurin R., Soronellas D., Vicent G.P. et al. Nucleosome-driven transcription factor binding and gene regulation. Mol. Cell. 2013; 49:67–79. [DOI] [PubMed] [Google Scholar]
- 29. Kasowski M., Grubert F., Heffelfinger C., Hariharan M., Asabere A., Waszak S.M., Habegger L., Rozowsky J., Shi M., Urban A.E. et al. Variation in transcription factor binding among humans. Science. 2010; 328:232–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Blackwell T.K., Weintraub H.. Differences and similarities in DNA-binding preferences of MyoD and E2A protein complexes revealed by binding site selection. Science. 1990; 250:1104–1110. [DOI] [PubMed] [Google Scholar]
- 31. Czernik P.J., Peterson C.A., Hurlburt B.K.. Preferential binding of MyoD-E12 versus myogenin-E12 to the murine sarcoma virus enhancer in vitro. J. Biol. Chem. 1996; 271:9141–9149. [DOI] [PubMed] [Google Scholar]
- 32. Cheng C., Gerstein M.. Modeling the relative relationship of transcription factor binding and histone modifications to gene expression levels in mouse embryonic stem cells. Nucleic Acids Res. 2012; 40:553–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cheng C., Alexander R., Min R., Leng J., Yip K.Y., Rozowsky J., Yan K.-K., Dong X., Djebali S., Ruan Y. et al. Understanding transcriptional regulation by integrative analysis of transcription factor binding data. Genome Res. 2012; 22:1658–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ramachandran P., Palidwor G.A., Perkins T.J.. BIDCHIPS: bias decomposition and removal from ChIP-seq data clarifies true binding signal and its functional correlates. Epigenetics Chromatin. 2015; 8:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cusanovich D.A., Pavlovic B., Pritchard J.K., Gilad Y.. The functional consequences of variation in transcription factor binding. PLoS Genet. 2014; 10:e1004226. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
ChIP-seq data for H3K4me1, H3K4me3, H3K27me3 and Histone H3 in primary myotubes and RNA-seq data for primary myoblasts and myotubes is available through series accession GSE80588. Sample accessions ChIP-seq: GSM2131164 H3K4me1-2DM, GSM2131165 H3K4me3-2DM, GSM2131166 H3K27me3-2DM, GSM2131167 Histone H3 2DM, GSM2131168. Sample accessions RNA-seq: EV-GM, GSM2131169 RNA-seq EV-2DM.