

Abstract
Living organisms regularly need to cope with fluctuating environments during their life cycle, including changes in temperature, pH, the accumulation of reactive oxygen species, and more. These fluctuations can lead to a widespread protein unfolding, aggregation, and cell death. Therefore, cells have evolved a dynamic and stress-specific network of molecular chaperones, which maintain a "healthy" proteome during stress conditions. ATP-independent chaperones constitute one major class of molecular chaperones, which serve as first-line defense molecules, protecting against protein aggregation in a stress-dependent manner. One feature these chaperones have in common is their ability to utilize structural plasticity for their stress-specific activation, recognition, and release of the misfolded client.
In this paper, we focus on the functional and structural analysis of one such intrinsically disordered chaperone, the bacterial redox-regulated Hsp33, which protects proteins against aggregation during oxidative stress. Here, we present a toolbox of diverse techniques for studying redox-regulated chaperone activity, as well as for mapping conformational changes of the chaperone, underlying its activity. Specifically, we describe a workflow which includes the preparation of fully reduced and fully oxidized proteins, followed by an analysis of the chaperone anti-aggregation activity in vitro using light-scattering, focusing on the degree of the anti-aggregation activity and its kinetics. To overcome frequent outliers accumulated during aggregation assays, we describe the usage of Kfits, a novel graphical tool which allows easy processing of kinetic measurements. This tool can be easily applied to other types of kinetic measurements for removing outliers and fitting kinetic parameters. To correlate the function with the protein structure, we describe the setup and workflow of a structural mass spectrometry technique, hydrogen-deuterium exchange mass spectrometry, that allows the mapping of conformational changes on the chaperone and substrate during different stages of Hsp33 activity. The same methodology can be applied to other protein-protein and protein-ligand interactions.
Keywords: Biochemistry, Issue 136, Chaperones, redox-regulated chaperone, Hsp33, ATP-independent chaperone, protein aggregation, hydrogen-deuterium exchange, HDX-MS, protein aggregation kinetics and analysis, protein oxidation and reduction.
Introduction
Cells frequently encounter an accumulation of reactive oxygen species (ROS) produced as byproducts of respiration1,2, protein and lipid oxidation3,4, and additional processes5,6,7. Despite ROS' beneficial role in diverse biological processes such as cellular signaling8,9 and immune response10, an imbalance between ROS production and its detoxification might occur, leading to oxidative stress7. The biological targets of ROS are proteins, lipids, and nucleic acids, the oxidation of which affect their structure and function. Therefore, the accumulation of cellular oxidants is strongly linked to a diverse range of pathologies including cancer9,11, inflammation12,13, and aging14,15, and have been found to be involved in the onset and progression of neurodegenerative disorders such as Alzheimer's, Parkinson's, and ALS disease16,17,18.
Both newly synthesized and mature proteins are highly sensitive to oxidation due to the potentially harmful modifications of their side chains, which shape protein structure and function19,20. Therefore, oxidative stress usually leads to a widespread protein inactivation, misfolding and aggregation, eventually leading to cell death. One of the elegant cellular strategies to cope with the potential damage of protein oxidation is to utilize redox-dependent chaperones, which inhibit the widespread protein aggregation, instead of forming stable complexes with misfolded client proteins21,22,23. These first-line defense chaperones are rapidly activated by a site-specific oxidation (usually on cysteine residues) that converts them into potent anti-aggregation molecules24. Since oxidative stress results in the inhibition of respiration and in decreases in the cellular ATP levels25, canonical ATP-dependent chaperones are less effective during oxidative stress conditions25,26,27. Therefore, redox-activated ATP-independent chaperones play a vital role in maintaining protein homeostasis upon the accumulation of oxidants in bacteria and eukaryotes (e.g., Hsp3328 and RidA29 in bacteria, Get330 in yeast, peroxiredoxins31 in eukaryotes). The activity of these chaperones strongly depends on reversible structural conformational changes induced by a site-specific oxidation that uncovers hydrophobic regions involved in the recognition of misfolded client proteins.
Research of the anti-aggregation mechanism and the principles governing the recognition of the client proteins by chaperones is not easy due to the dynamic and heterogenic nature of chaperone-substrate interactions32,33,34,35,36,37. However, stress-regulated chaperones have an opportunity to advance our understanding of the anti-aggregation function due to their ability to: 1) obtain two different forms of the chaperone, active (e.g., oxidized) and inactive (e.g., reduced), with the introduction or removal of a stress condition easily switching between them (e.g., oxidant and reducing agent), 2) have a broad range of substrates, 3) form highly stable complexes with the client proteins that may be evaluated by different structural methodologies, and 4) focus solely on the substrate recognition and release, mediated by redox-dependent conformational changes, as the majority of these chaperones lacks the folding capability.
Here, we analyze the bacterial redox-regulated chaperone Hsp33's anti-aggregation activity, a vital component of the bacterial defense system against oxidation-induced protein aggregation28. When reduced, Hsp33 is a tightly folded zinc-binding protein with no chaperone activity; however, when exposed to oxidative stress, Hsp33 undergoes extensive conformational changes which expose its substrate binding regions38,39. Upon oxidation, the zinc ion that is strongly bound to four highly conserved cysteine residues of the C-terminal domain is released40. This results in the formation of two disulfide bonds, an unfolding of the C-terminal domain, and a destabilization of the adjacent linker region41. The C-terminal and linker regions are highly flexible and are defined as intrinsically or partially disordered. Upon return to non-stress conditions, the cysteines become reduced and the chaperone returns to its native folded state with no anti-aggregation activity. The refolding of the chaperone leads to a further unfolding and destabilization of the bound client protein, which triggers its transfer to the canonical chaperone system, DnaK/J, for refolding38. Analysis of Hsp33's interaction sites suggests that Hsp33 uses both its charged disordered regions as well as the hydrophobic regions on the linker and N-terminal domain to capture misfolded client proteins and prevent their aggregation38,42. In the folded state, these regions are hidden by the folded linker and C-terminal domain. Interestingly, the linker region serves as a gatekeeper of Hsp33's folded and inactive state, "sensing" the folding status of its adjacent C-terminal domain34. Once destabilized by mutagenesis (either by a point mutation or a full sequence perturbation), Hsp33 is converted to a constitutively active chaperone regardless the redox state of its redox-sensitive cysteines43.
The protocols presented here allow monitoring of Hsp33's redox-dependent chaperone activity, as well as mapping conformational changes upon the activation and binding of client proteins. This methodology can be adapted to research other chaperone-client recognition models as well as non-chaperone protein-protein interactions. Moreover, we present protocols for the preparation of fully reduced and oxidized chaperones that can be used in studies of other redox-switch proteins, to reveal potential roles of protein oxidation on the protein activity.
Specifically, we describe a procedure to monitor chaperone activity in vitro and define its substrate specificity under different types of protein aggregation (chemically or thermally induced) using light scattering (LS) measured by a fluorospectrometer44. During aggregation, light scattering at 360 nm increases rapidly due to the increasing turbidity. Thus, aggregation can be monitored in a time-dependent manner at this wavelength. LS is a fast and sensitive method for testing protein aggregation and thus the anti-aggregation activity of a protein of interest using nanomolar concentrations, enabling the characterization of protein aggregation-related kinetic parameters under different conditions. Moreover, the LS protocol described here does not require expensive instrumentation, and can be easily established in any laboratory.
Nevertheless, it is quite challenging to obtain "clean" kinetic curves and to derive a protein's kinetic parameters from such light scattering experiments, due to noise and the large number of outliers generated by air bubbles and large aggregates. To overcome this obstacle, we present a novel graphical tool, Kfits45, used for reducing noise levels in different kinetic measurements, specifically fitted for protein aggregation kinetic data. This software provides preliminary kinetic parameters for an early assessment of the results and allows the user to "clean" large quantities of data quickly without affecting its kinetic properties. Kfits is implemented in Python and available in open source at 45.
One of the challenging questions in the field relates to mapping interaction sites between chaperones and their client proteins and understanding how chaperones recognize a wide range of misfolded substrates. This question is further complicated when studying highly dynamic protein complexes which involve intrinsically disordered chaperones and aggregation-prone substrates. Fortunately, structural mass spectrometry has dramatically advanced over the last decade and has successfully provided helpful approaches and tools to analyze the structural plasticity and map residues involved in protein recognition46,47,48,49. Here, we present one such technique-hydrogen-deuterium exchange mass spectrometry (HDX-MS)-which allows the mapping of residue-level changes in a structural conformation upon protein modification or protein/Ligand binding35,50,51,52,53,54,55. HDX-MS uses the continuous exchange of backbone hydrogens by deuterium, the rate of which is affected by the chemical environment, accessibility, and covalent and non-covalent bonds56. HDX-MS tracks these exchange processes using a deuterated solvent, commonly heavy water (D2O), and allows measurement based on the change in molecular weight following the hydrogen to deuterium exchange. Slower rates of hydrogen-deuterium exchange can result from hydrogens participating in hydrogen bonds or, simply, from steric hindrance, which indicates local changes in structure57. Changes upon a ligand binding or post-translational modifications can also lead to differences in the hydrogen environment, with a binding resulting in differences in the hydrogen-deuterium exchange (HDX) rates46,53.
We applied this technology to 1) map Hsp33 regions which rapidly unfold upon oxidation, leading to the activation of Hsp33, and 2) define the potential binding interface of Hsp33 with its full-length misfolded substrate, citrate synthase (CS)38.
The methods described in this manuscript can be applied to study redox-dependent functions of proteins in vitro, defining anti-aggregation activity and the role of structural changes (if any) in protein function. These methodologies can be easily adapted to diverse biological systems and applied in the laboratory.
Protocol
1. Preparation of Fully Reduced and Fully Oxidized Proteins
- Preparation of a fully reduced protein Note: Here, we describe the reduction of a zinc-containing protein, and use a ZnCl2 solution to restore the Zn-incorporated, reduced protein state. The ZnCl2 solution can be replaced or discarded. Note that the time and temperature of the reduction process depends on the protein stability and function, and is thus specific per protein.
- Thaw the protein sample on ice and spin it down to remove aggregates. Incubate the sample for at least 1.5 h at 37 °C with 5 mM DTT and 20 µM ZnCl2 (up to 70% of protein concentration). NOTE: The temperature at this step is protein-dependent and should be adjusted to the protein stability.
- Remove DTT using desalting columns. NOTE: There are different desalting columns available. The protocol below describes the procedure using specific desalting columns (see Table of Materials). Before using other desalting columns, we recommend examining their efficiency of DTT removal and protein recovery, since some desalting columns may partially absorb the protein of interest.
- Equilibrate the column with a potassium phosphate (KPi) buffer (40 mM, pH 7.5) by filling the column completely with the buffer and letting the buffer drip out. Repeat this process 2x.
- Remove the white disk filter in the column by gently pushing it down with tweezers and removing it; it is easiest to remove while the column is filled with the KPi buffer. Refill the column with KPi buffer and centrifuge it at 1,000 x g for 3 min.
- Transfer the column into a clean tube, add the protein sample slowly to the middle of the column and centrifuge it at 1,000 x g for 2 min. The DTT-free protein is now in the flow-through.
- Check its concentrations (e.g., use a UV/Vis spectrometer) and measure its absorbance at 280 nm. Calculate the protein concentration using Beer's law.
- Distribute half of the protein samples into aliquots. Incubate the aliquots in anaerobic conditions (e.g., using an anaerobic chamber) for 20 min for a complete removal of oxygen. Seal the tubes with plastic film and store the samples at -20 °C or -80 °C, depending on the protein. NOTE: Instead of using the anaerobic chamber, the tubes can also be flashed with argon gas to remove oxygen.
- Preparation of a fully oxidized protein NOTE: It is recommended to prepare oxidized protein samples from fully reduced proteins (described previously). This will reduce the heterogeneity in oxidation states of cysteines. It is possible to use different oxidizing reagents; here, we are focusing on hydrogen peroxide (H2O2). CAUTION: Avoid over-oxidation as it can lead to undesirable intramolecular disulfide bonds and irreversible oxidation of different amino acids, including cysteine, methionine, tyrosine, and others.
- To the remaining protein sample, add 5 mM H2O2 (freshly diluted) and incubate it for 3 h at 40 °C while shaking. NOTE: The temperature at this step is protein-dependent and should be adjusted to the protein stability.
- Equilibrate the column with KPi buffer (40 mM, pH 7.5) by filling the column completely with the buffer and letting the buffer drip out. Repeat this process 2x.
- Remove the white disk filter in the column by gently pushing it down with tweezers and removing it; it is easiest to remove while the column is filled with the KPi buffer.
- Refill the column with KPi buffer and centrifuge it at 1,000 x g for 3 min.
- Transfer the column into a clean tube, add the oxidized protein sample slowly to the middle of the column and centrifuge it at 1,000 x g for 2 min; the oxidized protein is now in the flow-through.
- Check the protein concentrations as in step 1.2.5, divide the oxidized proteins into aliquots, and store them at -20 °C or -80 °C, protein-dependent.
2. Light Scattering Aggregation Assay
Note: All concentrations in this assay are chaperone- and substrate-specific, and should be calibrated. All buffers should be 0.22 µm-filtered, as it is extremely important that the buffers are free of any particles or air bubbles and the cuvettes are clean and dust-free. It is very important to use a stirrer placed in the quartz cuvette. Check different stirrer sizes and shapes in order to ensure an efficient mixing of the entire solution without producing undesirable air bubbles. Moreover, there are different flouorospectrometers available in laboratories and facilities. Here, a specific fluorospectrometer (see Table of Materials) was used. Different instruments have a diverse sensitivity, measurement speed, and sampler parameters. Therefore, the exact measurement parameters (e.g., emission and excitation bandwidth, sensitivity, and others) should be optimized using a known aggregation-prone protein and its corresponding conditions. Using citrate synthase (CS) and/or luciferase as initial substrates in nanomolar concentrations is recommended.
- Chemical aggregation assay
- Prepare the denatured substrate by incubating 12 µM CS overnight in 40 mM HEPES (pH 7.5) and 4.5 M GdnCl. In order to preserve the pH, dissolve the GdnCl in the 40 mM HEPES (pH 7.5).
- Open the fluorospectrometer software and go to Time course measurement. Set the parameters to: Temperature: 25 °C; λem: 360; Em bandwidth: 5 nm; λex: 360; Ex bandwidth: 2.5 nm; and Data interval: 0.5 s.
- Prepare the sample by adding 1,600 µL of 40 mM HEPES to a quartz cuvette. Insert the cuvette into the sample holder and let the sample reach the desired temperature.
- Set the stirring to 600 rpm and begin the measurement until a baseline is established. Keep the stirring on for the entire measurement.
- To measure the CS aggregation in the absence of a chaperone, at 120 s into the measurement, add (gently, but quickly) 10 µL of denatured CS (final concentration of 75 nM). Continue the measurement for 1,200 s.
- To measure the CS aggregation in the presence of Hsp33, at 60 s into the measurement, add Hsp33 (final concentration of 300 nM). After an additional 60 s, add 10 µL of denatured CS (final concentration of 75 nM). Continue the measurement for 1,200 s. NOTE: When adding the substrate or chaperone, in order to avoid insertion of bubbles, use a 10-µL pipette.
- Thermal aggregation assay NOTE: The temperature for the thermal aggregation assay depends on the protein stability and should be adjusted to each protein independently.
- Open the fluorospectrometer software and go to Time course measurement. Set the parameters as follows: Temperature: 43 °C; λem: 360; Emission bandwidth: 5 nm; λex: 360; Excitation bandwidth: 2.5 nm; and Data interval: 0.5 s.
- Prepare the sample by adding 1,600 µL of pre-warmed 40 mM HEPES to a quartz cuvette. Insert the cuvette into the sample holder and let the sample reach 43 °C.
- Set the stirring to 600 rpm and begin the measurement until a baseline is established. Keep the stirring on for the entire measurement.
- To measure the CS aggregation in the absence of a chaperone, at 120 s into the measurement, gently add CS (final concentration of 125 nM) and continue measuring for 1,200 s.
- To measure the CS aggregation in the presence of Hsp33, at 60 s into the measurement, add Hsp33 (final concentration of 600 nM). After an additional 60 s, add CS (final concentration of 125 nM). Continue the measurement for 1,200 s. NOTE: When adding the substrate or chaperone, avoid the insertion of bubbles.
- Data analysis and noise removal by Kfits Note: Kfits is available at use of Kfits has previously been described in Rimon et al.45
- Upload the data file. NOTE: The input is the raw results from light scattering measurements in a textual comma-separated or tab-delimited format.
- To remove any noise, choose analysis parameters. Use Automatic best model (recommended), mark the Noise is always above signal flag.
- Manually remove obvious outliers using the green and red adjustable lines; this step will filter the noise above the green line and below the red line.
- Set the baseline and the fit curve. After applying the noise threshold, download the processed data.
3. Hydrogen-deuterium Exchange Mass Spectrometry
- Preparation of buffers and protein samples (Hsp33 and Hsp33-CS complex)
- Dilute the protein samples to a final concentration of 1 mg/mL in 25 mM Tris-HCl buffer at pH 7.5 and transfer them to 1.5-mL vials.
- Prepare protein-substrate complex samples by incubating Hsp33 with CS at a ratio of 1:1.5 at 43 °C.
- Add CS in a step-wise manner to avoid any rapid aggregation and ensure a fruitful binding between the Hsp33 and the thermally unfolded CS.
- Use at least four steps (i.e., each time add a quarter of the final volume) and incubate the samples for 15 min after every addition to allow CS protection by Hsp33.
- Remove any aggregates using a 30-min centrifugation at 16,000 x g at 4 °C. NOTE: New protein-substrate complexes must be prepared fresh. Substrate addition should be made gradually; otherwise, aggregation will occur. Temperature, substrate, and substrate concentrations are protein-specific.
- Prepare a buffer H (25 mM Tris-HCl, pH 7.5), which serves as deuteration control, and a buffer D (25 mM Tris-DCl, pH 7.09), which is the deuteration buffer. Also prepare a fresh quenching buffer (150 mM TCEP, 3 M GdnCl, 0.1% formic acid). NOTE: The quenching buffer should be optimized for the protein of interest in order to obtain its maximal sequence coverage after the pepsin digestion.
- Transfer all buffers and samples into vials, and place them in proper trays. Hold buffers H and D at 25 °C (tray 25 °C); on the other hand, hold the samples and quenching buffer at 0 - 2 °C (tray 0 °C). Place the samples in 150-µL glass inserts (see the Table of Materials) first, and then transfer them into vials.
- Preparation of the instrument NOTE: The mass spectrometer comes with two accompanying software programs: one controls the pumps, and the other controls the mass spectrometer (refer to the Table of Materials). The next steps will be described using these two software programs.
- Manually turn on all cooling units. Once all cooling systems reach their target temperatures, manually switch on both the high-performance liquid chromatography (HPLC) and loading pumps. NOTE: In our case, these consist of two cooling baths and a cooling box (which contains the trap and analytical columns). One cooling unit cools the tray at 0 °C, while the other keeps the tray at 25 °C; the temperature of the cooling box is 1 - 2 °C.
- Open the software which controls both pumps, as well as the software which controls the mass spectrometer; make sure that MS is on Standby.
- Disconnect the HPLC outlet valve from the MS source and wash the system first with a "triple cleaner" solution (1% formic acid, 33% acetonitrile, 33% isopropanol, 33% methanol), then with buffer B (80% acetonitrile, 0.1% formic acid), and finally with buffer A (0.1% formic acid, pH 2.25), and then insert the pepsin column into the system. If changing buffers, make sure to purge both pumps before proceeding.
- Ensure that the flow-rate for both pumps is 0.1 mL/min and the pressure is stable.
- After washing the system with a steady flow and steady pressure, insert the HPLC outlet into the MS source and turn the MS on.
- Mass spectrometer parameters
- Set the peptide ionization to electrospray ionization (ESI) at 175 °C, the sheath gas flow at 17, the aux gas glow at 2, and the spray voltage at 4.5 kV (Supplementary Figure 1).
- Set up the parameters as follows for the non-deuterated samples.
- Set the parameters: Scan Range m/z to 300 - 1500; Resolution to 70,000; Automatic gain control target (AGC) to 106; and the Maximum injection time (IT) at 100 ms. NOTE: The 5 most intense ions with charge states between +2 and +6 (their intensity higher than 1.3 x 104) and a dynamic window of 30 in will be isolated.
- Perform the ion fragmentation by higher-energy collisional dissociation (HCD) in the multiple-collision cell with normalized collision energy (NCE) equal to 28. Detect fragment ions by tandem MS at a resolution of 35,000, AGC target of 105, maximum IT at 60 ms, and an isolation window of 2.0 m/z.
- For the deuterated samples, employ MS1 only with a higher resolution of 140,000 and with otherwise similar parameters.
- Running the experiment
- Set the trays in the following manner.
- Place buffer H and D in Tray 25 °C, then place the samples and quenching buffer in the 0 °C tray.
- Place X empty vials, aligned in both trays, where X = the number of samples x the number of time points. NOTE: In the 25 °C tray, the empty vials serve as reaction vials, and in the tray 0 °C, they serve as quenching vials.
- Each of the empty vials contains an empty glass insert; discard and replace said insert between experiments. NOTE: In order to run the HDX experiment in the most efficient and least time-consuming way, a software which allows samples to be run simultaneously (Supplementary Figure 2) was used. The next steps will be described using this software.
- Open the software. Set up the program parameters to perform the following.
- Incubate each sample (5 µL) with 45 µL of buffer D for several minutes, depending on the time points selected (e.g., 1, 3, 18, 40, and 100 min) at 25 °C. Incubate the non-deuterated sample with 45 µL of buffer H instead.
- Immediately after the incubation, mix 50 µL of deuterated protein into 50 µL of ice-cold quenching buffer and inject them into an immobilized pepsin column (20 mm in length, 2.0 mm in diameter).
- Elute any peptides from the pepsin column into the pre-column by washing it with buffer A for 6 min at a rate of 50 µL/min. Keep buffer A in the cooling box at 0 °C.
- Elute the peptides out of the pre-column into the C18 analytical column (C18 Column, 130 Å, 1.7 µm, 2.1 mm x 50 mm, kept at 0 °C) and separate them by applying a linear gradient of acetonitrile at 100 µL/min. Run the acetonitrile gradient using acetonitrile 100% as buffer B: 7 min at 2%, 7 min at 10 - 30%, 2.5 min at 30 - 90%, 1.5 min at 90%, rapid gradient for 1 min at 90 - 8%, and finally equilibrate the column for 4 min at 2%.
- Build a running sequence (see Supplementary Figure 2): enter all time points, sample names, and locations in the trays, as well as the buffer names and locations. NOTE: The software allows the alignment of all the different running times into a program that runs several samples at once, efficiently decreasing running times.
- Data analysis NOTE: We work with several software programs in the process of data analysis, including Proteome Discoverer and MaxQuant (free software) for the peptide identification and analysis of sequence coverage, as well as HDX workbench, a free software that allows the analysis of the deuterium incorporation in proteins, and a comparison of the HDX rates between different sets of experiments. The next steps will be described using these software programs.
- Analyze the peptide coverage of the sample by running the non-deuterated control samples through the software (Supplementary Figure 3). Set up the software using the following parameters.
- Use the Sequest HT method with a No-enzyme (unspecific) cleave. Search for peptides of 4 - 144 amino acids with a mass tolerance of 7 ppm and 0.5 Da for the precursor's ion and fragment. Allow for a dynamic methionine oxidation.
- Create a database for the protein, through which the software can determine the peptide coverage. Export the identified peptides in textual format.
- Analyze the deuterated results using the HDX workbench free software58.
- Open the protein editor and define the protein by inserting the protein sequence and peptide coverage file.
- Next, open the Setup experiment wizard editor and enter all the inputs requested. NOTE: The program requires the number of samples, the number of time points, the number of replicates, the pH value of the buffers, and the temperature.
- Finally, input the parameters regarding the mass spectrometer used, after which the program generates a list of detected peptides. NOTE: The software detects the "HDX" peptides, inspects isotope clusters, and demonstrates a clear visualization of deuteration in the samples.
- Data presentation
- Label the regions with a deferential deuteration uptake on the structure using the PyMol software or any other visualization software.
- Introduce the deuterium uptake levels instead of the B-factor values. Note: A basic tutorial can be found at https://pymolwiki.org/index.php/Practical_Pymol_for_Beginners.

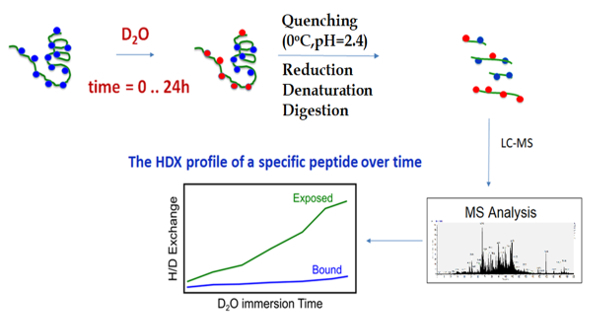
Representative Results
The two methods presented make it possible to follow the kinetic activity and dynamics of protein interactions between a chaperone and its substrate. Moreover, the reduction-oxidation protocol allows the preparation of a fully reduced and fully oxidized chaperone, giving a more in-depth understanding of the activation mechanism of redox-dependent disordered chaperones.
First, we used light scattering in order to examine the redox-dependent activity of the chaperone. Light scattering was performed on a chemically and thermally denatured CS protein in the presence of an oxidized (active) or reduced (inactive) chaperone (Figure 1). Chemical denaturation leads to a rapid CS aggregation induced by the refolding of a fully denaturized protein in unfavorable buffer conditions. On the other hand, the thermal-induced aggregation results from a relatively slow unfolding of the natively folded substrate. Therefore, these different types of aggregation processes vary in their kinetic parameters. Moreover, different chaperones can vary in their ability to prevent the aggregation of the same substrate in different aggregation modes.
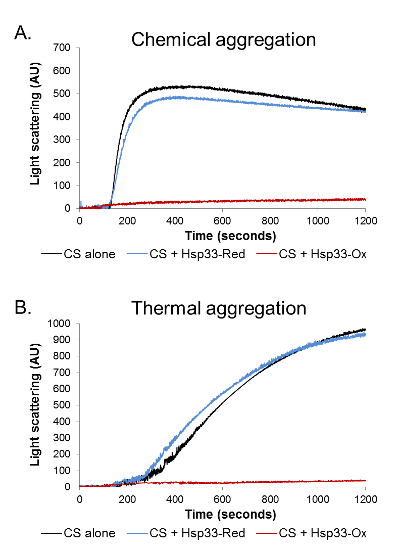
In both forms of denaturation, the addition of oxidized Hsp33 in a molar ratio of 1:4 (CS: Hsp33) completely abolished aggregation, lowering the 360 nm readings to negligible values. The presence of a reduced Hsp33, on the other hand, had no effect on the CS stability and gave an aggregation curve similar to the one detected in the absence of a chaperone (Figure 1).The results were analyzed and cleaned from background noise using Kfits, as schematically described in Figure 2.
The outline of the HDX-MS technique begins with the incubation of the chaperone with deuterium oxide, followed by quenching and digestion, and finally MS analysis and hydrogen-deuterium exchange profiling (Figure 3). During HDX-MS, the chaperone was incubated on the tray at 25 °C in a D2O-containing buffer. After a specific incubation time controlled by the sample handling robotic system, the protein solution was transferred to the quenching solution, acidic and denaturing, on the tray at 0 °C. The low pH and low temperature slows the hydrogen exchange and preserves the deuteration levels of all amide hydrogens, while the denaturing chemical unfolds the protein, enabling it to be properly digested. The robotic system transfers the sample from the tray at 0 °C to the cooling box where it flows into the online pepsin digestion column. Immediately after the protein digestion, the peptides proceed to the C18-trap column for the desalting and removal of fast exchangeable D2O (mainly located on the side chain atoms) and are then separated using the C18 column and analyzed using mass spectrometry. The sample flow is controlled by a two-valve system, which 1) ensures the separation of the pepsin and C18 column buffers in order to prevent the inactivation of the pepsin by organic solvents, and 2) enables the short desalting of the peptides to remove fast exchangeable D2O molecules and salts (Figure 4).

After the computational analysis, we receive a deuteration profile for each peptide, showing a change in mass as a function of the incubation time in D2O. The HDX Workbench software allows the comparison of experimental replicates in order to create statistical analyses of each condition (e.g., an active chaperone in an unbound form). The next step is a comparison of the deuteration curves between different samples; for example, a bound chaperone and an unbound chaperone. These comparisons of different chaperone states can reveal the structural perturbations that accompany substrate binding. Residues that exhibit a slower exchange in the bound state are likely interacting with a binding partner. It is important to note that decreased hydrogen-deuterium exchange rates can also indicate a refolding of a specific region upon binding without the actual direct interaction with the substrate (Figure 5A). The HDX Workbench software allows the examination of the results in the "perturbation" view by showing the deuteration with a standard error over the coverage of the whole protein.
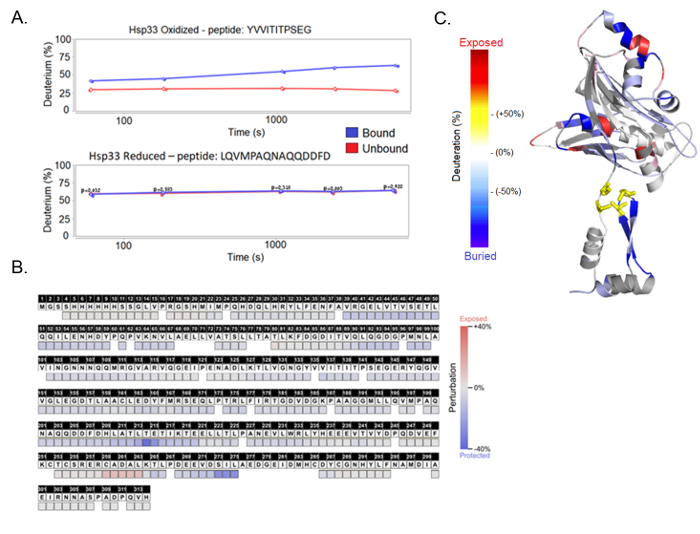
In this case, HDX-MS was used to compare the deuteration pattern between the bound and unbound chaperone Hsp33 to its substrate CS. The results reveal a client-interaction site within the C-terminal redox switch domain of the chaperone (Figure 5B). While reduced, Hsp33 has a completely ordered structure, where the interaction sites are buried. Hsp33 loses its structure and becomes functional when it senses oxidative stress; hence, we compared the reduced and oxidized Hsp33 under both bound and unbound conditions (Figure 5C). The results show that while the reduced Hsp33, whether bound or unbound, produces similar peptide curves, the oxidized Hsp33 displays a significant difference. The oxidized Hsp33 samples show a 30% deuteration difference between the bound and unbound chaperone, with specific areas of the bound protein indicating an allosteric hindrance. The analysis of additional peptides from the N-terminal and the thermobile linker regions are shown in Supplementary Figure 3.
Figure 1. Aggregation analysis by light scattering. The aggregation of chemically and thermally denatured CS was monitored in the presence of oxidized Hsp33 (red), reduced Hsp33 (blue), or in the absence of chaperone (black). A light scattering at 360 nm was monitored for 1,200 s. (A) During the chemical aggregation, in the absence of a chaperone or in the presence of a reduced Hsp33, the CS aggregated rapidly, reaching a plateau approximately 300 s into the measurement. (B) During the thermal aggregation, the CS aggregated gradually in the absence of a chaperone or in the presence of a reduced Hsp33. In both assays, the presence of oxidized Hsp33 completely abolished aggregation, as was demonstrated by negligible 360-nm readings. Please click here to view a larger version of this figure.
Figure 2. Removal of noise and data analysis using Kfits. This panel shows a schema summarizing the data analysis and noise removal by the Kfits tool, as described in the text and in Rimon et al.45. Please click here to view a larger version of this figure.
Figure 3. Workflow of the HDX-MS methodology. HDX-MS employs deuterated solvents to determine the chemical environment of amide hydrogens, uniformly distributed along the backbone of the protein. The protein of interest is incubated in a deuterating buffer in its native form for specific time periods, then the hydrogen-deuterium exchange is quenched by acidic conditions at a low temperature. The protein is digested by an enzyme stable under acidic conditions, such as pepsin. Afterward, the peptides are desalted and separated in a short time to avoid a back exchange. Finally, the peptides are analyzed by a mass spectrometer and the deuterium uptake is evaluated by an analysis software, for example by the HDX Workbench free software58. Please click here to view a larger version of this figure.
Figure 4. Components of the fully automated HDX-MS system. (A) This schematic diagram of the HDX-MS represents all the parts of the system: the loading pump and the HPLC pump control buffer flow and the sample transfer within the system. The robotic system contains two syringes and trays which are used to prepare the deuterated sample and transfer the proteins to the on-line digestion column via the input valve of the cooling box (LC compartment). Then, the digested peptides are directed to the C18 trap and separation columns, subsequently separated, and directed to the mass spectrometer. (B) This panel shows the actual HDX system setup as it was created in the lab. (C) This panel shows a schematic presentation of the online-digestion and the peptide separation fluidic configuration. The digestion and separation systems have one valve each that can be switched between load and inject modes. The first valve (on the left) is connected to the injection port and is controlled by the loading pump, directing samples into the pepsin column using the acidic buffer without any organic solvent that can damage the pepsin resin. The sample is transferred to the right valve into the trap column to remove residues of deuterium as well as fast exchanged deuterated atoms (mainly of the side chains). After a brief wash of 1 - 2 min in the trap column, the valve changes its mode to load and the sample is washed into the C18 separation column using the HPLC pump with increasing concentrations of acetonitrile. The sample is transferred to the mass spectrometer for a mass analysis. The length of the tubes is marked. Please click here to view a larger version of this figure.
Figure 5. HDX-MS results. (A) The results are presented as a deuteration percentage over time per protein fragment, generated by the HDX Workbench software. Each graph shows a protein sample, either oxidized or reduced, and compares between bound and unbound samples. The peptide fragments shown here are the peptide YVVITITPSEG found in the linker region and the peptide LQVMPAQNAQQDDFD found in the C-terminus. (B) The final results, presented as the deuteration difference between the bound and unbound complexes, show the perturbation per amino acid. (C) A structural model of the Hsp33 chaperone shows the deuteration uptake differences throughout the linker and C-terminal unstable regions. This panel is created using the PyMol software. Please click here to view a larger version of this figure.
 Supplementary Figure 1. Interface of the program operating mass spectrometer. The program is used to optimize and define mass spectrometer method parameters used for the HDX experiment. The method parameters (in the left panel) can be saved in the method, such that is recognized by other programs. Please click here to view a larger version of this figure.
Supplementary Figure 1. Interface of the program operating mass spectrometer. The program is used to optimize and define mass spectrometer method parameters used for the HDX experiment. The method parameters (in the left panel) can be saved in the method, such that is recognized by other programs. Please click here to view a larger version of this figure.
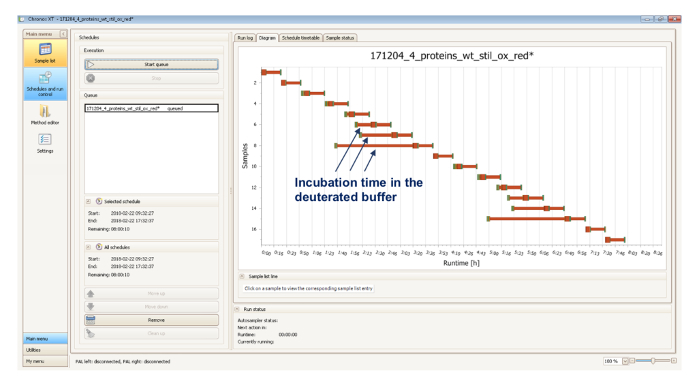 Supplementary Figure 2. This figure shows a screenshot of the software which serves as an integration platform of the robotic sampling handing system and the MS-LC system. The software optimizes the running time by the scheduling of the HDX experiment, following the defined incubation time in the deuterated buffer (blue arrows). For example, above is a diagram of 17 runs of two replicative analyses of the Hsp33 variant STIL, incubated in a deuterated buffer for different times, ranging from 0 - 100 min. Please click here to view a larger version of this figure.
Supplementary Figure 2. This figure shows a screenshot of the software which serves as an integration platform of the robotic sampling handing system and the MS-LC system. The software optimizes the running time by the scheduling of the HDX experiment, following the defined incubation time in the deuterated buffer (blue arrows). For example, above is a diagram of 17 runs of two replicative analyses of the Hsp33 variant STIL, incubated in a deuterated buffer for different times, ranging from 0 - 100 min. Please click here to view a larger version of this figure.
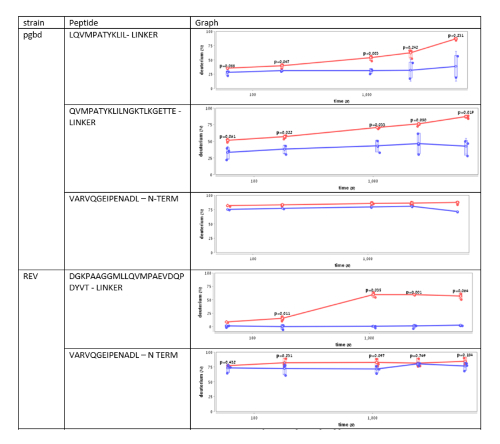 Supplementary Figure 3. This figure shows an analysis of the deuterium uptake of several Hsp33 peptides from different Hsp33 variants, named PGBD and REV34. The plots demonstrate changes in the deuterium uptake of the same region in Hsp33 in unbound (red) and bound (blue) states. Peptides from the N-terminal region show no significant difference in the HDX rates, while fragments from the linker region show a significant decrease in the HDX rates upon the interaction with the unfolded substrate, citrate synthase. Please click here to view a larger version of this figure.
Supplementary Figure 3. This figure shows an analysis of the deuterium uptake of several Hsp33 peptides from different Hsp33 variants, named PGBD and REV34. The plots demonstrate changes in the deuterium uptake of the same region in Hsp33 in unbound (red) and bound (blue) states. Peptides from the N-terminal region show no significant difference in the HDX rates, while fragments from the linker region show a significant decrease in the HDX rates upon the interaction with the unfolded substrate, citrate synthase. Please click here to view a larger version of this figure.
Discussion
In this paper, we provided protocols for the analysis of redox-dependent chaperone activity and the characterization of structural changes upon the binding of a client protein. These are complementary methodologies to define potential chaperone-substrate complexes and analyze potential interaction sites.
Here, we applied these protocols for the characterization of a complex between the redox-regulated chaperone Hsp33 with a well-studied chaperone substrate CS. We presented two different types of protein aggregation analyses, which differ in their kinetic and initial state: during a chemical denaturation, the substrate starts its unfolding process from the denatured form, while during a thermal aggregation, the unfolding is initiated from the native form of the substrate.
Light-scattering measurements are a useful tool to monitor the relative amount of protein aggregation. While we use this method to determine the active state of the chaperone, it can further be used to study general aggregation conditions, to compare the stability of a different protein, or to study destabilizing mutations both in the substrate and chaperone.
Despite its simplicity, the light scattering method might produce noisy data, with several outliers. To overcome this issue, we developed the Kfits software described above. The Kfits software makes it possible to simply and rapidly remove large quantities of noisy data, as well as calculate the kinetic parameters from the light scattering curve. Kfits is not restricted to protein aggregation and can be applied to any type of kinetic measurements.
Thus, the methodologies described here are simple, easy to perform, and not time-consuming. They can be applied to different molecular chaperones, enabling their redox dependence to be examined within a couple days. Moreover, the anti-aggregation activity of other, non-redox dependent chaperones can be evaluated using the described protocols of light scattering and Kfits. It is important to note that different parameters, such as temperature, protein concentration, and measurement time have to be adjusted to each protein system independently.
In order to gain structural information governing a chaperone working cycle, we presented the HDX-MS methodology. In contrast to the light scattering procedure, it is a more complex technology and requires specific instrumentation, including a mass spectrometer and, preferably, a robotic system for the sample preparation. However, this setup can be established, even without the robotic system, in an existing facility. Once it is established, the procedure is relatively simple and allows the analysis of challenging protein complexes which might be unreached by high-resolution methodologies (e.g., X-ray and NMR).
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors are thankful to Meytal Radzinski for her helpful discussions and critical reading of the article, and to Patrick Griffin and his lab members for their unlimited assistance while establishing the HDX analysis platform. The authors are grateful to the German-Israel Foundation (I-2332-1149.9/2012), the Binational Science Foundation (2015056), the Marie-Curie integration grant (618806), the Israel Science Foundation (1765/13 and 2629/16), and the Human Frontier Science Program (CDA00064/2014) for their financial support.
References
- Wong HS, Dighe PA, Mezera V, Monternier PA, Brand MD. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. Journal of Biological Chemistry. 2017;292(41):16804–16809. doi: 10.1074/jbc.R117.789271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP. How mitochondria produce reactive oxygen species. Biochemical Journal. 2009;417(1):1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker CL, Pomatto LCD, Tripathi DN, Davies KJA. Redox regulation of homeostasis and proteostasis in peroxisomes. Physiological Reviews. 2018;98(1):89–115. doi: 10.1152/physrev.00033.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohn A, Konig J, Jung T. Metabolic syndrome, redox state, and the proteasomal system. Antioxidants & Redox Signaling. 2016;25(16):902–917. doi: 10.1089/ars.2016.6815. [DOI] [PubMed] [Google Scholar]
- Holmstrom KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nature Reviews Molecular Cell Biology. 2014;15(6):411–421. doi: 10.1038/nrm3801. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JMC. Free radicals in biology and medicine. Oxford, UK: Oxford University Press; 2007. [Google Scholar]
- He L, et al. Antioxidants maintain cellular redox homeostasis by elimination of reactive oxygen species. Cellular Physiology and Biochemistry: International Journal of Experimental Cellular Physiology, Biochemistry, and Pharmacology. 2017;44(2):532–553. doi: 10.1159/000485089. [DOI] [PubMed] [Google Scholar]
- Bae YS, Oh H, Rhee SG, Yoo YD. Regulation of reactive oxygen species generation in cell signaling. Molecules and Cells. 2011;32(6):491–509. doi: 10.1007/s10059-011-0276-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles GI. The redox regulation of thiol dependent signaling pathways in cancer. Current Pharmaceutical Design. 2006;12(34):4427–4443. doi: 10.2174/138161206779010549. [DOI] [PubMed] [Google Scholar]
- Qiao J, et al. Regulation of platelet activation and thrombus formation by reactive oxygen species. Redox Biology. 2018;14:126–130. doi: 10.1016/j.redox.2017.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milkovic L, Siems W, Siems R, Zarkovic N. Oxidative stress and antioxidants in carcinogenesis and integrative therapy of cancer. Current Pharmaceutical Design. 2014;20(42):6529–6542. doi: 10.2174/1381612820666140826152822. [DOI] [PubMed] [Google Scholar]
- Duecker R, et al. Oxidative stress-driven pulmonary inflammation and fibrosis in a mouse model of human ataxia-telangiectasia. Redox Biology. 2018;14:645–655. doi: 10.1016/j.redox.2017.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterbourn CC, Kettle AJ. Redox reactions and microbial killing in the neutrophil phagosome. Antioxidants & Redox Signaling. 2013;18(6):642–660. doi: 10.1089/ars.2012.4827. [DOI] [PubMed] [Google Scholar]
- Jones DP. Redox theory of aging. Redox Biology. 2015;5:71–79. doi: 10.1016/j.redox.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labunskyy VM, Gladyshev VN. Role of reactive oxygen species-mediated signaling in aging. Antioxidants & Redox Signaling. 2013;19(12):1362–1372. doi: 10.1089/ars.2012.4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurian P, Obisesan TO, Craddock TJA. Oxidative species-induced excitonic transport in tubulin aromatic networks: potential implications for neurodegenerative disease. Journal of Photochemistry and Photobiology B: Biology. 2017;175:109–124. doi: 10.1016/j.jphotobiol.2017.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinelli P, et al. A single cysteine post-translational oxidation suffices to compromise globular proteins kinetic stability and promote amyloid formation. Redox Biology. 2018;14:566–575. doi: 10.1016/j.redox.2017.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozzo F, Mirra A, Carrì MT. Oxidative stress and mitochondrial damage in the pathogenesis of ALS: new perspectives. Neuroscience Letters. 2017;636:3–8. doi: 10.1016/j.neulet.2016.04.065. [DOI] [PubMed] [Google Scholar]
- Lo Conte M, Carroll KS. The redox biochemistry of protein sulfenylation and sulfinylation. Journal of Biological Chemistry. 2013;288(37):26480–26488. doi: 10.1074/jbc.R113.467738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichmann D, Jakob U. The roles of conditional disorder in redox proteins. Current Opinion in Structural Biology. 2013;23(3):436–442. doi: 10.1016/j.sbi.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suss O, Reichmann D. Protein plasticity underlines activation and function of ATP-independent chaperones. Frontiers in Molecular Biosciences. 2015;2:43. doi: 10.3389/fmolb.2015.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voth W, Jakob U. Stress-activated chaperones: a first line of defense. Trends in Biochemical Sciences. 2017;42(11):899–913. doi: 10.1016/j.tibs.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal N, Shapira M. HSP33 in eukaryotes - an evolutionary tale of a chaperone adapted to photosynthetic organisms. The Plant Journal. 2015;82(5):850–860. doi: 10.1111/tpj.12855. [DOI] [PubMed] [Google Scholar]
- Dahl JU, Gray MJ, Jakob U. Protein quality control under oxidative stress conditions. Journal of Molecular Biology. 2015;427(7):1549–1563. doi: 10.1016/j.jmb.2015.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter J, Linke K, Jatzek A, Jakob U. Severe oxidative stress causes inactivation of DnaK and activation of the redox-regulated chaperone Hsp33. Molecular Cell. 2005;17(3):381–392. doi: 10.1016/j.molcel.2004.12.027. [DOI] [PubMed] [Google Scholar]
- Wang J, Sevier CS. Formation and reversibility of BiP protein cysteine oxidation facilitate cell survival during and post oxidative stress. Journal of Biological Chemistry. 2016;291(14):7541–7557. doi: 10.1074/jbc.M115.694810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, et al. Glutathionylation of the bacterial Hsp70 chaperone DnaK provides a link between oxidative stress and the heat shock response. Journal of Biological Chemistry. 2016;291(13):6967–6981. doi: 10.1074/jbc.M115.673608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakob U, Muse W, Eser M, Bardwell JC. Chaperone activity with a redox switch. Cell. 1999;96(3):341–352. doi: 10.1016/s0092-8674(00)80547-4. [DOI] [PubMed] [Google Scholar]
- Muller A, et al. Activation of RidA chaperone function by N-chlorination. Nature Communications. 2014;5:5804. doi: 10.1038/ncomms6804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voth W, et al. The protein targeting factor Get3 functions as ATP-independent chaperone under oxidative stress conditions. Molecular Cell. 2014;56(1):116–127. doi: 10.1016/j.molcel.2014.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon JC, et al. Oxidative stress-dependent structural and functional switching of a human 2-Cys peroxiredoxin isotype II that enhances HeLa cell resistance to H2O2-induced cell death. Journal of Biological Chemistry. 2005;280(31):28775–28784. doi: 10.1074/jbc.M505362200. [DOI] [PubMed] [Google Scholar]
- Wright MA, et al. Biophysical approaches for the study of interactions between molecular chaperones and protein aggregates. Chemical Communications. 2015;51(51):14425–14434. doi: 10.1039/c5cc03689e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haslbeck M, Vierling E. A first line of stress defense: small heat shock proteins and their function in protein homeostasis. Journal of Molecular BIology. 2015;427(7):1537–1548. doi: 10.1016/j.jmb.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimon O, et al. A role of metastable regions and their connectivity in the inactivation of a redox-regulated chaperone and its inter-chaperone crosstalk. Antioxidants & Redox Signaling. 2017;27(15):1252–1267. doi: 10.1089/ars.2016.6900. [DOI] [PubMed] [Google Scholar]
- Daturpalli S, Kniess RA, Lee CT, Mayer MP. Large rotation of the N-terminal domain of Hsp90 is important for interaction with some but not all client proteins. Journal of Molecular Biology. 2017;429(9):1406–1423. doi: 10.1016/j.jmb.2017.03.025. [DOI] [PubMed] [Google Scholar]
- Rist W, Graf C, Bukau B, Mayer MP. Amide hydrogen exchange reveals conformational changes in hsp70 chaperones important for allosteric regulation. Journal of Biological Chemistry. 2006;281(24):16493–16501. doi: 10.1074/jbc.M600847200. [DOI] [PubMed] [Google Scholar]
- Koldewey P, Horowitz S, Bardwell JCA. Chaperone-client interactions: non-specificity engenders multifunctionality. Journal of Biological Chemistry. 2017;292(29):12010–12017. doi: 10.1074/jbc.R117.796862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichmann D, et al. Order out of disorder: working cycle of an intrinsically unfolded chaperone. Cell. 2012;148(5):947–957. doi: 10.1016/j.cell.2012.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter J, Ilbert M, Graf PC, Ozcelik D, Jakob U. Bleach activates a redox-regulated chaperone by oxidative protein unfolding. Cell. 2008;135(4):691–701. doi: 10.1016/j.cell.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakob U, Eser M, Bardwell JC. Redox switch of Hsp33 has a novel zinc-binding motif. Journal of Biological Chemistry. 2000;275(49):38302–38310. doi: 10.1074/jbc.M005957200. [DOI] [PubMed] [Google Scholar]
- Ilbert M, et al. The redox-switch domain of Hsp33 functions as dual stress sensor. Nature Structural & Molecular Biology. 2007;14(6):556–563. doi: 10.1038/nsmb1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groitl B, et al. Protein unfolding as a switch from self-recognition to high-affinity client binding. Nature Communications. 2016;7:10357. doi: 10.1038/ncomms10357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremers CM, Reichmann D, Hausmann J, Ilbert M, Jakob U. Unfolding of metastable linker region is at the core of Hsp33 activation as a redox-regulated chaperone. Journal of Biological Chemistry. 2010;285(15):11243–11251. doi: 10.1074/jbc.M109.084350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Mishra S, Khan E. Emerging methods for structural analysis of protein aggregation. Protein & Peptide Letters. 2017;24(4):331–339. doi: 10.2174/0929866524666170206123150. [DOI] [PubMed] [Google Scholar]
- Rimon O, Reichmann D. Kfits: a software framework for fitting and cleaning outliers in kinetic measurements. Bioinformatics. 2018;34(1):129–130. doi: 10.1093/bioinformatics/btx577. [DOI] [PubMed] [Google Scholar]
- Percy AJ, Rey M, Burns KM, Schriemer DC. Probing protein interactions with hydrogen/deuterium exchange and mass spectrometry - a review. Analytica Chimica Acta. 2012;721:7–21. doi: 10.1016/j.aca.2012.01.037. [DOI] [PubMed] [Google Scholar]
- Sinz A. Divide and conquer: cleavable cross-linkers to study protein conformation and protein-protein interactions. Analytical and Bioanalytical Chemistry. 2017;409(1):33–44. doi: 10.1007/s00216-016-9941-x. [DOI] [PubMed] [Google Scholar]
- Sinz A, Arlt C, Chorev D, Sharon M. Chemical cross-linking and native mass spectrometry: a fruitful combination for structural biology. Protein Science. 2015;24(8):1193–1209. doi: 10.1002/pro.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt C, Beilsten-Edmands V, Robinson CV. Insights into eukaryotic translation initiation from mass spectrometry of macromolecular protein assemblies. Journal of Molecular Biology. 2016;428:344–356. doi: 10.1016/j.jmb.2015.10.011. 2 Pt A. [DOI] [PubMed] [Google Scholar]
- Marciano DP, Dharmarajan V, Griffin PR. HDX-MS guided drug discovery: small molecules and biopharmaceuticals. Current Opinion in Structural Biology. 2014;28:105–111. doi: 10.1016/j.sbi.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistarz UH, Brown JM, Haselmann KF, Rand KD. Probing the binding interfaces of protein complexes using gas-phase H/D exchange mass spectrometry. Structure. 2016;24(2):310–318. doi: 10.1016/j.str.2015.11.013. [DOI] [PubMed] [Google Scholar]
- Harrison RA, Engen JR. Conformational insight into multi-protein signaling assemblies by hydrogen-deuterium exchange mass spectrometry. Current Opinion in Structural Biology. 2016;41:187–193. doi: 10.1016/j.sbi.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KA, Wilson DJ. Bottom-up hydrogen deuterium exchange mass spectrometry: data analysis and interpretation. Analyst. 2017;142(16):2874–2886. doi: 10.1039/c7an00662d. [DOI] [PubMed] [Google Scholar]
- Vadas O, Jenkins ML, Dornan GL, Burke JE. Using hydrogen-deuterium exchange mass spectrometry to examine protein-membrane interactions. Methods in Enzymology. 2017;583:143–172. doi: 10.1016/bs.mie.2016.09.008. [DOI] [PubMed] [Google Scholar]
- Zanphorlin LM, et al. Heat shock protein 90 kDa (Hsp90) has a second functional interaction site with the mitochondrial import receptor Tom70. Journal of Biological Chemistry. 2016;291(36):18620–18631. doi: 10.1074/jbc.M115.710137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hvidt A, Nielsen SO. Hydrogen exchange in proteins. Advances in Protein Chemistry. 1966;21:287–386. doi: 10.1016/s0065-3233(08)60129-1. [DOI] [PubMed] [Google Scholar]
- Roberts VA, Pique ME, Hsu S, Li S. Combining H/D exchange mass spectrometry and computational docking to derive the structure of protein-protein complexes. Biochemistry. 2017;56(48):6329–6342. doi: 10.1021/acs.biochem.7b00643. [DOI] [PubMed] [Google Scholar]
- Pascal BD, et al. HDX Workbench: software for the analysis of H/D exchange MS data. Journal of the American Society for Mass Spectrometry. 2012;23(9):1512–1521. doi: 10.1007/s13361-012-0419-6. [DOI] [PMC free article] [PubMed] [Google Scholar]