

Abstract
A useful technique for culturing cells in a self-assembling nanofiber three-dimensional (3D) scaffold is described. This culture system recreates an environment that closely mimics the structural features of non-polarized tissue. Furthermore, the particular intrinsic nanofiber structure of the scaffold makes it transparent to visual light, which allows for easy visualization of the sample under microscopy. This advantage was largely used to study cell migration, organization, proliferation, and differentiation and thus any development of their particular cellular function by staining with specific dyes or probes. Furthermore, in this work, we describe the good performance of this system to easily study the redifferentiation of expanded human articular chondrocytes into cartilaginous tissue. Cells were encapsulated into self-assembling peptide scaffolds and cultured under specific conditions to promote chondrogenesis. Three-dimensional cultures showed good viability during the 4 weeks of the experiment. As expected, samples cultured with chondrogenic inducers (compared to non-induced controls) stained strongly positive for toluidine blue (which stains glycosaminoglycans (GAGs) that are highly present in cartilage extracellular matrix) and expressed specific molecular markers, including collagen type I, II and X, according to Western Blot analysis. This protocol is easy to perform and can be used at research laboratories, industries and for educational purposes in laboratory courses.
Keywords: Bioengineering, Issue 136, Nanobiomaterials, Self-Assembling Peptide Scaffolds, Nanomaterials, Mammal Cells, Articular Chondrocytes, 3D-culture, Differentiation
Introduction
For many decades, mammalian cell culture has been performed under experimental conditions using classical two-dimensional (2D) culture systems due to practical and economic issues regardless of the non-physiological aspects. Although this culture system helps to study and understand most molecular and cellular mechanisms, we know today that new cell culture paradigms are needed to study more complex cellular systems. Therefore, three-dimensional (3D) culture systems are needed to recreate a microenvironment that is biophysically, biomechanically and biologically more similar to that of natural tissues. In recent years, 3D culture systems, in general, have become more prevalent among researchers and industry since they represent a new model of study or screening in which cells can grow in space, create cell-to-cell or cell-to-matrix interactions, migrate and eventually differentiate into specific cell lineages.
The overall goal of this methodology is to recreate an in vitro cellular microenvironment that is closer to the in vivo microenvironment. In particular, the synthetic self-assembling peptide scaffold (SAPS) is a type of biomaterial with unique properties; it forms a network of nanometer-sized pores made out of weak interactions among peptides with mechanical and structural properties similar those of natural extracellular matrices. In other words, the rationality behind the use of this material is that it creates a truly 3D-environment that is ideal for obtaining pseudo-3D tissues or organ units. However, most importantly, the 3D context allows the 3D structure to gain new biological functions that normally are not present in 2D culture platforms, such as properties related to tissue architecture, mass transfer phenomena, cell patterning and eventually tissue morphogenesis, which are key factors in future research and development of functional tissues and organs1,2. Moreover, an advantage of SAPS over their natural counterparts (Collagen, Matrigel) is that they are very stable at room temperature and do not require special conditions for post-production, distribution or storage3,4,5,6,7,8,9,10,11,12,13,14,15. SAPS is easy to handle, when desired; 3D gels can be simply obtained by increasing the ionic strength or by adjusting the pH to neutrality1,2. Finally, the methodology described here has been extensively used in vitro to promote maintenance, growth, and differentiation of a number of cell types, including chondrocytes, hepatocytes, endothelial cells, osteoblasts, neuronal cells as well as embryonic and somatic stem cells3,4,5,6,7,8,9,10,11,12,13,14,15. In the present work, we describe the use of a 3D-culture system to differentiate human expanded articular chondrocytes (hACh) into cartilage-like tissue as previously described11.
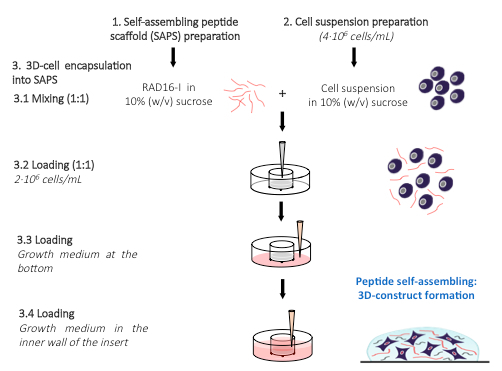
Here, a method to culture cells in a 3D system using SAPS is described. In this synthetic biomaterial, cells are first mixed with a peptide solution, which is subsequently induced to self-assemble, creating a network of nanometric dimensions around the cells and therefore creating a truly 3D environment (Figure 1). It is important to consider that cellular behavior is affected by matrix stiffness values (i.e., proliferation, migration and differentiation). Therefore, a common methodological control is to culture cells on top of the peptide scaffold with the same stiffness values (culture on two dimensions).
Protocol
1. Self-Assembling Peptide Scaffold (SAPS) Preparation
Prepare a 0.5% solution of peptide RAD16-I in 20% (w/v) sucrose, in a micro-centrifuge tube. Sonicate the peptide solution for 20 min in an ultrasonic bath at maximum power and at room temperature (RT) to ensure homogeneous mixing. NOTE: The volume of solution to be prepared will depend on the number of encapsulations desired. RAD16-I peptides do not suffer degradation under these sonication conditions.
Dilute the peptide solution prepared in step 1.1 with the necessary volume of 10% (w/v) sucrose to obtain the desired final concentration. NOTE: An important factor to take into consideration is the adjustment of the initial peptide concentration since the final matrix stiffness will depend on this parameter. The RAD16-I peptide solution should be 2x more concentrated than the desired final concentration of the 3D construct. Normally, the final RAD16-I peptide concentrations used are between 0.15–0.5% (w/v).
Place the micro-centrifuge tube containing the peptide solution into an ultrasonic bath at RT for 20 min to ensure homogeneous mixing. NOTE: The peptide solution, if not used immediately, can be stored for several months at 4 °C.
2. Cell Suspension Preparation
Centrifuge the cell suspension (obtained after trypsin treatment) at 60 x g and RT for 5 min.
Remove the supernatant using a Pasteur pipette connected to a vacuum line and suspend the cell pellet in 3–5 mL of 10% (w/v) sucrose using a micropipette. Proceed to count cells using a manual or automatic cell counter. NOTE: The 10% (w/v) sucrose solution is an isotonic and non-ionic medium that allows for cell maintenance and avoids peptide gelation during the mixing process.
Centrifuge the cell suspension at 60 x g and RT for 5 min. Remove the supernatant and suspend the cell pellet again in the necessary volume of 10% (w/v) sucrose to obtain double the desired final cell concentration (recommendable approximately 4 x 106 cells/mL).
Place a desired number of tissue culture inserts in each well of a 6-well plate (one insert per encapsulation).
Sonicate the peptide solution prepared in step 1 at maximum power and RT for 5 min.
Pipette and place 40 µL of the cell suspension (obtained in 2.3) for each encapsulation desired plus approximately 10–15% of extra volume into a standard micro-centrifuge tube.
3. 3D Cell Encapsulation into SAPS
Mix an equal volume of the RAD16-I solution in 10% sucrose (w/v) (40 µL per encapsulation) with an equal volume of the cell suspension (40 µL per encapsulation) prepared in step 2 to obtain a suspension of cells in the desired peptide concentration (in 10% sucrose). Mix by pipetting slowly up and down approximately 10 times. Avoid bubble formation during mixing. NOTE: This step is critical and must be performed very carefully since cells are under hostile conditions due to the low pH peptide (approximately 3.5–4.0).
Using a micropipette, carefully load 80 µL of the suspension into each insert.
Add 0.5 mL of culture media to the bottom of the well to wet the insert membrane, allowing media diffusion to promote peptide self-assembly and hydrogel formation. Let the peptide gel for approximately 20 min in the flow cabinet. NOTE: The manipulation of the samples during setting time could end in gel breaking.
Very slowly add 40 µL of medium to the insert and let it slide down the inner wall to the gel. NOTE: Normally, the loading of this volume should take between 20–30 s.
Place the plate in the incubator (37 °C, 5% CO2, humidified atmosphere) for 20 min. This step and successive medium additions will favor leaching of the sucrose and equilibration of cells with culture medium.
Add 60 µL of medium to the inner wall of the insert, allowing it to flow down the wall. Aspirate the medium of the well, which is rich in sucrose, and add 0.5 mL of fresh medium.
Add 60 µL of medium slowly to the insert and place the plate in the incubator (37 °C, 5% CO2, humidified atmosphere) for 10 min.
Aspirate the medium of the well and add 2.5 mL of fresh medium into the well. Add 200 µL of medium into the insert. Place the plate in the incubator (37 °C, 5% CO2, humidified atmosphere) to maintain the cells under the appropriate conditions.
Change the medium every day. Remove the old medium from the bottom of the well. Add 2.5 mL of fresh medium to the well and 200 µL to the insert. NOTE: If cell differentiation continues, the specific inducing media will be added to the cultures, resulting in the desired cell type commitment (see below, Cell differentiation).
4. Cell Viability in 3D-SAPS
NOTE: The cell viability assay is known as the Live/Dead — Viability/Cytotoxicity assay as well.
In a 15 mL tube, prepare the necessary volume of a fresh solution of 2 µM calcein AM and 2 µM of ethidium homodimer-1 (EthD-1) in PBS by vortexing for 10 s. Keep in the dark. NOTE: The volume to be used for each sample will depend on the well size according to the following: 2 mL for each well of a 6-multiwell plate; 1 mL for each well of a 12-multiwell plate and 0.5 mL for each well of a 24-multiwell plate.
Using a micropipette, rinse the 3D-SAPS samples 3 times with 2 mL of PBS, and then, cover it with the solution prepared in step 4.1. Incubate it at RT for 40 min in the dark.
Using a micropipette, rinse the constructs with 2 mL of PBS 5 times.
Visualize the samples under a fluorescence microscope using 10X or 20X magnification.
5. Cell Differentiation
Culture cells (37 °C, 5% CO2, humidified atmosphere) in Chondrocyte Basal Medium (CBM) with growth supplements. NOTE: The preparation of the medium is described in the manufacturers' instructions (See Materials Table).
Induce cell differentiation at day 2 with chondrogenic medium containing recombinant human transforming growth factor-b1 [TGFb1] (10 ng/mL), L-Ascorbic Acid 2-phosphate [AA2P] (25 µg/mL) and Dexamethasone (100 nM).
Maintain the culture for 4 weeks in an incubator with a humidified atmosphere at 37 °C and 5% CO2. Change chondrogenic medium every other day.
6. Toluidine Blue (TB) Staining
Wash the 3D-SAPS cultures twice by adding and then removing 2 mL of PBS with a micropipette. Fix them by adding 2 mL of 2% (w/v) p-formaldehyde in PBS for 2 h at RT.
Wash twice with 2 mL of 0.1% Triton X-100 in PBS (PBST).
Incubate with 2 mL of 0.05% TB in water for 20 min at RT.
Wash several times (at least 3 times) with 2 mL of PBS until the PBS solution removed is clear.
Analyze samples by visual inspection under a stereoscopic microscope11,13,14. NOTE: TB forms complexes with anionic glycoconjugates at the extracellular matrix, such as proteoglycans (PG) and glycosaminoglycans (GAG), which are mainly secreted by chondrocyte-like cells. Positive samples are stained blue.
7. Western Blot
NOTE: The procedure used in this protocol was previously described in reference11.
Lyse 3D-constructs in RIPA buffer containing protease inhibitor cocktail by pipetting.
Prepare acrylamide gel according to gel-volume requirements (of the specific gel running equipment) and run the cell lysates for 90 min at 150 V16. NOTE: Acrylamide gel is usually prepared at a concentration of 7.5–10% (w/v), depending on the protein's molecular weight.
Transfer the proteins contained in the gel into a polyvinylidene difluoride (PVDF) membrane by applying 40 V for 2 h at RT.
Incubate the PVDF membrane at RT for 2 h in blocking buffer (4% (w/v) nonfat powdered milk in PBST) in a separate container with enough volume to cover the membrane.
Incubate the membrane at RT for 1 h with primary antibodies at a final concentration of 1 mg/mL in PBST. See Materials Table.
Add secondary antibodies at a concentration of 1 mg/mL. See the Materials Table.
Prepare the membrane for hP detection with a Chemiluminescent substrate16. See Materials Table.
Take chemiluminescent images16.
Representative Results
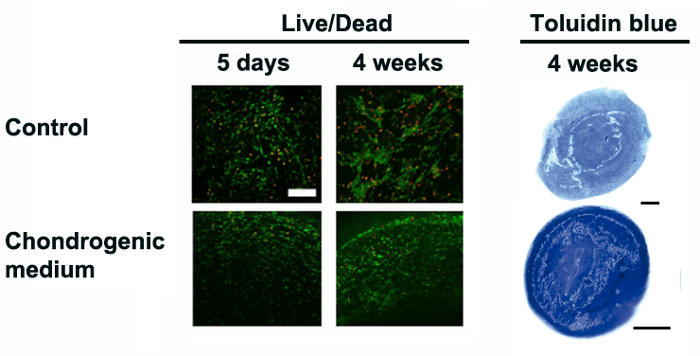
In the present work, a simple and faster protocol to culture cells in 3D using SAPS is described to obtain reliable qualitative and quantitative data. SAPS creates a 3D microenvironment with unique properties that allow cultured cells under desired conditions to proceed to cell maintenance, proliferation and/or differentiation3,4,5,6,7,8,9,10,11,12. In our group, several studies have been performed using human adipose tissue derived progenitor cells (hAPC), expanded human articular chondrocytes (hACh) and human normal dermal fibroblasts (hNDF) to culture, expand and differentiate them into cartilage-like tissue10,11,12. Here, we describe the 3D culture and re-differentiation of expanded hACh into cartilage-like tissue, as previously described11. Hence, we encapsulate cells in 3D-SAPS and culture them in chondrogenic induction medium, as described in Figure 1 (and in the Protocol). After 24 h, samples were assessed for viability with the Live/Dead Viability/Cytotoxicity Assay. The results indicate that a few percent of cells were dead (red) after 5 days of encapsulation and almost no dead cells were observed after 4 weeks of culture (Figure 2). Next, we stained them for glycosaminoglycan (GAG) deposition at the extracellular matrix using the Toluidine blue staining method. As expected, the induction group responded properly to the treatment, showing strong staining (Figure 2).
Finally, molecular markers of chondrogenesis and hypertrophy markers, including collagen type I, II and X, were analyzed by western blot (Figure 3). Collagen type I (COL1) was observed in the 2D and 3D systems but a lower band of ~130 kDa (representing the processed mature COL1 protein to be assembled at the extracellular matrix) was only observed in 3D. This result clearly indicates that cells undergoing differentiation in three dimensions do proceed in a more physiological way. Remarkably, collagen type II (COL2) was detected - as a single band of the expected molecular weight present at the extracellular matrix - only in 3D constructs cultured under chondrogenic conditions. Finally, collagen type X (COL10) was detected in both 2D and 3D constructs cultured under chondrogenic conditions, indicating some degree of hypertrophy after 4 weeks of culture.
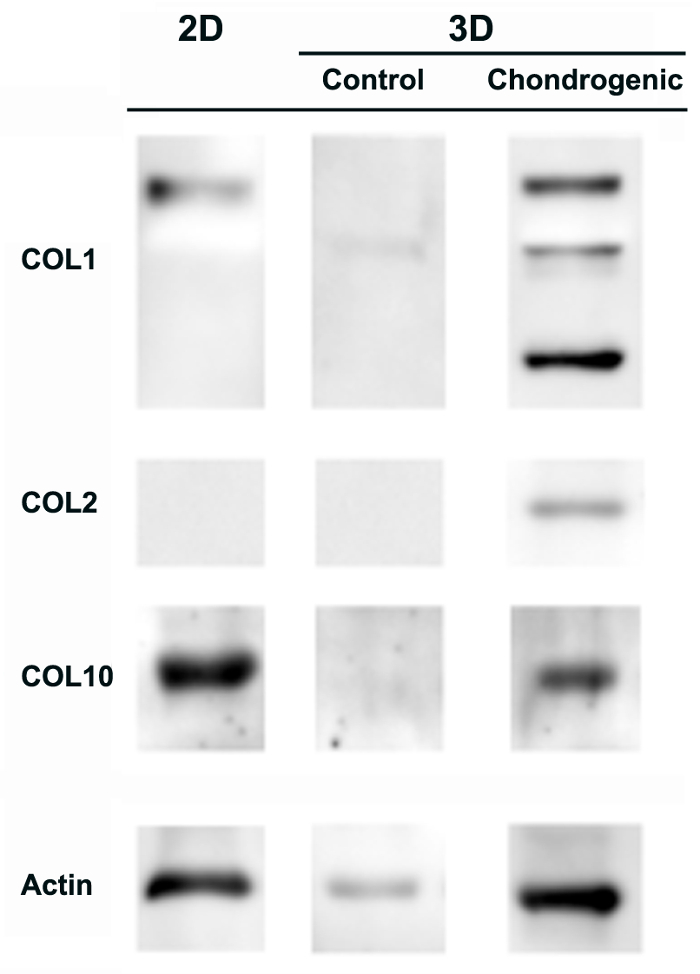
Figure 1: Schematic representation of cell encapsulation into 3D-SAPS. Schematic representation of the hACh encapsulation is described to obtain cells randomly dispersed within the 3D-SAPS. 1. Self-assembling peptide scaffold (SAPS) preparation; 2. cell suspension preparation; and 3. 3D-cell encapsulation into SAPS. Note that the schematic only shows steps 3.1 to 3.4.
Figure 2: Cell performance and chondrogenic differentiation in 3D-SAPS cultures. Cell viability staining performed at 5 days and 4 weeks of culture detected under a fluorescent microscope (live cells in green and dead cells in red). Scale bars = 200 µm. General chondrogenesis commitment assessed by Toluidine blue staining (sulfated glycosaminoglycans in blue) of 3D-SAPS constructs cultured in control and chondrogenic media. Scale bars = 500 µm. This figure has been modified from L. Recha-Sancho et al.11. Please click here to view a larger version of this figure.
Figure 3: Collagen I, II and X expression of hACh undergoing chondrogenesis. hACh cultured in a 2D-monolayer and in 3D-SAPS after 4 weeks in chondrogenic media and controls were assessed for the expression of collagen type I (COL1), collagen type II (COL2) and collagen type X (COL10). Actin expression was used as an internal control. This figure has been modified from L. Recha-Sancho et al.11. Please click here to view a larger version of this figure.
Discussion
Previously, our group and others have described the use of three-dimensional (3D) culture platforms with diverse cell systems3,4,5,6,7,8,9,10,11,12,13,14,15. In the present work, we describe an easy and reliable method of obtaining 3D culture systems that are applicable to any type of mammalian cells including any type of functional cell, embryonic or adult stem cells, or eventually, dysfunctional cells isolated from biopsies or tumors, and so on. In addition, independently, if stem cells are of embryonic or adult origin they would have a better lineage commitment capacity in the 3D environment than in classical 2D culture dishes10,11,12,13,14,15. Therefore, the cell culture in this system would lead to differentiation into functional tissue-like structures that could be used in different applications, from reparative or regenerative biomedicine to toxicological and pharmacological platforms.
We have shown a clear gain in cell function because the cellular microenvironment is similar to that of natural tissues in terms of the matrix structure and biomechanical, biophysical and biological parameters. Nevertheless, as the system becomes more complex, the number of parameters that need to be regulated also increases, which includes the need for an external supporting platform (such as an active perfusion system to avoid mass transfer phenomena-associated issues). Three-dimensionally cultured cells perform better in terms of regulating essential activities, such as migration, proliferation and differentiation. They could form complex networks allowing enhanced cellular crosstalk, which is by far an essential consequence of growing and differentiating in 3D. The fact that SAPS could create conditions in vitro similar to those extracellular matrix proteins represents an advantage since a rational study of the effect produced for each component added to the scaffold (growth factor, polysaccharide or signaling peptide) could be easily carried out.
The clear advantages of the use of SAPS compared to other natural scaffolds, such as collagen type I and Matrigel, are the following: 1) SAPS is a synthetic biomaterial with minimal variation from batch to batch production; 2) SAPS has the capacity to be functionalized with specific peptide motifs; and 3) SAPS presents low biodegradability in vitro, which permits the maintenance of the 3D construct with the same biophysical, biomechanical and structural properties over time. However, the limitation of using SAPS versus other scaffolds is found during the encapsulation step, where cells are in a hostile milieu due to the low pH. Therefore, this is a critical step in the described methodology. Moreover, it is important to set the SAPS concentration for each specific cell type before starting any experiment. This is, in fact, essential when cells are cultured on or into these biomaterials, since each particular cell type will present optimal biomechanical growth conditions.
Finally, we believe that the design and fabrication of this type of biomaterial scaffold would enhance the development of more physiological and reliable 3D tissue models to help the pharmaceutical industry to develop better therapeutic approaches for regenerative medicine, cancer or any medical treatment.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The research performed by the authors was supported in part by grants from the European Union Seventh Framework Programme (FP7/2007- 2013) under Grant Agreement No. 229239, and from the AO Foundation, Exploratory Research Collaborative Research Program Acute Cartilage Injury/lesion/Defect (CRP ACI) under the project Bioactive and Biomimetic Scaffolds for Cartilage Regeneration (BIOCART).
References
- Aggeli A, et al. Responsive gels formed by the spontaneous self-assembly of peptide into polymeric β-sheet tapes. Nature. 1997;386:259–262. doi: 10.1038/386259a0. [DOI] [PubMed] [Google Scholar]
- Semino CE. Can we build artificial stem cell compartments? Journal of Biomedicine and Biotechnology. 2003;3:164–169. doi: 10.1155/S1110724303208019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisiday J, et al. Self-assembling peptide hydrogel fosters chondrocyte extracellular matrix production and cell division: implications for cartilage tissue repair. Proceedings of the National Academy of Sciences, USA. 2002;99:9996–10001. doi: 10.1073/pnas.142309999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narmoneva DA, Vukmirovic R, Davis ME, Kamm RD, Lee RT. Endothelial cells promote cardiac myocyte survival and spatial reorganization: implications for cardiac regeneration. Circulation. 2004;110:962–968. doi: 10.1161/01.CIR.0000140667.37070.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genové E, Shen C, Zhang S, Semino CE. The effect of functionalized self-assembling peptide scaffolds on human aortic endothelial cell function. Biomaterials. 2005;26:3341–3351. doi: 10.1016/j.biomaterials.2004.08.012. [DOI] [PubMed] [Google Scholar]
- Garreta E, Genové E, Borrós S, Semino CE. Osteogenic differentiation of mouse embryonic stem cells and mouse embryonic fibroblasts in a three-dimensional self-assembling peptide scaffold. Tissue Engineering. 2006;12:2215–2228. doi: 10.1089/ten.2006.12.2215. [DOI] [PubMed] [Google Scholar]
- Sieminski AL, Semino CE, Gong H, Kamm RD. Primary sequence of ionic self-assembling peptide gels affects endothelial cell adhesion and capillary morphogenesis. Journal of Biomedical Materials Research Part A. 2008;87(2):494–504. doi: 10.1002/jbm.a.31785. [DOI] [PubMed] [Google Scholar]
- Semino CE. Self-assembling peptides: from bio-inspired materials to bone regeneration. Journal of Dental Research. 2008;87(2008):606–616. doi: 10.1177/154405910808700710. [DOI] [PubMed] [Google Scholar]
- Hernández Vera R, et al. Interstitial fluid flow intensity modulates endothelial sprouting in restricted Src-activated cell clusters during capillary morphogenesis. Tissue Engineering Part A. 2009;15(1):175–185. doi: 10.1089/ten.tea.2007.0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castells-Sala C, Martínez-Ramos C, Vallés-Lluch A, Monleón Pradas M, Semino C. In vitro development of bioimplants made up of elastomeric scaffolds with peptide gel filling seeded with human subcutaneous adipose tissue-derived progenitor cells. Journal of Biomedical Materials Research Part A. 2015;103(11):3419–3430. doi: 10.1002/jbm.a.35482. [DOI] [PubMed] [Google Scholar]
- Recha-Sancho L, Semino CE. Heparin-based self-assembling peptide scaffold reestablish chondrogenic phenotype of expanded de-differentiated human chondrocytes. Journal of Biomedical Materials Research Part A. 2016;104(7):1694–1706. doi: 10.1002/jbm.a.35699. [DOI] [PubMed] [Google Scholar]
- Bussmann BM, et al. Chondrogenic potential of human dermal fibroblasts in a contractile, soft, self-assembling, peptide hydrogel. Journal of Tissue Engineering and Regenerative Medicine. 2016;10(2):E54–E62. doi: 10.1002/term.1766. [DOI] [PubMed] [Google Scholar]
- Recha-Sancho L, Moutos FT, Abellà J, Guilak F, Semino CE. Dedifferentiated Human Articular Chondrocytes Redifferentiate to a Cartilage-Like Tissue Phenotype in a Poly (ε-Caprolactone)/Self-Assembling Peptide Composite Scaffold. Materials. 2016;9(6):472. doi: 10.3390/ma9060472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recha-Sancho L, Semino CE. Chondroitin Sulfate and Decorin based self-assembling scaffolds for cartilage tissue engineering. PlosOne. 2016;11(6):e0157603. doi: 10.1371/journal.pone.0157603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aloy-Reverté C, Moreno-Amador JL, Nacher M, Montanya E, Semino CE. Use of RGD-Functionalized Sandwich Cultures to Promote Redifferentiation of Human Pancreatic Beta Cells After In Vitro Expansion. Tissue Engineering Part A. 2017. [DOI] [PubMed]
- Kurien BT, Scofield RH. Protein blotting and detection. Methods in Molecular Biology. 2009;536 doi: 10.1007/978-1-59745-542-8_3. [DOI] [PubMed] [Google Scholar]