

Abstract
Human telomerase reverse transcriptase (TERT) is the catalytic subunit of telomerase, and it elongates telomere through RNA-dependent DNA polymerase activity. Although TERT is named as a reverse transcriptase, structural and phylogenetic analyses of TERT demonstrate that TERT is a member of right-handed polymerases, and relates to viral RNA-dependent RNA polymerases (RdRPs) as well as viral reverse transcriptase. We firstly identified RdRP activity of human TERT that generates complementary RNA stand to a template non-coding RNA and contributes to RNA silencing in cancer cells. To analyze this non-canonical enzymatic activity, we developed RdRP assay with recombinant TERT in 2009, thereafter established in vitro RdRP assay for endogenous TERT. In this manuscript, we describe the latter method. Briefly, TERT immune complexes are isolated from cells, and incubated with template RNA and rNTPs including radioactive rNTP for RdRP reaction. To eliminate single-stranded RNA, reaction products are treated with RNase I, and the final products are analyzed with polyacrylamide gel electrophoresis. Radiolabeled RdRP products can be detected by autoradiography after overnight exposure.
Keywords: Genetics, Issue 136, TERT, RNA-dependent RNA polymerase, double-stranded RNA, telomerase, immunoprecipitation, radioisotope
Introduction
Human telomerase reverse transcriptase (TERT) is well known as the catalytic subunit of telomerase, and it elongates telomere using telomerase RNA component (TERC), the specific RNA template1. Although TERT polymerizes telomeric DNA as a component of telomerase, the structural and phylogenetic analyses indicate that TERT is closely related with viral RNA-dependent RNA polymerases (RdRPs) as well as viral reverse transcriptase, and shares domains with these polymerases2,3,4. RdRP is the enzyme that generates complementary RNA strand to a template RNA. The enzyme is encoded not only in viruses but also in model organisms, such as plant, yeast and worm, and double-stranded RNA synthesis by RdRP contributes to transcriptional and post-transcriptional gene silencing in these organisms5,6. Although human RdRP had been missing for a long time, we found RdRP activity in human TERT in 20097.
We first confirmed RdRP activity of TERT with recombinant protein7, then established a sensitive in vitro assay to detect RdRP activity of endogenous TERT8. Here, we demonstrate the in vitro RdRP assay (IP-RdRP assay) for endogenous TERT. This method starts with immunoprecipitation (IP) of endogenous TERT, and is followed by in vitro RdRP reaction, in which radioactive ribonucleotides are incorporated into nascent RNA strands.
Protocol
1. Reagent Setup
- Reagents used for Cell Synchronization
- To generate Dulbecco's modified Eagle's medium (DMEM) containing 2.5 mM thymidine, dissolve 30.28 mg of thymidine per 1 mL of tissue culture grade water to prepare 125 mM thymidine. Filtrate the solution with a 0.22 μm syringe filter unit. Add 1 mL of freshly prepared 125 mM thymidine per 50 mL of DMEM supplemented with 10% (vol/vol) fetal bovine serum (FBS).
- To generate DMEM containing 0.1 μg/mL of nocodazole, dissolve nocodazole in dimethyl sulfoxide (DMSO) to prepare 5 μg/μL nocodazole. Add 1 µL of 5 µg/ µL nocodazole per 50 mL of DMEM supplemented with 10% (vol/vol) FBS.
- To generate DMEM containing 0.002% (vol/vol) of DMSO, add 1 µL of DMSO per 50 mL of DMEM supplemented with 10% (vol/vol) FBS.
- Reagents used for IP-RdRP assay
- Prepare lysis buffer A: 150 mM NaCl, 20 mM Tris-HCl (pH 7.4) and 0.5% (vol/vol) Nonidet P-40 (NP-40). Store the reagent at 4 °C.
- Prepare 5x acetate buffer: 50 mM 2-[4-(2-Hydroxyethyl)-1-piperazinyl]ethanesulfonic acid (HEPES)-KOH (pH 7.8), 500 mM potassium acetate and 20 mM MgCl2. Store the reagent at room temperature.
- Prepare Wash buffer 1: 1x acetate buffer, 10% (vol/vol) glycerol, 0.1% (vol/vol) Triton X-100 and 0.06x protease inhibitor cocktail, ethylenediamenetetraacetic acid (EDTA)-free. Prepare Wash buffer 1 on the day of use or the day before use. Store the reagent at 4 °C.
- Prepare AGC solution: 1x acetate buffer, 10% (vol/vol) glycerol and 0.02% (wt/vol) 3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonat (CHAPS). Store the reagent at 4 °C.
- To generate AGC solution containing 2 mM CaCl2, add 100 µL of 1 M CaCl2 per 50 mL of AGC solution. Store the reagent at 4 °C.
- To generate AGC solution containing 3 mM ethylene glycol bis (b-aminoethylether)-N,N,N',N'-tetraacetic acid (EGTA), add 375 µL of 0.4 M EGTA (pH 7.5) per 50 mL of AGC solution. Store the reagent at 4 °C.
- Prepare Wash buffer 2: 1x acetate buffer and 0.02% (wt/vol) CHAPS. Store the reagent at 4 °C.
- Prepare HMD solution: 0.2 M HEPES-KOH (pH 7.8), 40 mM MgCl2 and 2 mM dithiothreitol (DTT). Store the reagent at -20 °C. NOTE: HMD solution should be renewed at least in three months.
- Prepare 2x Proteinase K buffer: 20 mM Tris-HCl (pH 7.6), 20 mM EDTA and 1% (wt/vol) sodium dodecyl sulfate (SDS). Store the reagent at -20 °C.
- Prepare 2x loading buffer: 95% (vol/vol) formamide, 20 mM EDTA and 1% (wt/vol) Orange G. Store the reagent at -20 °C.
2. Preparation of HeLa cells
NOTE: Prepare HeLa cells synchronized in mitotic phase (manipulated) or dividing asynchronously (unmanipulated). Culture the cells in the presence of 5% CO2 at 37 °C.
- Synchronization of HeLa cells in mitosis
- Plate 1 x 106 of HeLa cells with 10 mL of DMEM supplemented with 10% (vol/vol) FBS in a 10 cm culture dish.
- Two days after plating, replace the medium with 10 mL of DMEM containing 2.5 mM thymidine and 10% (vol/vol) FBS. Culture the cells for 24 h.
- Wash the cells three times with 10 mL of phosphate buffered saline (PBS) (-), and culture the cells with 10 mL of DMEM containing 10% (vol/vol) FBS for 6 h.
- Replace the medium with 10 mL of DMEM containing 0.1 µg/mL of nocodazole and 10% (vol/vol) FBS, and culture the cells for 14 h.
- Shake the culture dish gently, and retrieve the supernatant containing the detached mitotic cells. Count the cell number for the IP-RdRP assay.
- Centrifuge the sample at 490 x g for 5 min at 4 °C, and discard the supernatant. NOTE: The cell pellet can be stored at -80 °C.
- Preparation of unmanipulated HeLa cells
- Plate 1 x 106 of HeLa cells with 10 mL of DMEM supplemented with 10% (vol/vol) FBS in a 10 cm culture dish.
- Two days after plating, replace the medium with 10 mL of DMEM containing 10% (vol/vol) FBS. Culture the cells for 30 h.
- Replace the medium with 10 mL of DMEM containing 0.002% (vol/vol) DMSO and 10% (vol/vol) FBS, and culture the cells for 14 h.
- Collect the cells by trypsinization using 0.7 mL of trypsin (2.5 g/L) containing 1 mM EDTA. Count the cell number for the IP-RdRP assay.
- Centrifuge the sample at 490 x g for 5 min at 4 °C, and discard the supernatant. NOTE: The cell pellet can be stored at -80 °C.
3. IP-RdRP Assay
- Protein A-agarose bead preparation
- Transfer 1 mL (bed volume 500 µL) of affinity resin to a 1.5 mL microcentrifuge tube. Centrifuge at 800 x g for 5 min at 4 °C, and discard the supernatant.
- Add 800 µL of Lysis buffer A to the beads, and mix well by inverting the tube several times. Centrifuge at 800 x g for 3 min at 4 °C, and discard the supernatant. Repeat this step twice (total three washes).
- Add 800 µL of Lysis buffer A to the beads, mix well by inverting the tube several times. Rotate the tube overnight (or for at least 4 h) at 4 °C on a rotary shaker.
- Centrifuge the tube at 800 x g for 3 min at 4 °C, and discard the supernatant.
- Add 500 µL of Lysis buffer A to the beads, and mix well by inverting the tube several times. Store the beads at 4 °C.
- Preparation of cell lysate
- (Optional) If the cells are frozen in step 2, place and thaw the frozen cells on ice until the cells become loose.
- Wash the cells once with 10 mL of PBS (-). Centrifuge the sample at 1,450 x g for 5 min at 4 °C, and discard the supernatant.
- Lyse the cells with ice-cold Lysis buffer A (1 mL of Lysis buffer A per 1 x 107 of the cells) by gentle pipetting.
- Sonicate the sample in a 1.5 mL tube with following conditions; set amplification of the sonicator at 25%, and sonicate for 10 s.
- Centrifuge the sample at 20,400 x g for 15 min at 4 °C.
- Collect the supernatant. Transfer 1 mL each of the supernatant into 1.5 mL microcentrifuge tubes. CAUTION: Perform all procedures under RNase-free conditions.
- Immunoprecipitation
- Pre-clear the lysate from step 3.2.6 by adding 40 µL (bead volume 20 µL) of prewashed protein A-agarose beads per 1 mL of the lysate. Mix well by inverting the tube several times and rotate the sample for 30 min at 4 °C.
- Centrifuge the sample at 13,000 x g for 1 min at 4 °C, and recover the supernatant.
- Add 40 µL (bed volume 20 µL) of prewashed protein A-agarose beads and 10 µg of anti-human TERT antibody (clone: 10E9-2)8 into the pre-cleared lysate. Mix well by inverting the tube several times and rotate the sample at 4 °C over night.
- Centrifuge the sample at 3,300 x g for 0.5 min at 4 °C, and remove the supernatant.
- Wash the beads with Wash buffer 1: add 1 mL of Wash buffer 1 to the beads, mix well by inverting the tube, and rotate the sample for 5 min at 4 °C. Centrifuge the sample at 3,300 x g for 0.5 min at 4 °C, and remove the supernatant. Repeat this step three additional times.
- Wash the beads once with AGC solution containing 2 mM CaCl2: add 1 mL of AGC solution containing 2 mM CaCl2 to the beads, mix well by inverting the tube, and rotate the sample for 5 min at 4 °C. Centrifuge the sample at 3,300 x g for 0.5 min at 4 °C, and remove the supernatant.
- Treat the beads with Micrococcal nuclease (MNase): prepare the MNase reaction mixture described in Table 1. Resuspend the beads in 60 µL of the MNase reaction mixture by gentle pipetting. Shake the sample gently (20 to 25 rpm) on a turntable of a shaker set in a Peltier-type incubator for 15 min at 25 °C. NOTE: It is very important to use an undulating shaker and strictly follow the speed limit in this step. Other type of shakers, such as rotating mixers, are not recommended.
- Centrifuge the sample at 3,300 x g for 0.5 min at 4 °C, and remove the supernatant.
- Wash the beads twice with AGC solution containing 3 mM EGTA: add 1 mL of AGC solution containing 3 mM EGTA to the beads, mix well by inverting the tube, and rotate the sample for 5 min at 4 °C. Centrifuge the sample at 3,300 x g for 0.5 min at 4 °C, and remove the supernatant. Repeat this step once.
- Wash the beads once with Wash buffer 2: add 1 mL of Wash buffer 2to the beads, mix well by inverting the tube, and rotate the sample for 5 min at 4 °C. Centrifuge the sample at 3,300 x g for 0.5 min at 4 °C, and remove the supernatant. Keep the beads on ice while setting up RdRP reaction. CAUTION: Perform all procedures under RNase-free conditions.
- RdRP reaction
- Prepare a RdRP reaction mixture in a new tube as described in Table 2.
- Add 6 µL of [α-32P]UTP (3,000 Ci/mmol) to the mixture and mix well by pipetting. NOTE: [α-32P]ATP, [α-32P]CTP, or [α-32P]GTP can be used for the reaction instead of [α-32P]UTP.
- Add 1 µL of template RNA (1 µg/µL) to the mixture and mix well by pipetting. NOTE: To establish the RdRP assay, the following RNA template is recommended: 5'-GGGAUCAUGUGGGUCCUAUUACAUUUUAAACCCA-3' 9.
- Resuspend the beads with 20 µL of the RdRP reaction mixture from step 3.4.3 by pipetting.
- Shake the sample gently (20 to 25 rpm) on a turntable of a shaker set in a peltier-type incubator for 2 h at 32 °C.
- Add 5.4 µL of Proteinase K (20 mg/mL) and 45.4 µL of 2x Proteinase K buffer to the reaction mixture. Mix by pipetting and shake (20 to 25 rpm) the sample on a turntable set in a Peltier-type incubator for 30 min at 37 °C.
- Add 109.2 µL of RNase-free water to the sample.
- Add 200 µL of acid phenol-chloroform to the sample. Mix well by vortex, and then centrifuge the sample at 21,100 x g for 5 min at room temperature. Transfer the aqueous phase to a new tube. Repeat this step once.
- Add 20 µL of 3 M sodium acetate (pH = 5.2), 4 µL of precipitation carrier and 250 µL of ethanol to the aqueous phase. Shake the tube well for mixing, then centrifuge the sample at 21,100 x g for 20 min at 4 °C. Discard the supernatant.
- Wash the pellet with 300 µL of cold 70% (vol/vol) ethanol stored at -20 °C. Centrifuge the sample at 21,100 x g for 15 min at 4 °C.
- Discard the supernatant and air-dry the pellet. CAUTION: Perform all procedures under RNase-free conditions. NOTE: The pellet from step 3.4.11 can be stored at -20 °C for at least 2 days.
- RNase treatment
- Resuspend the pellet from step 3.4.11 in 20 µL of RNase-free water.
- Prepare a RNase reaction mixture in a new tube as described in Table 3.
- Add 180 µL of the RNase reaction mixture to the sample, and incubate the tube for 2 h at 37 °C.
- Add 2.3 µL of 10% (vol/vol) SDS to the sample and incubate for 15 min at 37 °C.
- Add 2 µL of Proteinase K (20 mg/mL) to the sample and incubate for 15 min at 37 °C.
- Add 205 µL of acid phenol-chloroform to the sample. Mix well by vortex, then centrifuge the sample at 21,100 x g for 5 min at room temperature. Transfer the aqueous phase to a new tube.
- Add 20 µL of 3 M sodium acetate (pH 5.2), 4 µL of precipitation carrier and 250 µL of ethanol to the aqueous phase. Shake the tube well for mixing, then centrifuge the sample at 21,100 x g for 20 min at 4 °C. Discard the supernatant.
- Wash the pellet with 300 µL of cold 70% (vol/vol) ethanol stored at -20 °C. Centrifuge the sample at 21,100 x g for 15 min at 4 °C.
- Discard the supernatant and air-dry the pellet. NOTE: The pellet from step 3.5.9 can be stored at -20 °C for at least 2 days.
- Gel electrophoresis and autoradiography
- Resuspend the sample with 20 µL of RNase-free water.
- Add 20 µL of 2x loading buffer.
- Boil the sample for 10 min at 95 °C. Crush the sample on the ice.
- Run the gel. In case the template size is 34 nts (see also step 3.4.3), 7 M Urea-10% polyacrylamide gel is used for denaturing gel electrophoresis.
- Dry the gel with a gel dryer.
- Expose the film to the dried-gel within an X-ray cassette with an intensifying screen at -80 °C.
Representative Results
With the recommended RNA template, radioactive RdRP products are observed between 20 to 30 nucleotides (nt) specifically in HeLa cells in mitotic phase after overnight exposure (Figure 1A). Typically, signal intensity of the RdRP products demonstrates two peaks that are positioned around 25 nt and 30 nt in consonance with the size distribution of the products confirmed by next generation sequencing9. In contrast to mitotic cells, HeLa cells without synchronization demonstrate almost absence of the 20–30 nt radioactive products. Oval signals, that are placed below 20 nt in all samples, are nonspecific drifts.
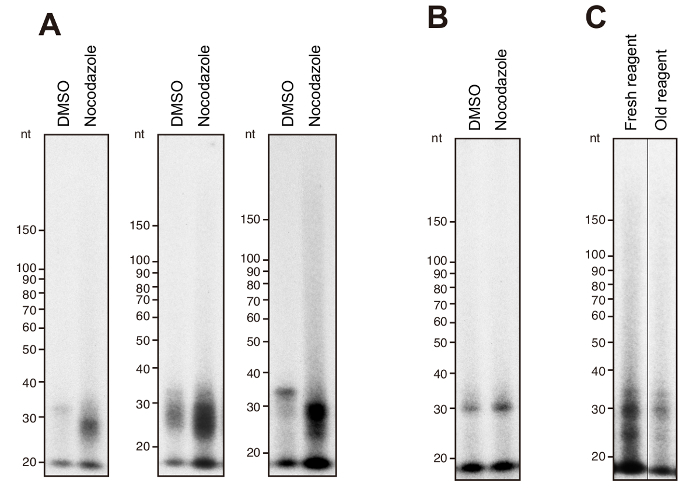
In case that there is only a ~30 nt product with weak signal in mitotic HeLa cells, it indicates that the RdRP reaction did not go well. For example, if MNase treatment is performed roughly and inappropriately, the signal intensity in mitotic HeLa cells decreases significantly (Figure 1B). RdRP reaction performed with old HMD solution also reduces the products (Figure 1C).
Figure 1: Representative RdRP products of endogenous TERT in mitotic HeLa cells. (A) Three representative results of the IP-RdRP assay in HeLa cells treated with nocodazole (manipulated) or DMSO (unmanipulated). The chemically synthesized 34 nt RNA was used as a template. (B) IP-RdRP assay in mitotic HeLa cells performed with inappropriate MNase treatment. (C) IP-RdRP assay in mitotic HeLa cells performed with either fresh or old HMD solution. Please click here to view a larger version of this figure.
| Reagents | Volume |
| Micrococcal Nuclease, 20 U/µL | 1 µL |
| Micrococcal Nuclease Buffer, 10x | 8 µL |
| RNase-free water | 51 µL |
Table 1: MNase reaction mix component.
| Reagents | Volume |
| CHAPS, 0.07% (wt/vol) | 6 µL |
| HMD solution | 2 µL |
| rATP, 80 mM | 0.5 µL |
| rGTP, 8 mM | 1 µL |
| rUTP, 0.4 mM | 1 µL |
| rCTP, 8 mM | 1 µL |
| RNase inhibitor, 40 U/µL | 0.5 µL |
| RNase-free water | 1 µL |
Table 2: RdRP reaction mix component.
| Reagents | Volume |
| RNase ONE Ribonuclease, 10 U/µL | 0.2 µL |
| Reaction Buffer, 10x | 20 µL |
| RNase-free water | 159.8 µL |
Table 3: Ribonuclease reaction mix component.
Discussion
The IP-RdRP assay is a sensitive method to detect RdRP activity of human TERT. TERT protein is highly expressed in mitotic HeLa cells, in which TERT forms the RdRP complex8,9,10. This suggests that mitotic HeLa cells are an optimal material to detect RdRP activity. In the protocol described above, mitotic and non-synchronized HeLa cells are included as a positive and a negative example, respectively. As shown in Figure 1A, RdRP assay products from the recommended RNA template in mitotic HeLa cells show broad radioactive signals between 20 to 30 nt. In addition to HeLa cells, we have performed the assay with different types of cell lines, and found that the signal pattern can be changed in different cell types9: some show a strong signal only around 30 nt, some show a similar pattern with HeLa cells. We have also performed the assay with RNA templates other than the 34 nt template8, short and long (~300 nt) RNAs with various sequences, and successfully obtained RdRP products from those templates although there may be some preference. For the first trial, however, we recommend using HeLa cells in mitotic phase and the 34 nt RNA template. RdRPs can generate double-stranded RNA both in a primer-dependent and in a primer-independent manner11. We have reported that TERT preserves this property as human RdRP7,9; TERT synthesizes dsRNA from an RNA template with 3'-foldback structure through a back-priming mechanism7. For the 34 nt RNA template, TERT synthesizes complementary strands without using primers9. To specifically detect RdRP products synthesized in a primer-independent manner, i.e. de novo synthesized RNA products, [α-32P]NTP can be replaced with [γ-32P]NTP in the RdRP reaction9.
MNase treatment of TERT immune complexes on beads is a critical step to achieve desired results. If the MNase treatment is performed too long or with intensive shaking, the RdRP products would be reduced remarkably. To avoid such a trouble, we strongly recommend to strictly follow the protocol carefully. HMD solution is another critical factor for success. If you find that the signals are very weak, replace the HMD solution to a newly prepared one.
Throughout the protocol, one should take great care to avoid RNase contamination. Ribonuclease treatment should be performed with dedicated equipment. After manipulating the Ribonuclease, one should discard tips and tubes with RNase immediately, eliminate RNase with specialized solution (e.g. RNase Quiet), and change groves.
TERT interacts with not only TERC but also many kinds of endogenous RNAs, and we have reported part of them7. Modification in the IP-RdRP assay, such as the IP-RdRP assay without MNase treatment, will provide a full list of endogenous RNA templates for TERT-associated RdRP activity and bioinformatic guide on their sequence features. We have successfully applied this protocol to tissue lysate. Because TERT is expressed in a variety of normal and tumor tissues, this assay might be useful to investigate non-canonical enzymatic function of TERT in human.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported in part by the Project for Development of Innovative Research on Cancer Therapeutics (P-DIRECT) (15cm0106102h0002, K.M.) and the Project for Cancer Research and Therapeutic Evolution (P-CREATE) (16cm01061150001, K.M.) from Japan Agency for Medical Research and Development, AMED; the Takeda Science Foundation (Y.M.); Grant of the Princess Takamatsu Cancer Research Fund (13-24520, Y.M.); and JSPS KAKENHI Grant Number JP16K07133 (Y.M.).
References
- Martinez P, Blasco MA. Telomeric and extra-telomeric roles for telomerase and the telomere-binding proteins. Nat Rev Cancer. 2011;11(3):161–176. doi: 10.1038/nrc3025. [DOI] [PubMed] [Google Scholar]
- Nakamura TM, et al. Telomerase catalytic subunit homologs from fission yeast and human. Science. 1997;277(5328):955–959. doi: 10.1126/science.277.5328.955. [DOI] [PubMed] [Google Scholar]
- Gillis AJ, Schuller AP, Skordalakes E. Structure of the Tribolium castaneum telomerase catalytic subunit TERT. Nature. 2008;455(7213):633–637. doi: 10.1038/nature07283. [DOI] [PubMed] [Google Scholar]
- Salgado PS, et al. The structure of an RNAi polymerase links RNA silencing and transcription. PLoS Biol. 2006;4(12):e434. doi: 10.1371/journal.pbio.0040434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castel SE, Martienssen RA. RNA interference in the nucleus: roles for small RNAs in transcription, epigenetics and beyond. Nat Rev Genet. 2013;14(2):100–112. doi: 10.1038/nrg3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama T, Cam H, Verdel A, Moazed D, Grewal SI. RNA-dependent RNA polymerase is an essential component of a self-enforcing loop coupling heterochromatin assembly to siRNA production. Proc Natl Acad Sci U S A. 2005;102(1):152–157. doi: 10.1073/pnas.0407641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maida Y, et al. An RNA-dependent RNA polymerase formed by TERT and the RMRP RNA. Nature. 2009;461(7261):230–235. doi: 10.1038/nature08283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maida Y, et al. Involvement of telomerase reverse transcriptase in heterochromatin maintenance. Mol Cell Biol. 2014;34(9):1576–1593. doi: 10.1128/MCB.00093-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maida Y, Yasukawa M, Masutomi K. De Novo RNA Synthesis by RNA-Dependent RNA Polymerase Activity of Telomerase Reverse Transcriptase. Mol Cell Biol. 2016;36(8):1248–1259. doi: 10.1128/MCB.01021-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto N, et al. Maintenance of tumor initiating cells of defined genetic composition by nucleostemin. Proc Natl Acad Sci U S A. 2011;108(51):20388–20393. doi: 10.1073/pnas.1015171108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk AA, Makeyev EV, Bamford DH. Initiation of viral RNA-dependent RNA polymerization. J Gen Virol. 2004;85(Pt 5):1077–1093. doi: 10.1099/vir.0.19731-0. [DOI] [PubMed] [Google Scholar]