

Abstract
Li-Fraumeni syndrome (LFS) is an autosomal dominant hereditary cancer disorder. Patients with LFS are predisposed to a various type of tumors, including osteosarcoma--one of the most frequent primary non-hematologic malignancies in the childhood and adolescence. Therefore, LFS provides an ideal model to study this malignancy. Taking advantage of iPSC methodologies, LFS-associated osteosarcoma can be successfully modeled by differentiating LFS patient iPSCs to mesenchymal stem cells (MSCs), and then to osteoblasts--the cells of origin for osteosarcomas. These LFS osteoblasts recapitulate oncogenic properties of osteosarcoma, providing an attractive model system for delineating the pathogenesis of osteosarcoma. This manuscript demonstrates a protocol for the generation of iPSCs from LFS patient fibroblasts, differentiation of iPSCs to MSCs, differentiation of MSCs to osteoblasts, and in vivo tumorigenesis using LFS osteoblasts. This iPSC disease model can be extended to identify potential biomarkers or therapeutic targets for LFS-associated osteosarcoma.
Keywords: Cancer Research, Issue 136, iPSCs, Li-Fraumeni syndrome, reprogramming, differentiation, mesenchymal stem cells, osteoblasts, osteosarcoma, xenograft
Introduction
Between 2006 and 2007, several breakthrough findings from the laboratories of Drs. Shinya Yamanaka and James A. Thomson led to the development of induced pluripotent stem cells (iPSCs)1,2,3. By reprogramming somatic cells with defined transcriptional factors to form iPSCs, researchers were able to generate cells with key characteristics namely, pluripotency, and self-renewal, which was previously thought to only exist in human embryonic stem cells (hESCs). iPSCs could be generated from any individual or patient and did not have to be derived from embryos, vastly expanding the repertoire of available diseases and backgrounds for the study. Since then, patient-derived iPSCs have been used to recapitulate the phenotype of various human diseases, from Alzheimer's disease4 and amyotrophic lateral sclerosis5 to long QT syndrome6,7,8.
These advances in iPSC research also have opened new avenues for the cancer research. Several groups recently have used patient iPSCs to model cancer development under a susceptible genetic background9,10,11, with successful application demonstrated to date in osteosarcoma9, leukemia10,11,12, and colorectal cancer13. Although iPSC-derived cancer models are still in their infancy, they have demonstrated great potential in phenocopying disease-associated malignancies, elucidating pathological mechanisms, and identifying therapeutic compounds14.
Li-Fraumeni syndrome (LFS) is an autosomal dominant hereditary cancer disorder caused by TP53 germline mutation15. Patients with LFS are predisposed to a various type of malignancies including osteosarcoma, making LFS iPSCs and their derived cells particularly well-suited to studying this malignancy16. An iPSC-based osteosarcoma model was first established in 2015 using LFS patient-derived iPSCs9 subsequently differentiated into mesenchymal stem cells (MSCs) and then to osteoblasts, the originating cells of osteosarcoma. These LFS osteoblasts recapitulate osteosarcoma-associated osteogenic differentiation defects and oncogenic properties, demonstrating the model potential as a "bone tumor in a dish" platform. Interestingly, genome-wide transcriptome analyses reveal aspects of an osteosarcoma gene signature in LFS osteoblasts and that features of this LFS gene expression profile are correlated with poor prognosis in osteosarcoma9, indicating the potential of LFS iPSCs disease models to reveal features of clinical relevance.
This manuscript provides a detailed description of how to use LFS patient-derived iPSCs to model osteosarcoma. It details the generation of LFS iPSCs, differentiation of iPSCs to MSCs and then to osteoblasts, and use of an in vivo xenograft model using LFS osteoblasts. The LFS disease model comprises several advantages, most notably the ability to generate unlimited cells at all stages of osteosarcoma development for mechanistic studies, biomarker identification, and drug screening9,14,16.
In summary, the LFS iPSC-based osteosarcoma model offers an attractive complementary system for advancing osteosarcoma research. This platform also provides a proof-of-concept for cancer modeling using patient-derived iPSCs. This strategy described below can be readily extended to model malignancies associated with other genetic disorders with cancer predispositions.
Protocol
This work was approved by The University of Texas Health Science Center at Houston (UTHealth) Animal Welfare Committee. The experiments are performed in strict accordance with the standards established by the UTHealth Center for Laboratory Animal Medicine & Care (CLAMC) which is accredited by American Association for Laboratory Animal Care (AAALAC International).The human subjects in this study fall under Scenario A ("No Human Subjects Research") as defined by the NIH SF424 documentation. Therefore, it does not need any approval by UTHealth human research ethics committee.
1. Generation of LFS iPSCs (Figure 1A)
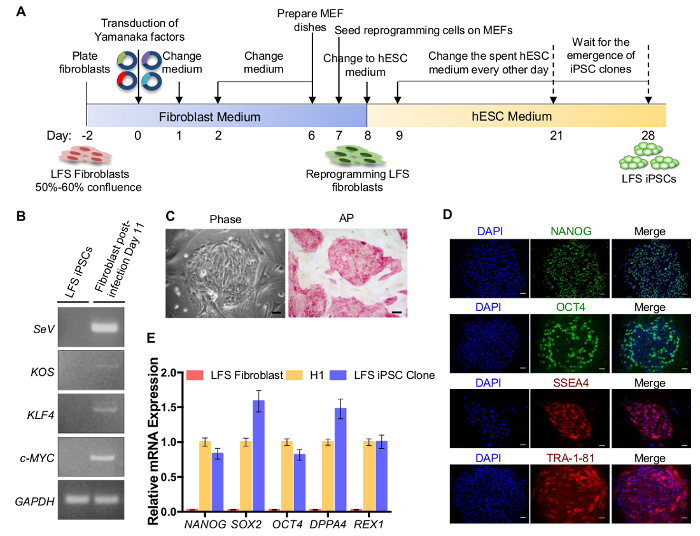
On day -2, plate 2 x 104 LFS patient fibroblasts into one well of a 6-well plate, and culture the cells with fibroblast medium (Table 1) at 37 °C in a humidified 5% CO2 incubator. Ensure that the fibroblasts reach 50 - 60% confluence on the day of transduction (Day 0).
Perform Sendai virus transduction on Day 0. To do this, replace the spent medium with pre-warmed fibroblast medium. Thaw the commercial Sendai virus reprogramming kit (see Table of Materials) on ice. Infect LFS fibroblasts with mixed Sendai viruses according to the manufacturer's instructions.
- Maintenance of the transduced cells in fibroblast medium (Day 1 - 6):
- On the day 1, 24 h after transduction, remove the fibroblast medium containing remaining Sendai virus and replace it with 2 mL fresh fibroblast medium. Culture the transduced fibroblasts at 37 °C in a humidified 5% CO2 incubator.
- Change the medium with the fibroblast medium every other day from the Day 2 - 6.
- Maintenance of the transduced cells in the feeder culture condition (Day 7 - 28):
- Prepare irradiated CF1 mouse embryonic fibroblast (MEF) culture dishes on Day 6. To do this coat six 100 mm tissue culture dishes by adding 5 mL of 0.1% gelatin to each plate at room temperature for 30 min. Aspirate the 0.1% gelatin after coating.
- Remove MEFs from liquid nitrogen container. Thaw MEFs using the commercially available thawing system following the manufacturer's instructions. Plate the cells on the gelatin-coated plates with MEF/MSC culture medium (Table 1) at a seeding density of around 6.7 x 105 cells per 100 mm tissue culture dish.
- Inoculate the transduced cells on MEF plates (Day 7): Trypsinize the transduced cells using 1 mL of 0.25% Trypsin-EDTA per 100 mm dish for 2 - 4 min at room temperature. Neutralize trypsin with 9 mL of fibroblast medium. Transfer cells to conical tubes and centrifuge cells at 230 x g at 37 °C for 4 min. Plate the transduced fibroblasts on the six MEF dishes prepared on Day 6 and culture them in 8 mL of fibroblast medium per dish. NOTE: The confluency of MEFs is around 20% before seeding transduced cell on MEF plates.
- On the day 8, discard the fibroblast medium and replace with hESC medium (Table 1).
- Wait for the emergence of iPS clones from Days 9 - 28 (Figure 1C). Change the spent hESC medium every other day and monitor the emergence of iPS clones under the microscope. Wait for iPSCs clones big enough to be observed by the naked eye and then proceed to expand iPS clones in feeder-free culture condition.
- Expansion of the emerged iPS clones in feeder-free culture condition (Day 29 - 79~99):
- To prepare the matrix-coated plate, coat a 48-well plate with 120 µL of the basement membrane matrix coating solution (Table 1) per well. Make sure the basement membrane matrix coating solution covers the well and coats the culture dish surface. Leave it at room temperature for 1 h. Aspirate the coating solution immediately before use and add 250 µL of hESC medium per well with 2 µM ROCK inhibitor Thiazovivin.
- On the Day 28, aspirate the spent hESC medium and wash iPS clones with 1x DPBS. Pick up visible iPS clones with a 1 mL pipette tip and transfer into a well of a treated 48-well plate (step 1.4.1). Seed one colony per well of 48-well plate.
- Centrifuge the plate at 230 x g for 4 min to facilitate the cell attachment. Culture iPS clones at 37 °C in a humidified 5% CO2 incubator.
- The following day (Day 29), remove the spent hESC medium and add 250 µL of the commercially available iPSC medium in each well.
- Maintain iPS clones at 37 °C in a humidified 5% CO2 incubator, and change the iPSC medium every other day.
- When iPS clones reach 85% confluence, subculture cells at a 1:10 ratio by culture area. Add 60 µL of the commercial passaging solution to detach the cells and incubate at 37 °C for 3 min.
- Seed half of the detached iPSCs on a basement membrane matrix-coated 12-well plate in 1 mL of hESC medium with 2 µM ROCK inhibitor Thiazovivin. Culture iPS clones in a 12-well plate with 1 mL of the commercially available iPSC medium per well and subculture iPSCs at a 1:10 ratio until they reach passage 10. NOTE: iPSCs are sub cultured every 5 - 7 days at a 1:10 ratio. It takes up to 10 weeks for iPS clones to reach passage 10. iPSC characterizations including cell morphology, alkaline phosphatase (AP) activity, and the expression of pluripotency factors and hESC markers, can be examined after confirming the removal of Sendai virus in iPSCs (usually demonstrated after around 10 passages) (Figure 1B-E). Antibody and primer information for iPSC characterization are included in Table 2 and 4.
2. Differentiation of LFS iPSCs to Mesenchymal Stem Cells (MSCs) (Figure 2A)
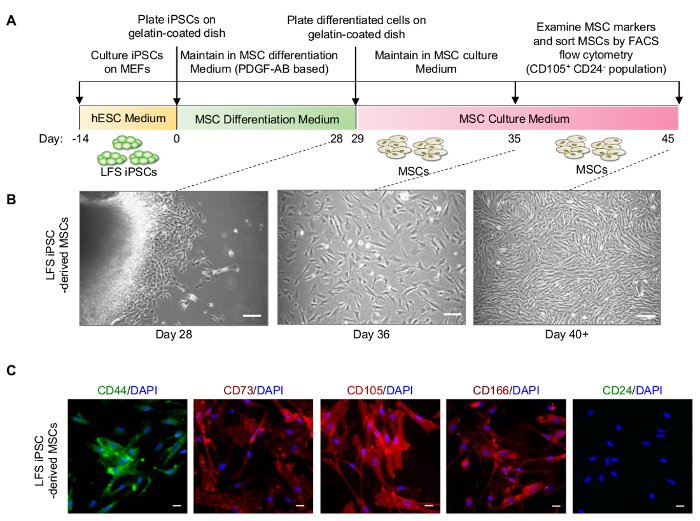
Culture iPSC clones on MEF plates for at least 2 weeks (Day ~ -14 - 0): Prepare the MEF dishes (100 mm tissue culture dishes) as previously described (1.3.1 - 1.3.2). Maintain iPSCs in hESC medium and change medium every other day. Whenever iPSCs reach 90% confluence, subculture iPSCs by 1:10 dilution.
- Removal of MEFs from iPSCs (Day 0):
- After maintaining iPSCs on MEFs for 2 weeks, culture iPSCs in 100 mm tissue culture dishes until cells reach 80 - 90% confluence. Remove the spent hESC medium and wash iPSCs with 5 mL of 1x DPBS. Add 1 mL of the cell detachment solution to detach the cells and incubate at room temperature for 3 min.
- Add 9 mL of MEF/MSC culture medium to the plate to neutralize the cell detachment solution activity and transfer the detached cells to a new 100 mm tissue culture dish without any coating.
- Incubate the cells at room temperature for 30 min to allow the MEFs to attach to the plate.
- Carefully collect the supernatant which contains unattached iPSCs, and centrifuge at 230 x g at 4 °C for 4 min. NOTE: Carelessly flushing the plate bottom leads to detachment of MEFs.
- Differentiation of iPSCs toward MSCs (Day 0 - 28):
- Coat three wells of a 6-well plate with 1 mL of 0.1% gelatin per well at room temperature for 30 min.
- Remove the supernatant of the collected iPSCs from 2.2.4.
- Resuspend iPSCs in 6 mL of MSC differentiation medium (Table 1) and seed cells in the three coated wells with 2 mL per well (Day 0).
- Change the MSC differentiation medium every two days to remove unattached and/or dead cells (Day 1 - 28).
- Maturation of iPSC-derived MSCs (Day 28 - 45):
- Remove the spent MSC differentiation medium and wash cells with 1 mL of 1x DPBS.
- Trypsinize MSCs with 0.5 mL of 0.25% Trypsin-EDTA per well at room temperature for 3 min.
- Neutralize trypsin by 1.5 mL of the MEF/MSC culture medium and collect MSCs from three wells into one 15 mL conical tube. Centrifuge at 230 x g at 4 °C for 4 min.
- Remove the supernatant and resuspend MSCs in 3 mL of MEF/MSC culture medium and plate on one 60 mm 0.1% gelatin-coated plate (Day 28).
- Culture MSCs in MEF/MSC culture medium and change medium every two days. When the cells reach 100% confluence, subculture MSCs at a 1:3 ratio using 0.25% Trypsin-EDTA. NOTE: The MSCs display a fibroblast-like morphology (Figure 2B). When MSCs grow in a high density, elongated MSCs can form a swirl-like pattern (Figure 2B).
- Perform the immunofluorescent staining to evaluate iPSC-derived MSCs by examining MSC surface markers CD44, CD73, CD105, and CD166 (Figure 2C). NOTE: The dilution of antibodies are shown in Table 2. Immunofluorescent staining of live cells is performed according to manufacturer's instructions.
- After confirmation, expand MSCs at a 1:3 ratio in 100 mm tissue culture dishes. Freeze down vials (3 vials per 100 mm tissue culture dish) using the freezing medium containing 10% DMSO and 90% FBS. NOTE: To enrich the MSC population, MSCs can be sorted using CD105+, CD24- criteria by flow cytometry9,17. iPSC-derived MSCs differentiated using the PDGF-AB-based method commonly can be maintained for 2 - 3 months in culture without loss of MSC identity9. Freeze MSCs from an early passage number after confirming MSC characteristics.
3. Differentiation of LFS MSCs to Osteoblasts (Figure 3A)
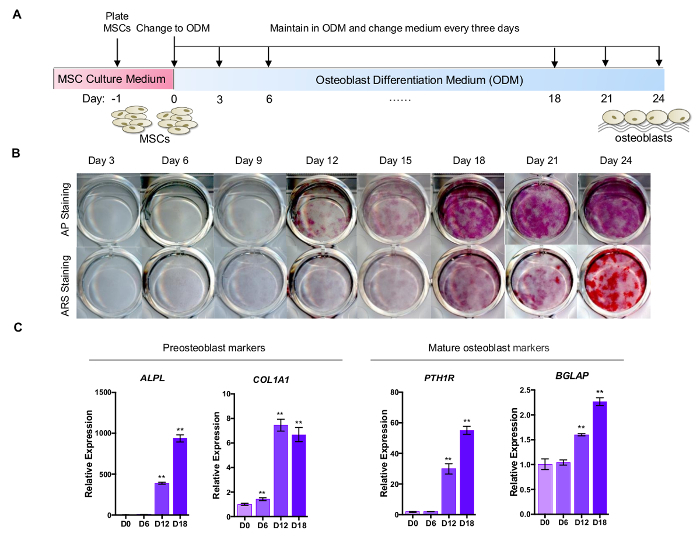
- Preparation of MSCs for the osteogenic differentiation (Day -1)
- To ensure an adequate quantity of cells to complete the osteogenic differentiation protocol, begin with MSCs cultured in 100 mm tissue culture dishes to 95% confluence. Remove the spent culture medium and wash MSCs with 5 mL of 1x DPBS.
- Trypsinize MSCs with 1 mL of 0.25% trypsin-EDTA per 100 mm dish at room temperature for 3 min. To ensure the cells are not over trypsinized, stop trypsinization when 50% of the cell detachment is observed under the microscope.
- Neutralize trypsin by 9 mL of MEF/MSC culture medium and collect MSCs from 3 wells into one 15 mL conical tube. Centrifuge at 230 x g at 4 °C for 4 min.
- Aspirate the supernatant, resuspend MSCs in 5 mL of MSC medium, and count the number of cells by a hemocytometer.
- Plate proper number of MSCs on a culture plate. NOTE: The detail of cell numbers of 12-well plate, 6 well-plate, and 100 mm tissue culture dishes are summarized in Table 3.
- Induction of osteogenic differentiation by osteoblast differentiation medium (ODM) (Table 1) (Day 0 - 24):
- Aspirate MEF/MSC culture medium and replace with the proper volume of ODM (1 mL, 2 mL, and 8 mL for each well of 12-well plate, each well of 6-well plate, and 100 mm tissue culture dish, respectively) to differentiate MSCs to osteoblasts (Day 0).
- Aspirate the spent ODM and replace with the proper volume of ODM every two days during the osteogenic differentiation process.
- Perform the alkaline phosphatase (AP) and the Alizarin Red S (ARS) staining to detect bone-associated alkaline phosphatase produced by preosteoblasts and mineral deposition produced by mature osteoblasts, respectively, on Day 3, 6, 9, 12, 15, 18, 21, and 24 (Figure 3B). NOTE: AP staining and ARS staining are performed using commercially available kit following manufacturer protocol.
- Examine the expression levels of preosteoblast and mature osteoblast markers (ALPL and COL1A1 for preosteoblasts; PTH1R and BGLAP for mature osteoblasts) by qRT-PCR (Figure 3C). NOTE: Positive AP staining is expected after Day 9 of osteogenic differentiation and positive ARS staining can be expected after Day 21 of osteogenic differentiation. The MSCs growing in MEF/MSC culture medium can be served as a negative control of AP staining and ARS staining.
4. The Xenograft Model to Study In Vivo Tumorigenesis of LFS MSC-derived Osteoblasts (Figure 4A)
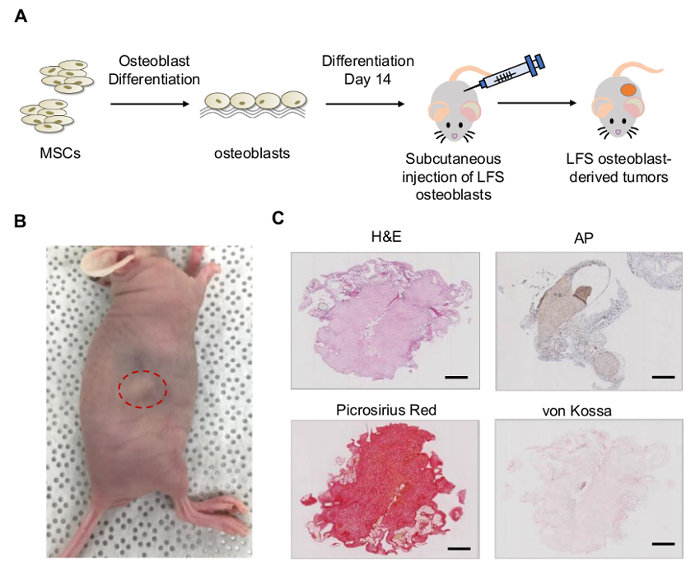
- Differentiation of LFS MSCs to osteoblasts:
- Seed 3 - 4.5 x 105 MSCs in 100 mm tissue culture dish. Prepare five dishes for one subcutaneous injection.
- Culture MSCs in ODM to differentiate MSCs to osteoblasts as described in 3.2 until Day 14.
- Preparation of differentiated osteoblasts for subcutaneous injection:
- To prepare osteoblast detachment solution (Table 1), dissolve 1 g type II collagenase in 1,000 mL of αMEM medium to make the collagenase II solution (1 mg/mL). Keep the collagenase II solution at -20 °C. Mix 0.25% Trypsin-EDTA and the collagenase II solution at a 1:1 ratio to make the osteoblasts detachment solution.
- Remove the spent ODM and wash differentiated osteoblasts with 5 mL of 1x DPBS per 100 mm dish.
- Add 1.5 mL of osteoblast detachment solution per 100 mm dish and incubate dishes at 37 °C for 30 min. Examine and ensure the osteoblast detachment by microscopy.
- Neutralize the osteoblasts detachment solution by ODM and collect the detached osteoblasts in a 50 mL conical tube. Centrifuge at 230 x g at 4 °C for 5 min.
- Remove the supernatant and resuspend cells in 25 mL of ODM medium. Pass the cells through a 70 µm cell strainer to remove aggregated cells. Count the cell numbers using a hemocytometer.
- Prepare 1 x 107 osteoblasts per subcutaneous injection, and centrifuge cells at 230 x g at 4 °C for 4 min.
- Resuspend the 1 x 107 osteoblasts in 50 µL ice-cold 1x DPBS. Mix differentiated osteoblasts with 50 µL of phenol-red free basement membrane matrix (osteoblasts suspension and basement membrane matrix were mixed at a 1:1 ratio) and maintain osteoblasts on ice before injection.
- In vivo xenograft tumor model of LFS MSC-derived osteoblasts:
- Anesthetize 8-week-old female immunocompromised NU/NU mice in a chamber supplying 5% (v/v) inhaled isoflurane in 1 L/min of oxygen (provided by CLAMC). After the animal becomes recumbent, switch the anesthetic delivery system to the nose cone, supplying 2% (v/v) inhaled isoflurane in 200mL/min of oxygen (provided by CLAMC). Monitor the depth of anesthesia by checking the corneal and pedal reflexes to make the mice are sufficiently anesthetized and are not suffering from discomfort during the procedure.
- Disinfect the area to be injected with alternating swabs of chlorhexidine and 70% isopropanol. Use 26 G needle to perform subcutaneous injection of basement membrane matrix-mixed LFS osteoblasts into one side of hind legs of 8-week-old immunocompromised NU/NU mice (Figure 4A).
- Monitor mice by performing weekly palpation at injection sites for growth of subcutaneous nodular densities. Tumor formation can be observed around 6-10 weeks after injection (Figure 4B).
- Euthanize the mice by carbon dioxide asphyxiation method followed by cervical dislocation. Dissect the developed tumors. Determine the tumor grade and the differentiation index, preosteoblasts, and mature osteoblasts by various staining methods (e.g. Hematoxylin and Eosin (H&E) staining, Picro Sirius Red staining, and von Kossa staining respectively).
Representative Results
This protocol presents the procedures including LFS iPSC generation, MSC differentiation, osteoblast differentiation, and in vivo tumorigenesis assay using LFS MSC-derived osteoblasts.
Scheme for the generation of LFS iPSCs from fibroblasts by using a commercially available Sendai virus reprogramming kit is shown in Figure 1A. Sendai virus-based delivery of the Yamanaka four factors is a non-integrating reprogramming method. Therefore, stable LFS iPSC clones should be free of Sendai virus genome and loss of exogenous OCT4, SOX2, KLF4, and c-MYC transgenes, which can be examined by RT-PCR using specific primers (Figure 1B). As it is suggested in the manufacturer's instructions, generation of Sendai virus-free iPSCs depends on both culture and passage conditions. Under the described culture conditions in this protocol, removal of Sendai virus in iPSCs usually can be detected after 10 passages (Figure 1B). The established LFS iPSC clones should exhibit the typical hESC morphology and show the positive AP activity (Figure 1C). LFS iPSCs highly express pluripotency factor mRNAs (NANOG, OCT4, SOX2, DPPA4, and REX1), comparable to hESC H1 line and much higher than parental fibroblasts (Figure 1D). The expression of pluripotency factors (NANOG, OCT4) and hESC markers (SSEA4 and TRA-1-81) can also be examined by immunofluorescent staining (Figure 1E).
iPSCs are maintained on MEFs for at least 14 days before initiating MSC differentiation (Figure 2A). Although a lot of cell death happen during MSC differentiation, masses of differentiated cells are visible on the cell culture plate and fibroblast-like MSCs at the edge of the cell masses can be observed at Day 28. (Figure 2B). After sub culturing differentiated cells in MEF/MSC culture medium, differentiated MSCs proliferate quickly and show fibroblast-like morphology around Day 35, and then gradually represent an elongated shape and form a swirl-like pattern at Day 40 (Figure 2B). Differentiated LFS MSCs express MSC markers, including CD44, CD73, CD105, and CD166 by immunofluorescence staining (Figure 2C).
Figure 3A outlines osteoblastic differentiation. When MSCs are subjected to osteogenic differentiation signals, the differentiated cells start to show positive alkaline phosphatase activity at Day 9 (Figure 3B). ARS staining can be used to detect mineral deposition produced by mature osteoblasts. The bright red color staining instead of brown color staining indicates the positive result of ARS staining, which can be observed after Day 21 (Figure 3B). The differentiating osteoblasts show increasing expression level of preosteoblast genes (ALPL and COL1A1) and mature osteoblast genes (BGLAP and PTH1R) during osteogenic differentiation.
In vivo xenograft model can be established by subcutaneously injecting LFS MSC-derived osteoblasts in NU/NU mice (Figure 4A). Tumors can be observed 6 - 10 weeks after subcutaneous injection (Figure 4B). The LFS osteoblast-derived tumors demonstrated immature osteoblast characteristics, positive AP activity (AP staining), positive collagen matrix deposition (picrosirius red staining) but negative mineralization (von Kossa staining) (Figure 4C).
Figure 1: iPSC generation from LFS patient fibroblasts. (A) Schematic diagram of iPSC generation. (B) RT-PCR detection of Sendai virus genome and transgenes (KOS (KLF4, OCT4, and SOX2), KLF4, and c-MYC) in the reprogrammed iPSC clone after 10 passages (left) and fibroblast post-infection Day 11 (right, positive control). LFS iPSC clone is Sendai virus free after 10 passages. GAPDH is shown as internal control. (C) Cell morphology of LFS iPSCs and AP staining. Scale bar, 50 µm (D) qRT-PCR of NANOG, SOX2, OCT4, DPPA4, and REX1 mRNA expression in LFS iPSCs. hESC H1 line and parental LFS fibroblasts are used as a positive and a negative control, respectively. The mRNA expression is normalized to GAPDH expression. The relative mRNA expression is adjusted to hESC H1 line as 1. (E) Immunostaining of hESC pluripotent transcription factors (NANOG and OCT4) and hESC surface markers (SSEA4 and TRA-1-81) in LFS iPSCs. Scale bar, 50 µm. The dilution of antibodies used in this study are shown in Table 2.The primer sequences are shown in Table 4. Please click here to view a larger version of this figure.
Figure 2: Differentiation of LFS iPSCs to MSCs. (A) Schematic diagram of PDGF-AB-induced MSC differentiation. (B) Cell morphology of LFS iPSC-derived MSCs at differentiation Day 28, Day 36, and Day 40+. Scale bar, 100 µm. (C) Immunofluorescence staining demonstrates that differentiated LFS MSCs exhibit CD44+, CD73+, CD105+, CD166+, and CD24- signature. Scale bar, 30 µm. The dilution of antibodies used in this study are shown in Table 2. Please click here to view a larger version of this figure.
Figure 3: Differentiation of LFS MSCs to osteoblasts. (A) Schematic diagram of osteogenic differentiation. (B) AP and ARS staining are performed at different time points (Day 3, 6, 9, 12, 15, 18, 21, and 24). Osteoblastic differentiation of LFS MSC-derived osteoblasts is expected to lead to positive AP staining around Day 9 and positive ARS staining at Day 21. (C) The qRT-PCR analysis demonstrates the increased expression of pre-osteoblast (ALPL and COL1A1) and mature osteoblast (PTH1R and BGLAP) genes during osteogenic differentiation. The mRNA expression is normalized to GAPDH expression. Expression levels were relative to cells at differentiation Day 0. The primer sequences are shown in Table 4. Please click here to view a larger version of this figure.
Figure 4:In vivo tumorigenesis of LFS MSC-derived osteoblasts. (A) Schematic diagram of tumor xenograft of LFS osteoblasts. (B) NU/NU mice bear LFS osteoblast-derived tumors after 10 weeks of subcutaneous injection. (C) LFS osteoblast-derived tumors were examined by H&E, AP, picrosirius red, and von Kossa staining for morphology, bone-associated AP, collagen, and mineral deposits, respectively. The LFS-derived tumors represent immature osteoblast characteristics showing positive AP activity, positive collagen, and negative mineral mineralization. Scale bar, 1 mm. Please click here to view a larger version of this figure.
| Fibroblast Medium (500 mL) | |
| DMEM | 440 mL |
| Heat-inactivated FBS | 50 mL |
| Antibiotics(Pen/Strep) (100x) | 5 mL |
| Nonessential Amino Acid (100x) | 5 mL |
| 2-Mercaptoethanol | 3.5 µL |
| MEF/MSC Culture Medium (500 mL) | |
| DMEM | 440 mL |
| Heat-inactivated FBS | 50 mL |
| Antibiotics(Pen/Strep) (100x) | 5 mL |
| L-Glutamine (100x) | 5 mL |
| hESC medium (500 mL) | |
| DMEM/F-12 | 384.5 mL |
| KnockOut Serum Replacement | 100 mL (total: 20%) |
| Nonessential Amino Acid (100x) | 5 mL |
| Antibiotics (Pen/Strep) (100x) | 5 mL |
| L-Glutamine (100x) | 5 mL |
| bFGF (10 µg/mL) | 500 µL |
| 2-Mercaptoethanol | 3.5 µL |
| MSC Differentiation Medium (500 mL) | |
| KnockOut DMEM/F-12 or DMEM/F-12 | 445 mL |
| KnockOut Serum Replacement | 50 mL |
| bFGF (10 µg/mL) | 500 µL (10 ng/mL) |
| PDGF-AB (25 µg/mL) | 200 µL (10 ng/mL) |
| Nonessential Amino Acid (100x) | 5 mL |
| 2-Mercaptoethanol | 3.5 µL |
| Osteoblast Differentiation Medium (ODM) (500 mL) | |
| αMEM | 395 mL |
| Heat-inactivated FBS | 50 mL |
| 10 mM β-Glycerophosphate | 50 mL (1.08 g in 50 mL αMEM) |
| 0.1 µM Dexamethasone (Light Sensitive) | 10 µL of 5 mM dexamethasone |
| 200 µM Ascorbic Acid (Light Sensitive) | 100 µL of 1 mM ascorbic acid |
| Antibiotics (Pen/Strep) (100x) | 5 mL |
| Matrigel coating solution (50 mL) | |
| Basement Membrane Matrix | 2 mL |
| DMEM/F-12 (pre-cold 4 °C) | 48 mL |
| Osteoblast Detachment Solution (50 mL) | |
| 0.25% Trypsin-EDTA | 25 mL |
| Collagenase II Solution (1 mg/mL) | 25 mL |
| Note: Prepare osteoblast detachment solution fresh right before use. Collagenase II solution is stored at -20 °C for up to 6 months. |
Table 1: Compositions of fibroblast medium, MEF/MSC culture medium, hESC medium, MSC differentiation medium, ODM, and osteoblast detachment solution.
| Antibody Name | Dilution |
| NANOG | 1:500 |
| OCT4 | 1:300 |
| SSEA-4 PE-conjugated | 1:600 |
| TRI-1-81 | 1:600 |
| Donkey Anti-Goat IgG | 1:500 |
| Goat Anti-Rabbit IgG | 1:500 |
| CD105 | 1:500 |
| CD44 | 1:500 |
| CD73 | 1:500 |
| CD166 | 1:500 |
| CD24 | 1:500 |
Table 2: Antibody dilution.
| Culture Plate | Seeding Density | Assay |
| 12-well Plate | 0.67x104 cells per well | Alkaline phosphatase staining (AP staining) |
| Alizarin red S staining (ARS staining) | ||
| 6-well Plate | 2x104 cells per well | RT-PCR detection |
| Preosteoblast markers: ALPL, COL1A1 | ||
| Mature osteoblast markers: PTH1R, BGLAP | ||
| 100 mm Dish | 3 - 4.5x105 cells per plate | In vivo tumorigenesis |
Table 3: Seeding density of MSCs for osteoblastic differentiation.
| Sendai Virus Primers | |
| Target | Primer Sets |
| SeV | Forward: GGATCACTAGGTGATATCGAGC |
| Reverse: ACCAGACAAGAGTTTAAGAGATATGTATC | |
| KOS (KLF4/OCT4/SOX2) | Forward: ATGCACCGCTACGACGTGAGCGC |
| Reverse: ACCTTGACAATCCTGATGTGG | |
| KLF4 | Forward: TTCCTGCATGCCAGAGGAGCCC |
| Reverse: AATGTATCGAAGGTGCTCAA | |
| c-MYC | Forward: TAACTGACTAGCAGGCTTGTCG |
| Reverse: TCCACATACAGTCCTGGATGATGATG | |
| RT-PCR Primers | |
| Target | Primer Sets |
| NANOG | Forward: TTTGTGGGCCTGAAGAAAACT |
| Reverse: AGGGCTGTCCTGAATAAGCAG | |
| SOX2 | Forward: AGAAGAGGAGAGAGAAAGAAAGGGAGAGA |
| Reverse: GAGAGAGGCAAACTGGAATCAGGATCAAA | |
| OCT4 | Forward: AACCTGGAGTTTGTGCCAGGGTTT |
| Reverse: TGAACTTCACCTTCCCTCCAACCA | |
| DPPA4 | Forward: GACCTCCACAGAGAAGTCGAG |
| Reverse: TGCCTTTTTCTTAGGGCAGAG | |
| REX1 | Forward: GCCTTATGTGATGGCTATGTGT |
| Reverse: ACCCCTTATGACGCATTCTATGT | |
| ALPL | Forward: GGGACTGGTACTCAGACAACG |
| Reverse: GTAGGCGATGTCCTTACAGCC | |
| COL1A1 | Forward: GTGCGATGACGTGATCTGTGA |
| Reverse: CGGTGGTTTCTTGGTCGGT | |
| PTH1R | Forward: AGTGCGAAAAACGGCTCAAG |
| Reverse: GATGCCTTATCTTTCCTGGGC | |
| BGLAP | Forward: GGCGCTACCTGTATCAATGG |
| Reverse: GGCGCTACCTGTATCAATGG |
Table 4: Primer information.
Discussion
To achieve higher MSC differentiation efficiency, several aspects are critical. One is the culture condition of iPSCs before initiating MSC differentiation. The protocol presented in the manuscript is based on previous studies 9,17. iPSCs need to be cultured on MEFs for at least 2 weeks. Maintaining iPSCs in good conditions on MEFs are critical for cells to attach on the gelatin-coated plate for MSC differentiation. Another important aspect is the density of iPSCs on MEFs before differentiation. 80 - 90% confluency of iPSCs is preferred to initiate MSC differentiation. The overgrowth of iPSCs will impair cell survival during the differentiation process. The last critical point is the seeding density when starting MSC differentiation. In our hands, high cell density promotes iPSC attachment to the gelatin-coated plate. One 100 mm dish iPSCs can be seeded into three wells of 6-well plate. Initiation of MSC differentiation by directly resuspending and plating iPSCs in MSC differentiation medium produces great stresses on iPSCs, therefore occasionally resulting in severe cell death after seeding. High density of iPSCs increases the success rate of MSC differentiation.
Unlike MSC differentiation, the osteoblastic differentiation process is more straightforward. To achieve reproducible differentiation results, ensure the initiating MSC numbers are consistent. Results of osteoblastic differentiation from the same cell line may vary from batch to batch, therefore, setting up differentiation for different lines at the same time will give more comparative results among groups. The increase of MSC numbers shorten the differentiation process indicated by positive AP and ARS staining at an earlier time point. Also, ensure the change of ODM medium is handled in a gentle way and powerful vacuum suction system is not recommended in the late differentiation stage (Day 18 - 24) due to the potential detachment of aggregated osteoblasts during culture process.
The differentiated osteoblasts used for in vivo xenograft experiment are osteoblasts at the Day 14 differentiation time point. The osteoblasts later than Day 14 may aggregate and be difficult to dissociate due to the huge accumulations of collagens and other bone matrix materials produced by osteoblasts. To prevent the difficulty of osteoblast dissociation, LFS MSCs can be seeded in 2 - 3-fold higher density in the initiation step of osteoblastic differentiation to facilitate osteoblast differentiation. LFS MSC-derived osteoblasts can be dissociated and collected at the Day 6 - 10 differentiation time point for in vivo tumorigenesis assay at the Day 6 - 7 differentiation time point. The LFS xenograft tumor model demonstrates LFS MSC-derived osteoblasts recapitulate in vivo tumorigenic ability, which provides an alternative platform to study LFS-associated osteosarcoma.
In summary, an LFS iPSC-based osteosarcoma model provides a valuable recourse for osteosarcoma research. In addition to osteosarcoma, LFS patients suffer from various other types of cancers such as soft tissue sarcoma, breast cancer, and brain tumor. Therefore, LFS iPSC-based disease models can be extended to model other LFS related malignancies. Combining LFS patient-derived iPSCs and engineered mutant p53 (mutp53) hESCs18, LFS disease model also has great value in delineating pathogenesis of mutp53 associated malignancies and developing novel therapeutic strategies targeting oncogenic p5316.
Disclosures
The authors have nothing to disclose.
Acknowledgments
R. Z. is supported by UTHealth Innovation for Cancer Prevention Research Training Program Pre-Doctoral Fellowship (Cancer Prevention and Research Institute of Texas grant RP160015). J.T. is supported by the Ke Lin Program of the First Affiliated Hospital of Sun Yat-sen University. D.-F.L. is the CPRIT scholar in Cancer Research and supported by NIH Pathway to Independence Award R00 CA181496 and CPRIT Award RR160019.
References
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Yu J, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318(5858):1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- Yagi T, et al. Modeling familial Alzheimer's disease with induced pluripotent stem cells. Hum Mol Genet. 2011;20(23):4530–4539. doi: 10.1093/hmg/ddr394. [DOI] [PubMed] [Google Scholar]
- Dimos JT, et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. 2008;321(5893):1218–1221. doi: 10.1126/science.1158799. [DOI] [PubMed] [Google Scholar]
- Moretti A, et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med. 2010;363(15):1397–1409. doi: 10.1056/NEJMoa0908679. [DOI] [PubMed] [Google Scholar]
- Itzhaki I, et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature. 2011;471(7337):225–229. doi: 10.1038/nature09747. [DOI] [PubMed] [Google Scholar]
- Carvajal-Vergara X, et al. Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature. 2010;465(7299):808–812. doi: 10.1038/nature09005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DF, et al. Modeling familial cancer with induced pluripotent stem cells. Cell. 2015;161(2):240–254. doi: 10.1016/j.cell.2015.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulero-Navarro S, et al. Myeloid Dysregulation in a Human Induced Pluripotent Stem Cell Model of PTPN11-Associated Juvenile Myelomonocytic Leukemia. Cell Rep. 2015;13(3):504–515. doi: 10.1016/j.celrep.2015.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotini AG, et al. Functional analysis of a chromosomal deletion associated with myelodysplastic syndromes using isogenic human induced pluripotent stem cells. Nat Biotechnol. 2015;33(6):646–655. doi: 10.1038/nbt.3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotini AG, et al. Stage-Specific Human Induced Pluripotent Stem Cells Map the Progression of Myeloid Transformation to Transplantable Leukemia. Cell Stem Cell. 2017;20(3):315–328. doi: 10.1016/j.stem.2017.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo M, et al. Colonic organoids derived from human induced pluripotent stem cells for modeling colorectal cancer and drug testing. Nat Med. 2017;23(7):878–884. doi: 10.1038/nm.4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingold J, Zhou R, Lemischka IR, Lee DF. Modeling Cancer with Pluripotent Stem Cells. Trends Cancer. 2016;2(9):485–494. doi: 10.1016/j.trecan.2016.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YH, et al. Osteosarcoma: Molecular Pathogenesis and iPSC Modeling. Trends Mol Med. 2017;23(8):737–755. doi: 10.1016/j.molmed.2017.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, et al. Li-Fraumeni Syndrome Disease Model: A Platform to Develop Precision Cancer Therapy Targeting Oncogenic p53. Trends Pharmacol Sci. 2017;38(10):908–927. doi: 10.1016/j.tips.2017.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian Q, et al. Derivation of clinically compliant MSCs from CD105+, CD24- differentiated human ESCs. Stem Cells. 2007;25(2):425–436. doi: 10.1634/stemcells.2006-0420. [DOI] [PubMed] [Google Scholar]
- Zhou R, et al. A homozygous p53 R282W mutant human embryonic stem cell line generated using TALEN-mediated precise gene editing. Stem Cell Res. 2018;27:131–135. doi: 10.1016/j.scr.2018.01.035. [DOI] [PubMed] [Google Scholar]