

Abstract
Cells need to be able to regenerate their parts to recover from external perturbations. The unicellular ciliate Stentor coeruleus is an excellent model organism to study wound healing and subsequent cell regeneration. The Stentor genome became available recently, along with modern molecular biology methods, such as RNAi. These tools make it possible to study single-cell regeneration at the molecular level. The first section of the protocol covers establishing Stentor cell cultures from single cells or cell fragments, along with general guidelines for maintaining Stentor cultures. Culturing Stentor in large quantities allows for the use of valuable tools like biochemistry, sequencing, and mass spectrometry. Subsequent sections of the protocol cover different approaches to inducing regeneration in Stentor. Manually cutting cells with a glass needle allows studying the regeneration of large cell parts, while treating cells with either sucrose or urea allows studying the regeneration of specific structures located at the anterior end of the cell. A method for imaging individual regenerating cells is provided, along with a rubric for staging and analyzing the dynamics of regeneration. The entire process of regeneration is divided in three stages. By visualizing the dynamics of the progression of a population of cells through the stages, the heterogeneity in regeneration timing is demonstrated.
Keywords: Developmental Biology, Issue 136, Stentor coeruleus, ciliates, protists, membranellar band, oral apparatus, regeneration, single-cell assays
Introduction
Cells are not simple bags of enzymes, but rather highly complex machines whose components are carefully scaled to the correct size and arranged in well-defined positions. The morphogenesis of individual cells represents a key process in cell and developmental biology, but its molecular mechanism is unknown1,2. While some cultured cells resemble blobs, unicellular organisms can have extremely complicated architectures, exemplified by the complex cortical patterns seen in ciliates3,4.
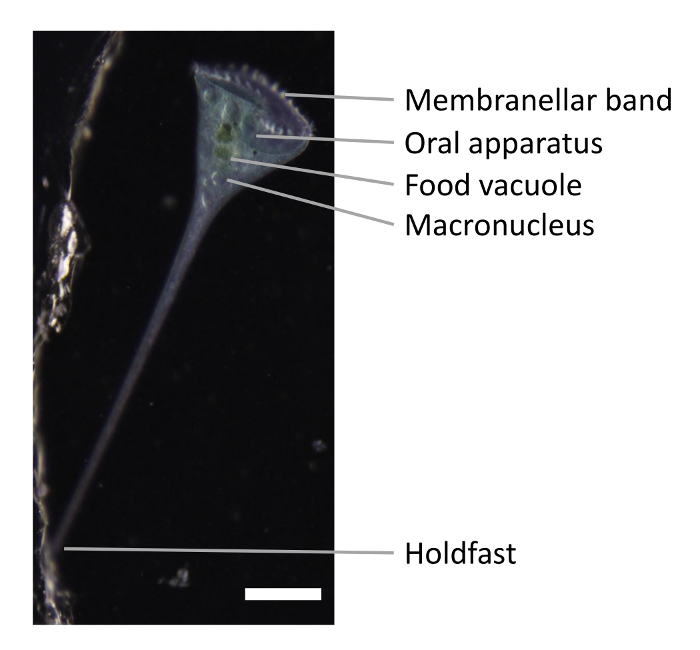
Perhaps the most extreme example of a highly structured cell is Stentor coeruleus, a giant heterotrichous ciliate distantly related to Tetrahymena and Paramecium. Stentor is 1 mm long and is covered with more than 100 longitudinal stripes of blue pigment alternating with rows of cilia organized by parallel stacks of microtubule ribbons that run the length of the entire cell. The cell is trumpet-shaped (Figure 1), with a membranellar band and an oral apparatus (OA) at its anterior end, and a holdfast that attaches the cell to the substrate at its posterior end. In addition to the clear anterior-posterior polarity, the cell also shows a distinctive chiral patterning, such that spacing between ciliary rows gradually increases in a clockwise direction. This results in a discontinuity where the narrowest row meets the widest row, and this region of the cell surface, known as the locus of stripe contrast, can induce the formation of the second set of anterior end structures when grafted onto another cell5, making it formally equivalent to Spemann's Organizer. Thus, all key processes of developmental biology have their analogs in Stentor: axiation, pattern formation, and induction. In an embryo, these processes are driven by fate differences between different cells, but in Stentor, they must be driven by fate differences between different regions within a single cell. What defines the differences between the regions within Stentor is a mystery.
If any part of Stentor is cut off, the missing piece of the cell can regenerate to yield a normal cell in a matter of hours. If a cell is cut in half, or even into much smaller pieces, each piece reorganizes into a normal-looking but smaller cell and restores proper proportionality between cell parts6,7. Even tiny fragments, 1/64th the size of the original cell, are able to regenerate into a small but normally proportioned cell, and then grow to the full size6. Stentor thus presents a unique opportunity to study the mechanisms of organelle size scaling and cell growth regulation using surgical methods that are usually applied at the level of tissues or whole organisms.
One of the properties of Stentor that allows it to regenerate from a wide range of surgical operations is that it contains a single nodulated macronucleus (Figure 1) with about 50,000 copies of the entire genome8. As long as a cell fragment contains at least one macronuclear node, it has the ability to regenerate fully. Another property underlying Stentor's regeneration ability is its prodigious wound-healing ability. Although many cell types are capable of healing their wounds9, Stentor is able to recover from an extraordinary range of physical perturbations. An example of Stentor recovery from a drastic perturbation, along with the methods for visualizing cytoplasmic flow in Stentor10 were previously reported. These methods allow the study of how wounding and subsequent regeneration affect the physical state of the cytoplasm.
Stentor's huge size, extraordinary regeneration ability, and the fact that it manifests many of the developmental phenomena seen in multicellular embryos (such as organizers, axiation, and patterning) attracted many developmental biologists during the turn of the last century, including Thomas Hunt Morgan7. During the 50's and 60's, microsurgical approaches demonstrated a startling array of regenerative and morphogenetic processes in this single-celled organism11. However, Stentor has been developed as a molecular biology model system only recently. During the past several years, the genome of Stentor was sequenced and assembled8, and the method to perturb gene expression using RNAi by feeding was developed12.
One of the reasons that Stentor was developed into a model organism for modern molecular biology only recently was the difficulty of growing large cultures due to its long cell cycle (3 to 5 days). However, modern genomic and proteomic methods require less material than they used to, and the volume of a single Stentor cell is sufficient for these methods, even without resorting to ultrasensitive methods that were developed for the analysis of single cells that are much smaller than Stentor. Section 1 of the protocol details the procedure for establishing a large culture from a single Stentor cell. The same approach can be used to establish a large culture from a cell fragment obtained by cutting a cell. Section 1 also provides the guidelines for maintaining healthy Stentor cultures over long periods of time. Section 2 of the protocol provides the methodology for inducing cell regeneration by cutting the cells manually with a glass needle. Section 3 of the protocol is dedicated to two methods of inducing the regeneration of specific cell structures (membranellar band and oral apparatus): treating the cells with either sucrose or urea leads to the shedding of these structures, followed by their regeneration. Section 4 of the protocol details a method for the imaging of individual regenerating cells over long periods of time. Section 4 ends with the description of the stages of regeneration and tips on the analysis of regeneration dynamics.
Protocol
1. Culturing Stentor and Establishing Stentor Cultures from Single Cells or Cell Fragments
- Prepare Chlamydomonas reinhardtii culture to be used as food for Stentor.
- Obtain Chlamydomonas reinhardtii cells from a commercial supplier (Table of Materials).
- Establish a 500 mL liquid culture of Chlamydomonas reinhardtii in commercially available TAP media using sterile technique13.
- Keep the Chlamydomonas culture under a lamp at a concentration near saturation (at O.D. of about 1) by diluting it with TAP media twice a week. Note: The Chlamydomonas culture can be grown on a shaker.
- Regularly check whether the Chlamydomonas culture is healthy by placing a drop of culture on a slide, covering it with a coverslip, and checking it under a microscope at 40X magnification. Note: Do not use the culture for Stentor feeding if it is contaminated with bacteria or if Chlamydomonas cells are aggregated into clusters. If either of these problems occurs, start a new Chlamydomonas culture.
Obtain Stentor coeruleus cells from a commercial supplier (Table of Materials).
- If Stentor cells are needed from their natural habitat, collect them from a pond, lake, or river11.
- To collect Stentor from a pond, lake, or river, find an area with some vegetation where the water is relatively calm, shady, and clear. Note: There is a higher probability of finding Stentor at the locations where duckweed grows.
- Collect at least 2 L of water in a container that is easy to pour from.
- After detritus and particulate matter have settled to the bottom of the container, gently fill a few smaller containers to examine for Stentor, waiting for a few seconds after each collection for the detritus to settle in the large container.
- Once the collection of samples is completed at a location, move to a new location that is at least 10 m away and repeat the collection of samples there. Note: Not every pond has a Stentor population, so multiple ponds may have to be sampled.
- Return with the samples to the lab and transfer the water from the containers into individual Petri dishes.
- In each Petri dish, search for Stentor under a stereomicroscope with oblique light at 5X magnification. Transfer individual Stentor cells using a 1 mL pipette into a well of a glass spot plate containing at least 100 µL of commercially available pasteurized spring water (PSW). Note: Stentor cells have a trumpet-like shape when they are attached to a substrate (Figure 1). Swimming Stentor cells are less extended than the cells attached to a substrate (Video 1).
- Wash Stentor in PSW at least 3x. Perform the washes by removing about 90% of the water from the well while keeping Stentor cells in the well, followed by adding 500 µL of PSW into the well. Note: Before handling Stentor using pipettes with pipette tips of 10 µL or smaller, cut about 0.5 mm off the end of the pipette tip with scissors to avoid wounding the cells due to shear forces that are generated as a large cell flows through too small of a tip opening.
- Prepare Chlamydomonas before each feeding of Stentor.
- Transfer 1 mL of Chlamydomonas culture prepared as in Step 1.1 into a 1.5 mL microcentrifuge tube. Centrifuge at 2,095 x g for 3 min.
- Remove the supernatant and resuspended the pellet in 1 mL of PSW. Centrifuge at 2,095 x g for 3 min. Remove the supernatant and resuspend the pellet in 500 µL of PSW. Note: Thus, washed and concentrated Chlamydomonas will be referred to as “prepared Chlamydomonas” in the following steps. TAP media is detrimental to Stentor, thus washing Chlamydomonas before feeding Stentor is important.
- If a clonal Stentor culture is needed, start the culture from an individual Stentor cell or a cell fragment.
- Since single Stentor cells do not grow well in PSW, prepare conditioned media by filter sterilizing 500 µL of media from an existing, healthy Stentor culture.
- Transfer 500 µL of conditioned media to one of the wells of a glass spot plate.
- Transfer one Stentor into the well of the glass spot plate containing conditioned media. Use as little medium as possible to make the transfer.
- Feed each Stentor 5 µL of prepared Chlamydomonas every 48 h. When Stentor divide, count the number of cells in the well and add 5 µL of prepared Chlamydomonas per cell.
- Keep Stentor in a shaded place since the cells are sensitive to light (for example, in clear plastic boxes covered with paper towels).
- Exchange Stentor medium in the well with fresh conditioned medium every 96 h.
- Prepare fresh conditioned media as in Step 1.5.1.
- Make all Stentor cells detach from the bottom of the well by gently pipetting the liquid up and down in the well.
- Carefully aspirate the liquid from the well using a 1 mL pipette, making sure all the cells remain in the well. Add 500 µL of fresh conditioned media to the well.
- When the number of cells in the well exceeds 20, move the cells to a larger container, for example, a wide-mouth glass jar.
- Add 20 mL of PSW into an autoclaved wide-mouth glass jar. Note: Autoclaving of glassware for Stentor can be replaced with careful washing and rinsing.
- Carefully pipette the media up and down in the well to detach all the cells from the bottom of the well. Collect all Stentor cells using a 1 mL pipette tip and gently transfer them to the glass jar.
- Do not tighten the lids on the jars with Stentor cultures, to allow sufficient access to air.
- Add PSW to the jar every 48 h to keep Stentor density at about 20 cells/mL. Estimate the density of cells by eye. Note: Alternatively, use a 1 mL pipette to bubble air into the jar to make cells detach from the walls, pipette up 1 mL of the culture, and count the number of cells in the pipette tip.
- Every 48 h, feed prepared Chlamydomonas to Stentor cultures in jars (see Step 1.4). Start with feeding the culture with 200 µL of prepared Chlamydomonas. As the volume of the culture increases, gradually increase the amount of prepared Chlamydomonas used for feeding up to 1 mL.
- When the culture volume reaches about 90% of the jar’s capacity, transfer the culture to a bigger container.
- Pipette the culture inside the jar, up and down, with a 1 mL pipette to detach Stentor from the glass.
- Pour the entire contents of the jar into a 2-cup glass container.
- Rinse the jar with about 25 mL of PSW into the 2-cup glass container to collect the remaining Stentor.
- Maintain healthy cultures in 2-cup glass containers.
- Feed Stentor cultures 2 mL of prepared Chlamydomonas per 100 mL of culture every 4 - 5 days. Add PSW to the glass container every 4 - 5 days to keep Stentor density at about 20 cells/mL. Note: 450 mL is the maximum volume a 2-cup container holds.
- Once a week, inspect the cultures under a 5X dissecting microscope for rotifers, fungus, and other growth. To prolong the health of the culture, remove contaminating microorganisms along with abnormally shaped and colorless Stentor cells using a 1 mL pipette.
- When the glass container is about 90% full, split the culture.
- Add 25 mL of PSW to the glass container. Use a 25 mL pipette to pipette up and down to detach Stentor from the glass. Move about 50% of the culture into a new 2-cup glass container.
- Add 25 mL of PSW to both cultures and continue maintaining the two cultures as described in this section of the protocol. Note: Since Stentor cells are sensitive to high temperature, maintain the temperature at 25 °C or lower in the room where the cultures are kept. Alternatively, the cultures can be kept in an incubator. Refer to Table 1 for troubleshooting of Stentor culturing.
2. Inducing Regeneration by Cutting Stentor Cells
Use a needle puller to make several needles from capillary tubes using the program as follows: heat - 735, pull - 100, velocity - 110, time - 150, pressure - 400.
- Prepare a 4% methylcellulose solution in 50 mL of PSW.
- Add 1 g of methylcellulose (viscosity: 1500 cP) to 50 mL of PSW.
- Incubate at 4 °C for at least 8 h to facilitate the dissolution of methylcellulose.
- Keep 4% methylcellulose solution at room temperature.
- Prepare a glass spot plate for storing the cell fragments after performing the cuts.
- Filter sterilize media from a healthy culture, 500 mL per glass spot plate well needed.
- Transfer 500 mL of sterilized media in each of the wells needed.
Collect one healthy Stentor (having a defined trumpet shape, vibrant blue-green color, and no large vacuoles) in a 2 µL droplet and place it on a coverslip or slide. Add 2 µL of 4% methylcellulose (Figure 2). Let Stentor slow down before cutting it (Video 2).
Hold the glass needle as parallel to the cutting surface as possible to prevent breaking the needle.
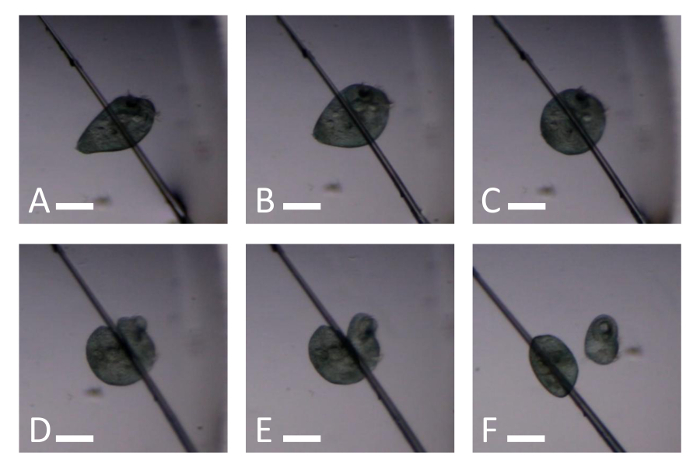
Using a stereo dissecting microscope, locate the tip of the glass needle and move it closer to the cell (Figure 3A). Observe the Stentor contract upon contact with the needle (Figure 3B and C). Note: If the cell is in an orientation that makes separating the anterior end from the posterior end difficult, rotate it very gently with the side of the needle.
Gently press on the contracted Stentor with the side of the glass needle to cut the cell in two (Figure 3D and E, Video 3). Move the two fragments apart ensuring that there is no cytoplasmic connection between them, to avoid fragment fusion (Figure 3F).
Check whether both fragments have at least one macronuclear node each by examining the fragments under a dissecting microscope with oblique illumination. Note: Having at least one macronuclear node is essential for cell survival. The cell may shed its pellicle (transparent shell) along with the blue-green pigment during or after the cut. In most cases, this will not affect the long-term viability of the cell.
Move the fragments into wells of a glass spot plate prepared in Step 2.3.
Cut multiple cells because a fraction of the cut cells will not regenerate.
If performing multiple cuts in one session, replace the glass needle when it becomes heavily coated with Stentor residue or when its tip is broken. Alternatively, clean the needle by wiping it gently on a piece of silicon spacer. Note: Glass needles can be used for multiple cell cutting sessions.
Keep the glass spot plate with cell fragments in a humidity chamber.
Proceed to Section 4 of the protocol for details on imaging and interpretation of Stentor regeneration results.

3. Inducing Regeneration of Membranellar Band and Oral Apparatus by Sucrose or Urea Treatment
- Membranellar band and oral apparatus removal using sucrose
- Prepare a 25% solution of sucrose in PSW.
- Add 500 µL of 25% sucrose in PSW to a microcentrifuge tube with a snap cap.
- Prepare 3 microcentrifuge tubes with snap caps with 1 mL of PSW in each tube for washing the cells after sucrose treatment.
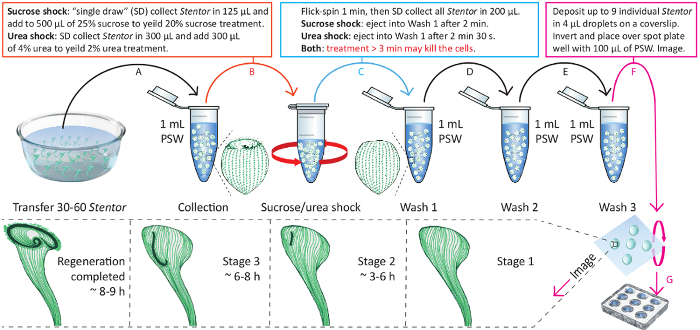
- Collect 30 - 60 Stentor cells in 1 mL of their culture media into a separate microcentrifuge tube using a 1 mL pipette (Figure 4, arrow A).
- Collect all the cells from the tube in 125 µL final volume using a 200 µL pipette in a single draw. Transfer them to the tube with 500 µL of 25% sucrose (prepared in Step 3.1.2) to obtain 625 µL of 20% sucrose solution (Figure 4, arrow B). Start a stopwatch.
- Incubate Stentor in this 20% sucrose solution and flick-spin the microcentrifuge tube in the rack for 1 min.
- Collect all the cells in a single draw (adjust the pipette to 200 µL max capacity for easier collection).
- Keep the cells in the pipette tip until the stopwatch shows 2 min of sucrose treatment.
- Eject Stentor into one of the microcentrifuge tubes prepared in Step 3.1.3 (Figure 4, arrow C). Flick-spin the tube in the rack.
- Wash the cells two more times, once in each of the two remaining microcentrifuge tubes containing PSW prepared in Step 3.1.3 (Figure 4, arrows D and E). Note: Single draw cell collection technique is not important in between the washes.
- Proceed to Section 4 of the protocol for details on imaging and interpretation of Stentor regeneration dynamics (Figure 4, arrows F and G).
- Membranellar band and oral apparatus removal using urea
- Prepare a solution of 4% urea in PSW.
- Add 300 µL of 4% urea in PSW to a microcentrifuge tube with a snap cap.
- Prepare 3 microcentrifuge tubes with snap caps containing 1 mL of PSW in each tube for washing the cells after urea treatment.
- Collect 30 - 60 Stentor cells in 1 mL of their culture media into a separate microcentrifuge tube using a 1 mL pipette (Figure 4, arrow A).
- Collect all the cells from the tube in 300 µL final volume using a 1 mL pipette in a single draw. Transfer them to the tube with 300 µL of 4% urea (prepared in Step 3.2.2) to obtain 600 µL of 2% urea solution (Figure 4, arrow B). Start a stopwatch.
- Incubate Stentor in this 2% urea solution and flick-spin the microcentrifuge tube in the rack for 1 min.
- Collect all the cells in a single draw.
- Keep the cells in the pipette tip until the stopwatch shows 2 min of urea treatment.
- Eject Stentor into one of the microcentrifuge tubes prepared in Step 3.2.3 (Figure 4, arrow C). Flick-spin the tube in the rack.
- Wash the cells two more times, once in each of the two remaining microcentrifuge tubes containing PSW prepared in Step 3.2.3 (Figure 4, arrows D and E). Note: Single draw cell collection technique is not important in between the washes.
- Proceed to Section 4 of the protocol for details on imaging and interpretation of Stentor regeneration dynamics (Figure 4, arrows F and G).
4. Imaging and Analyzing Cell Regeneration
- If using an upright microscope, use the hanging droplet method to image regeneration of individual cells.
- Put 100 µL of PSW in a well of a glass spot plate.
- Isolate 1 Stentor cell in 4 µL of culture media (the media that they are in), using a 10 or 20 µL pipette. Deposit the droplet in the middle of a 22 x 22 mm2 coverslip (Figure 5). Note: If the cells that are 1) unhealthy (abnormally shaped or heavily vacuolated) or 2) still have membranellar bands or oral apparatuses (for chemical treatment experiments), do not use for imaging.
- Arrange 4 more droplets of Stentor in the culture media around the previous droplet, while leaving enough space between the droplets.
- Invert the coverslip with droplets and gently place it over the well of the glass spot plate to which PSW was added in Step 4.1.
- Note: PSW in the well will minimize the evaporation of the droplets (Figure 5).
- Prepare the remaining cells for imaging by following Steps 4.1.1 - 4.1.4.
- Image the regenerating cells under a microscope with the desired time resolution. Alternatively, observe the regeneration using a dissecting stereo microscope. Note: Regeneration will begin immediately after cell cutting or treatment with sucrose or urea. However, visible new oral structures will form about 3 h after the beginning of regeneration.
- For each time point, assign one of the three regeneration stages to each of the cells. To do this, compare each cell to the representative images illustrating the stages (Figure 4 and Figure 6).
- Assign Stage 1 to the cells that do not have a membranellar band yet (Figure 6A).
- Assign Stage 3 to the cells with an oral primordium. Note: An oral primordium is an invagination appearing 6 - 8 h after treatment at the posterior end of the membranellar band (Figure 4, Figure 6C and 6D). Regeneration is completed when the cells have a membranellar band at their anterior end and the characteristic trumpet cell shape (Figure 4, Figure 6E). Most Stentor cells are fully regenerated within 8-9 h since the start of regeneration.
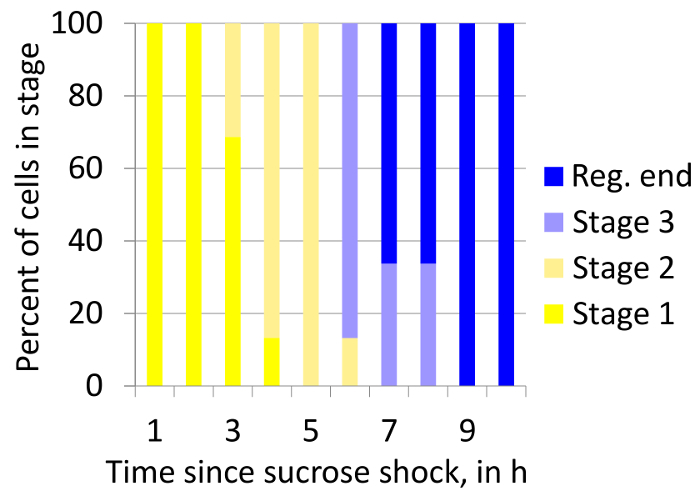
Plot the percentage of cells in each of the regeneration stages for each time point in the form of a stacked box plot (Figure 7). Note: This type of plot allows the visualization of regeneration dynamics in a population of cells.


Representative Results
Stentor cultures have been reliably established and maintained from individual cells or cell fragments using Section 1 of the protocol. A representative example of a large healthy culture is shown in Video 1.
The time course of regeneration was measured in Stentor using the sucrose treatment method for initiating regeneration outlined in Step 3.1, combined with the imaging and analysis method discussed in Section 4 (Figure 7). This plot indicates that there is a one-hour-long spread in the time taken by the population of cells to reach any particular stage. This type of analysis allows the study of temporal heterogeneity in the regeneration process in a population of regenerating cells.
The following is a summary of regeneration timing that has been observed thus far after dozens of sucrose treatments (Figure 4 and Figure 6). Stage 1 is when Stentor cells look like teardrops without any membranellar band (this stage starts immediately after sucrose washout). This stage lasts for 3 - 6 h. Stage 2 is when a membranellar band appears and grows (3 - 6 h after sucrose treatment). This stage lasts for 3 - 4 h. Stage 3 is when an oral primordium appears at the posterior end of the membranellar band (6 - 8 h after sucrose treatment), and both structures are moved toward the anterior end of the cell. This stage lasts for 1 - 2 h. When both the membranellar band and the oral apparatus reached the anterior end of the cell, this indicated the completion of regeneration. The cell has adopted characteristic Stentor trumpet-like shape. Cells were completely regenerated 8 - 9 h after sucrose treatment.
Figure 1. Snapshot of Stentor. The membranellar band and the oral apparatus are shown at the anterior end of the cell. The holdfast is at the posterior end of the cell. Stentor macronucleus is nodulated. A well-fed Stentor has green food vacuoles containing mostly Chlamydomonas. Scale bar is 0.5 mm. Please click here to view a larger version of this figure.
Figure 2. Stentor cutting set-up. Cell cutting is performed by manually manipulating a glass needle while looking at the cell using a commercially available dissecting stereo microscope. The purpose of the tissue paper is to provide the white background to see the cells better. Please click here to view a larger version of this figure.
Figure 3. Snapshots of Video 3 illustrating how to cut a Stentor with a glass needle. (A) A Stentor immediately before the needle touches it. (B) A Stentor gently squeezed between a needle and glass slide. (C) A contracted Stentor reacting to the force of the needle. (D) A Stentor being cut by gently pressing on the cell with the side of the needle. (E) A Stentor now cut in two but not separated. (F) Two Stentor fragments. Scale bar is 0.25 mm. Please click here to view a larger version of this figure.
Figure 4. Schematic visualization of Section 3 and Section 4 of the protocol. This is an illustrated protocol for performing sucrose or urea treatment and observation of cell regeneration. The time indicated in the bottom panel is measured from the beginning of the Wash 1. Please click here to view a larger version of this figure.
Figure 5. The setup for imaging and/or direct observation of regeneration with an upright microscope. In each 4 µL droplet hanging from the coverslip, there is one cell undergoing regeneration. Water in the wells of the glass spot plate limits evaporation of the droplets. This setup allows following the regeneration of multiple cells in parallel. Please click here to view a larger version of this figure.
Figure 6. Snapshots of Stentor in each stage of the membranellar band and the oral apparatus regeneration. (A) Stage 1 is characterized by a teardrop-like cell shape and the absence of the membranellar band. (B) Stage 2 is characterized by the appearance of the membranellar band, a cilia-based structure that beats continuously. Membranellar bands are marked with white dashed lines in panels B - D. (C and D) Stage 3 is characterized by the appearance of oral primordium (marked with an arrow). Oral primordium looks like an invagination at the posterior end of the membranellar band. The oral primordium will then move up towards the anterior of the cell to become the oral apparatus. (E) Regeneration is completed when the cell has adopted the characteristic Stentor trumpet-like shape, and the oral apparatus (marked with an arrow) has widened at the anterior end of the cell. Scale bar is 0.25 mm. Please click here to view a larger version of this figure.
Figure 7. Stacked box plot showing how proportions of cells in all stages of regeneration after sucrose treatment changes over over time. The plot shows heterogeneity in the timing of regeneration stages within a population of regenerating cells. "Reg. end" indicates the completion of regeneration. The number of cells: 28. Please click here to view a larger version of this figure.
 Video 1. Example of a healthy Stentor culture. Generally, if a culture is this concentrated, splitting the culture is recommended. Please click here to view this video. (Right-click to download.)
Video 1. Example of a healthy Stentor culture. Generally, if a culture is this concentrated, splitting the culture is recommended. Please click here to view this video. (Right-click to download.)
 Video 2. Slowing down Stentor with methylcellulose.
Please click here to view this video. (Right-click to download.)
Video 2. Slowing down Stentor with methylcellulose.
Please click here to view this video. (Right-click to download.)
 Video 3. Cutting Stentor. Individual cells can be manually cut into multiple pieces under a stereo dissecting microscope by pressing a glass needle through the cell. Please click here to view this video. (Right-click to download.)
Video 3. Cutting Stentor. Individual cells can be manually cut into multiple pieces under a stereo dissecting microscope by pressing a glass needle through the cell. Please click here to view this video. (Right-click to download.)
| Problem with culture | Possible remedy | Possible prevention |
| Culture is overwhelmed by rotifers or other large organisms. | Remove as many Stentor as possible into a new culture dish. Then wash the cells three times. | Wash Stentor thoroughly when receiving them, even when purchasing them from a commercial supplier. |
| Culture is overwhelmed by fungus or other small organisms. | Identify a corner where there are the least Stentor. In that area, remove as much media as possible without removing many Stentor and add in new PSW. Mix gently but throughly to distrubute the invading organisms. Skip the next feeding and the Stentor will eat them. | Observe the culture regularly and remove small contaminants by exchanging media for fresh PSW. |
| Stentor are laying on top of each other. | Split the culture so that the concentration is less than 20 cells/mL. | Split cultures more often. |
| Stentor cells are mating. | Feed every 4 days instead of every 5 days. | |
| Culture has a lot of dark waste material. | Remove as much of the dark material, Stentor waste, as possible without removing the cells. If Stentor are attached to the dark material, pipette up and down to release the Stentor cells from their waste and then remove the media with the waste without removing the cells. Or, remove those Stentor and feed accordingly less. | Feed Stentor 1 mL less food per 100 mL of culture. |
| Unhealthy-looking (vacuolated/bloated or rounded) Stentor. | Remove as much media as possible without removing many Stentor cells and add new PSW. | Exchange the media regularly. |
Table 1. Troubleshooting guide for culturing Stentor.
Discussion
Culturing Stentor presents a number of challenges. First, to perform experiments that require large numbers of cells, one needs to maintain a large number of Stentor cultures, as cultures become unhealthy when Stentor concentration exceeds 20 cells/mL. Second, the organisms that can contaminate Stentor cultures often divide faster than Stentor and overwhelm the culture (a common contaminant is rotifers). Thus, it is necessary to inspect the culture under a microscope periodically and remove the contaminants. Occasionally, a new culture needs to be started from a small number of cells rescued manually from a contaminated culture. This increases the time required to maintain healthy Stentor cultures. Third, expanding a single cell into a 400 mL culture requires at least one month because Stentor cell cycle is 3 - 5 days. Fourth, unlike other model ciliates, Stentor rarely go into cyst form and they cannot be frozen.
An important aspect of culturing Stentor is the selection of an appropriate food organism. Various methods for culturing Stentor were previously described. One of them suggests to use skim milk to culture bacteria which then feed Stentor. Such a technique is effective in culturing Stentor; however, application of genomic techniques requires pure samples to avoid confusion resulting from genomic reads from undefined food organisms. Using the current protocol, Stentor can be grown in mass and, because their food, Chlamydomonas, has been sequenced, the presence of genomic contamination from the food organism can be detected and controlled for. For unknown reasons, Tartar had to replenish his stocks by returning to where he found Stentor before. With the current protocol, we have been able to keep Stentor for years.
Regeneration experiments with Stentor are generally straightforward, but there are a few critical details to keep in mind. In regard to Section 2, cutting Stentor cells in half can be mastered in minutes. Mastering more advanced microsurgery procedures of Stentor may require a week of practice. While performing a sucrose or urea treatment (Section 3), if at the beginning of imaging most cells still have their membranellar bands, increase incubation time by 10 - 30 s for when sucrose treatment is performed next. Do not increase the time of sucrose treatment beyond 3 min incubation because it will result in cell death.
Imaging of Stentor regeneration requires methods to image large cells over long periods of time. The imaging method detailed in Section 4 can only be used with an upright microscope. If an inverted microscope is available instead, then cells can be placed on a slide or coverslip in small chambers. One method is to create a chamber out of petroleum jelly and cover with another coverslip to prevent evaporation. Alternatively, a chamber for an individual cell can be made by making a hole with a hole punch of the desired size in a 1 x 1 cm2 square of silicon spacer (Table of Materials), placing the spacer on a coverslip, putting cells in the chamber, and covering the chamber with another coverslip. When imaging, if Stentor are not in the correct orientation to observe the characteristic features of each stage of regeneration, tap the plate firmly to make the cell contract. Watch the cell extend to identify the stage of regeneration (full extension takes about 45 s). If the cell is still in the wrong orientation, repeat the tapping. The images shown in Figure 1 and Figure 6 were taken by a stereo zoom microscope; however, all experiments detailed can be performed using a 5X dissecting stereo microscope.
Another challenge besides culturing and imaging Stentor is the tracking of regeneration, specifically the amount of time necessary to identify stages. If the regenerating cell is perfectly oriented with the region of the oral primordium clearly visible, identification of the stage takes a few seconds. Sometimes, however, the Stentor cell is in an orientation preventing clear assignment of regeneration stage, thus taking more time to identify. The significant amount of time required to stage individual regenerating cells might delay the quantification of all regenerating cells, thus decreasing the temporal precision of staging and requiring the observer to wait and re-image the cell after it has moved to a new orientation. Consequently, this experiment is both time and labor intensive. For these reasons, it would be highly desirable to develop automated methods for detecting Stentor cells and assigning stages of regeneration in video microscopy data. These would also allow for more reproducible experiments, increases in the sample size, and the removal of human bias.
The emergence of highly sensitive genomic and proteomic methods has begun to put studies of single cells into reach. For such single-cell analyses, the giant size of a Stentor cell makes it a desirable test subject for proof-of-concept experiments. For such experiments to be possible, culturing Stentor is fundamental, and so the methods described here should play a role in further development of more advanced single-cell techniques.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by NIH grant R01 GM113602 (WFM) and NSF 1144247 (AL). We acknowledge Mark Slabodnick and Natalie Kirkland for developing the initial versions of some of these protocols and for informative discussions. We thank Karina Perlaza and Greyson Lewis for critical reading of the manuscript.
References
- Kirschner M, Gerhart J, Mitchison T. Molecular "vitalism". Cell. 2000;100(1):79–88. doi: 10.1016/s0092-8674(00)81685-2. [DOI] [PubMed] [Google Scholar]
- Shulman JM, St Johnston D. Pattern formation in single cells. Trends in Cell Biology. 1999;9(12):M60–M64. [PubMed] [Google Scholar]
- Wloga D, Frankel J. From molecules to morphology: cellular organization of Tetrahymena thermophila. Methods in Cell Biology. 2012;109:83–140. doi: 10.1016/B978-0-12-385967-9.00005-0. [DOI] [PubMed] [Google Scholar]
- Frankel J. Pattern Formation: Ciliate Studies and Models. Oxford: Oxford University Press; 1989. [Google Scholar]
- Tartar V. Grafting experiments concerning primordium formation in Stentor coeruleus. Journal of Experimental Zoology Part A. 1956;131(1):75–121. [Google Scholar]
- Lillie FR. On the smallest parts of Stentor capable of regeneration; a contribution on the limits of divisibility of living matter. Journal of Morphology. 1896;12(1):239–249. [Google Scholar]
- Morgan TH. Regeneration of proportionate structures in Stentor. The Biological Bulletin. 1901;2(6):311–328. [Google Scholar]
- Slabodnick MM, et al. The macronuclear genome of Stentor coeruleus reveals tiny introns in a giant cell. Current Biology. 2017;27(4):569–575. doi: 10.1016/j.cub.2016.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang SKY, Marshall WF. Self-repairing cells: How single cells heal membrane ruptures and restore lost structures. Science. 2017;356(6342):1022–1025. doi: 10.1126/science.aam6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slabodnick M, Prevo B, Gross P, Sheung J, Marshall W. Visualizing cytoplasmic flow during single-cell wound healing in Stentor coeruleus. Journal of Visualized Experiments. 2013;82(82):50848. doi: 10.3791/50848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartar V. Biology of Stentor. Oxford: Pergammon Press; 1961. [Google Scholar]
- Slabodnick MM, et al. The kinase regulator mob1 acts as a patterning protein for stentor morphogenesis. PLOS Biology. 2014;12(5):e1001861. doi: 10.1371/journal.pbio.1001861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris E. The Chlamydomonas sourcebook: Introduction to Chlamydomonas and its laboratory use. Vol. 1. Cambridge: Academic Press; 2009. [Google Scholar]