

Abstract
The assembly of protein complexes is a central mechanism underlying the regulation of many cell signaling pathways. A major focus of biomedical research is deciphering how these dynamic protein complexes act to integrate signals from multiple sources in order to direct a specific biological response, and how this becomes deregulated in many disease settings. Despite the importance of this key biochemical mechanism, there is a lack of experimental techniques that can facilitate the specific and sensitive deconvolution of these multi-molecular signaling complexes.
Here this shortcoming is addressed through the combination of a protein complementation assay with a conformation-specific nanobody, which we have termed Bimolecular Complementation Affinity Purification (BiCAP). This novel technique facilitates the specific isolation and downstream proteomic characterization of any pair of interacting proteins, to the exclusion of un-complexed individual proteins and complexes formed with competing binding partners.
The BiCAP technique is adaptable to a wide array of downstream experimental assays, and the high degree of specificity afforded by this technique allows more nuanced investigations into the mechanics of protein complex assembly than is currently possible using standard affinity purification techniques.
Keywords: Immunology and Infection, Issue 136, Protein-protein interactions, Proteomics, Interactomics, BiCAP, Signal Transduction, Bimolecular fluorescence complementation, Nanobody
Introduction
Protein complex assembly is a key process in maintaining the spatiotemporal specificity of many signalling pathways1,2. While the critical nature of this regulatory role is widely recognized, there is a lack of experimental techniques available to scrutinize these complexes. Most interactomics studies focus upon interactions with individual proteins, or the sequential enrichment of individual complex components. Here we present a technique for the isolation of a specific protein dimer while excluding the individual moieties of the component proteins as well as complexes formed with competing binding partners3. We have called this technique Bimolecular Complementation Affinity Purification (BiCAP), as it is a combination of a previously existing protein fragment complementation assay, Bimolecular Fluorescence Complementation (BiFC), with the novel use of a conformation-specific recombinant nanobody towards GFP and its derivatives (see Table of Materials).
A typical protein-fragment complementation assay relies on the expression of "bait" and "prey" proteins fused to split fragments of reporters such as luciferase4, β-galactosidase5, or green fluorescent protein (GFP)6 (Figure 1A). Through the interaction of the bait and prey proteins, the split reporter domains are encouraged to refold into a functional structure, allowing the interaction of the bait and prey proteins to be visualized or quantified. BiCAP was adapted from a version of this technique that made use of fragments of the GFP variant Venus. Fluorescent protein complementation assays are a popular method for visualizing protein-protein interactions in a live cell, but until now have been limited to this one function7. BiCAP represents a significant advance in this regard, as this technique not only allows for visualization, but also the isolation and interrogation of the resulting protein-protein interaction.
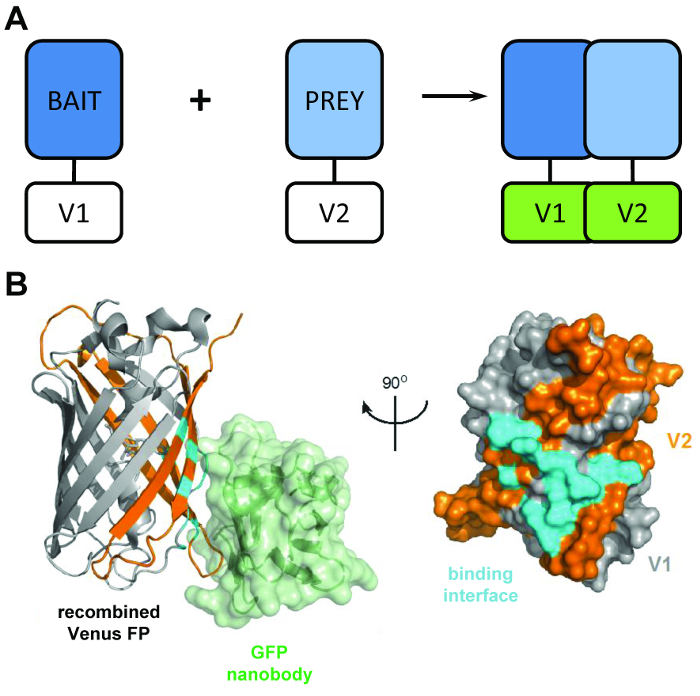
Figure 1: The structural principal behind the BiCAP technique. (A) A schematic outlining the principal behind bimolecular fluorescence complementation showing the 'bait' and 'prey' proteins tagged with the N-terminal V1 or C-terminal V2 fragments of the full-length Venus protein. (B) Structural analysis of the interaction interface (cyan) between the GFP nanobody (green) and recombined Venus, showing the position of the V1 (grey) and V2 (orange) fragments (PDB accession 3OGO). This figure is republished fromCroucher et al.3 Reprinted with permission from AAAS. Please click here to view a larger version of this figure.
The BiCAP technique makes use of two non-fluorescent fragments of Venus (named V1 and V2) which associate with a low degree of affinity unless an interaction occurs between their fusion partners. In this instance, the two split domains refold into the functional β-barrel structure of the fluorophore (Figure 1B)6. The key innovation of BiCAP comes from the introduction of the recombinant GFP nanobody, which recognizes a three-dimensional epitope on the β-barrel of GFP (and variants such as Venus) which is only present on the correctly recombined and folded fluorophore (Figure 1B)8. Crucially, the GFP nanobody does not bind to either of the individual Venus fragments. This facilitates the isolation of protein dimers only after the two proteins have formed a complex of their own volition, leading to more representative results than those acquired from methods that make use of chemically-induced, forced interactions9.
BiCAP is a powerful technique that specifically focuses on multi-protein complexes, which can potentially be combined with a number of downstream applications to improve the granularity of our understanding of the role these complexes play in signal transduction. It also encompasses the important feature of allowing visualization of protein interactions in situ. To date, BiCAP has been demonstrated as an effective method of analyzing the interactome of receptor tyrosine kinase (RTK) dimers3, but the adaptability of this method means that it can be adopted in almost any protein interaction context.
Protocol
1. Plasmid Cloning
NOTE: To generate plasmid vectors with the V1 or V2 tags fused to either the N-terminus or C-terminus of the gene of interest, BiFC destination vectors have been deposited with Addgene [N-terminal tag: pDEST-V1-ORF (#73635), pDEST-V2-ORF (#73636). C-terminal tag: pDEST-ORF-V1 (#73637), pDEST-ORF-V2 (#73638)]. The gene/s of interest will need to be in specific recombination cloning compatible entry vectors (i.e., pDONR223 or pENTR221), without stop codons, to proceed with cloning. Many compatible clones are already available within various plasmid collections, including the Mammalian Genome Collection (https://genecollections.nci.nih.gov/MGC/).
- Perform a recombination reaction to insert the gene of interest between the attR sites of the appropriate BiFC destination vector:
- In a 0.2 mL tube, add 150 ng entry vector, 150 ng BiFC destination vector, and 8 µL TE buffer (pH 8.0).
- Add 2 µL recombinase enzyme mix (see Table of Materials). Mix well and centrifuge briefly.
- Incubate the reaction for 1 h at room temperature or at 16 °C overnight.
- Stop the reaction by adding 1 µL Proteinase-K solution and incubating at 37 °C for 10 min.
- Transform heat-shock-competent cells (see Table of Materials) with the LR reaction product: NOTE: Any basic strain can conceivably be used at this point, as long as it does not contain the Ccdb antitoxin, which would prevent the specific selection of plasmids containing the insert.
- Thaw competent cells on ice and transfer 50 µL of cells into a 14 mL round-bottomed polypropylene tube.
- Add 1 µL of reaction product to the cells and gently mix. Incubate for 20 min on ice.
- Heat shock in 42 °C water bath for 45 s. Immediately return to ice for 2 min.
- Add 1 mL of LB media and incubate with shaking at 37 °C for 1 h.
- Plate the transformation onto a 10 cm agar plate containing Ampicillin (100 µg/mL) and incubate at 37 °C overnight.
- Purification of BiFC plasmid.
- To purify plasmid from an individual colony, add 100 mL of LB media to a large conical flask and add Ampicillin (100 µg/mL). To screen multiple colonies, add 5 mL of LB media to 14 mL round-bottomed polypropylene tubes and add Ampicillin (100 µg/mL).
- Using a sterile inoculation loop, pick a single colony from the agar plate. Place the inoculation loop into the LB media and mix briefly.
- Cover the top of the conical flask with aluminum foil and incubate overnight at 37 °C with shaking.
- Produce transfection grade plasmid DNA using a standard maxiprep or midiprep plasmid DNA purification kit (see Table of Materials)10. Assess quality of DNA by absorbance spectra. NOTE: DNA should have an A260/A280 ratio >1.8 and an A260/A230 ratio >2.0. At this point, it is recommended to screen multiple individual colonies to check for efficient expression of the insert and tag.
2. Cell Culture and Transfection
NOTE: For transfection of the BiFC vectors it is important to achieve a high efficiency, and relatively homogenous transfection. The vectors will likely be compatible with any standard transfection reagent, and the transfection conditions should be optimized accordingly. To perform mass spectrometry, we usually culture cells within 10 cm dishes, although this can also be proportionally scaled down to smaller dishes or plates for experiments that require less material.
Seed 1.0 × 106 HEK293T cells in a 10 cm dish with 10 mL of DMEM media, supplemented with 10% FBS and Penicillin/Streptomycin (1:100).
Dilute 2.5 µg of each BiFC vector into 500 µL of transfection buffer (see Table of Materials) in a 1.5 mL microcentrifuge tube.
Add 10 µL of transfection reagent (see Table of Materials).
Vortex mixture for 10 s, then briefly centrifuge. Incubate for 10 min at room temperature.
Add the DNA transfection mixture to the plate, dropwise. Incubate the cells for a sufficient length of time for the BiFC fusion proteins to interact and for Venus folding and fluorophore maturation, generally ~8 - 24 h. NOTE: The peak excitation for Venus is 515 nm, and its peak emission is 528 nm, although this is readily viewed using a standard fluorescent microscope set up to visualize GFP fluorescence.
3. Sample Preparation
- Harvesting lysates
- Prior to lysate collection, prepare Cell Lysis Buffer [50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% (v/v) non-ionic detergent (see Table of Materials)]. Store at 4 °C.
- Immediately prior to harvesting, supplement 10 mL of Cell Lysis Buffer with protease inhibitor and phosphatase inhibitor (see Table of Materials). Keep supplemented Cell Lysis Buffer on ice.
- Wash cells twice in ice-cold PBS. Aspirate the PBS and add 1 mL of ice-cold supplemented Cell Lysis Buffer. Place the dish on ice, ensuring the buffer is spread over the entire surface area.
- Incubate on ice for approximately 5 min, then use a cell scraper (see Table of Materials) to scrape the cells and transfer to a pre-chilled 1.5 mL microcentrifuge tube.
- Remove cellular debris by centrifuging the collected lysate at 18,000 × g for 5 min at 4 °C and transfer cleared supernatant into fresh microcentrifuge tubes. NOTE: At this point the lysate can be used immediately, or stored at -80 °C. It is also recommended to keep an aliquot of crude lysate so that the efficiency of transfection and the affinity purification can be validated.
- Affinity purification NOTE: The BiCAP isolation step is performed using the GFP nanobody conjugated to agarose beads (see Table of Materials).
- Prepare the agarose beads by washing an appropriate volume (20 µL per sample + 10 µL excess) in 1 mL PBS. Centrifuge the beads at 300 x g and remove the supernatant.
- Add 20 µL to each lysate sample.
- Incubate the samples for 2 h at 4 °C with end-to-end rotation. NOTE: At this point the samples can be prepared for either SDS-PAGE and western blotting, or analysis by mass spectrometry.
- Preparation of BiCAP eluant for western blotting.
- Centrifuge the beads at 300 x g and wash three times in Cell Lysis Buffer.
- Resuspend the washed beads in 50 µL of appropriately diluted sample buffer (see Table of Materials) and heat the samples at 95 °C for 2-3 min. NOTE: Samples prepared in this manner can be stored at -20 °C for several months.
- Perform SDS-PAGE and western blotting11 for both the V1 tag and V2 tags (see Table of Materials), as well as any other proteins of interest.
- Preparation of BiCAP eluant for mass spectrometry. NOTE: For analysis by label-free quantitative (LFQ) mass spectrometry it is recommended to prepare the samples in at least quadruplicate to ensure robust statistical power.
- Centrifuge the beads at 300 x g and wash six times in Cell Lysis Buffer without detergent. This is necessary to remove both the excess protein and also the detergent that will interfere with mass spectrometry analysis. Proceed to the next step immediately, or store the beads at -80 °C.
- Trypsinize beads in 60 µL Buffer 1 [2 M urea, 50 mM Tris-HCl (pH 7.5), 5 µg/mL trypsin]. Allow the beads to digest for 30 min at 27 °C in a thermomixer shaking at 800 RPM.
- Briefly centrifuge beads, then collect supernatant and transfer to microcentrifuge tubes (see Table of Materials).
- Wash the beads in 25 µL Buffer 2 [2 M urea, 50 mM Tris-HCl (pH 7.5), 1 mM dithiothreitol], in order to reduce the bound proteins.
- Pool the beads and supernatant into one microcentrifuge tube. Allow the digestion to occur overnight at room temperature.
- Alkylate the samples by adding 20 µL of iodoacetamide (5 mg/mL in ultrapure water) to each sample and incubate in the dark for 30 min.
- Inhibit digestion by treating each sample with 1 µL trifluoroacetic acid (TFA). This step will also acidify the samples in preparation for stage tipping.
- Construct C18 stage tips by stacking 6 layers of 1 mm solid phase extraction C18 (Octadecyl) membrane disks (see Table of Materials) into a 200 µL micropipette tip. Prepare a single tip for each sample.
- Wet the stage tips with methanol, and equilibrate with 50 µL 0.1% (v/v) trifluoroacetic acid (TFA), 80% (v/v) acetonitrile.
- Wash the tips with 50 µL 0.1% (v/v) TFA.
- Load the acidified peptides onto the stage tips and then elute using 0.1% (v/v) TFA, 80% (v/v) acetonitrile in two steps.
- Evaporate the samples using a vacuum concentrator.
- If needed, store dried peptides at -80 °C.
4. Mass Spectrometry.
Resuspend samples in 15 µL of 5% formic acid, 2% acetonitrile (in ultrapure water).
Carefully load 6 µL onto an LC plate and place into a nanoLC HPLC system (see Table of Materials).
Pack a 20 cm, 75 µM inner diameter column with 1.9 µm C18 stationary phase particles (see Table of Materials). Load 5 µL peptide onto each column.
Elute peptides using a linear gradient of acetonitrile at 250 nL/min over 140 min and introduce by nanoelectrospray into a Linear Trap Quadropole (LTQ) hybrid mass spectrometer coupled to the nanoLC HPLC system.
Collect tandem MS data for the top 10 most abundant ions per scan over a 140-minute time gradient. Randomize the order of data collection and interchange with BSA run between each sample to minimize temporal bias.
5. Analysis
Process raw MS data using default setting within MaxQuant software version 1.2.7.4 and analyze MaxQuant output using modified version of the Perseus statistical analysis workflow in the R software environment3. NOTE: The statistical analysis workflow from this point is summarized in Figure 2. Briefly, LFQ intensities of proteins identified using MaxQuant are transformed and filtered followed by normalization and imputation. Interacting proteins of dimer pairs are identified by comparison with a Venus control, to exclude nonspecific background binders, with Student's t test and Benjamini-Hochberg correction for multiple comparisons. Data quality is confirmed by comparisons of individual histograms, multiple regression, and hierarchical clustering.
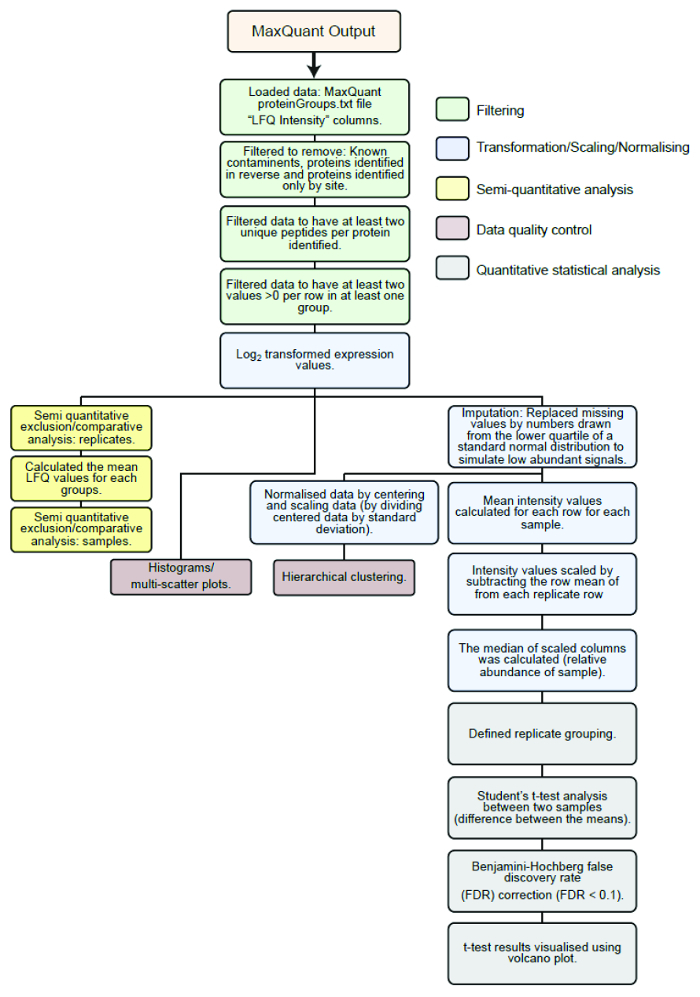
Figure 2: Outline of the statistical analysis workflow. Flow diagram of statistical analysis pipeline used to analyze the LFQ intensities of proteins identified from raw mass spectrometry data processed using MaxQuant. Green boxes: filtering, blue boxes: transformation/normalising/scaling, brown boxes: data quality control, yellow boxes: semi-quantitative exclusion/comparative analysis, grey boxes: statistical analysis. This figure is republished fromCroucher et al.3 Reprinted with permission from AAAS. Please click here to view a larger version of this figure.
Representative Results
Following the use of recombination cloning to generate of V1 and V2 tagged genes of interest with the BiFC pDEST plasmids, co-transfection of two plasmids containing an interacting pair of proteins will result in the generation of a Venus fluorescent signal after approximately 8-24 hours. In the absence of a positive signal it is possible that the protein interaction may not be occurring due to the choice of cell line, a low transfection efficiency or that the orientation of the BiFC tags is not optimal. Troubleshooting for each of these scenarios is discussed below.
We have previously observed a positive interaction for a number of different protein types ~16 hours after the co-transfection of HEK-293T cells. This includes the dimerisation of the RTK ERBB2 at the plasma membrane (Figure 3A)3 and the binding of Ubiquitin to the E3 ligase UBR5, occurring predominantly within the nucleus (Figure 3A)12. The ability to visualize the specific sub-cellular localization of these protein-protein interactions, as opposed to the whole-cell fluorescence of the full-length Venus protein (Figure 3A), is the primary function of the existing BiFC technique. However the key innovation of the BiCAP technique is the ability to specifically purify and characterize these interacting proteins.
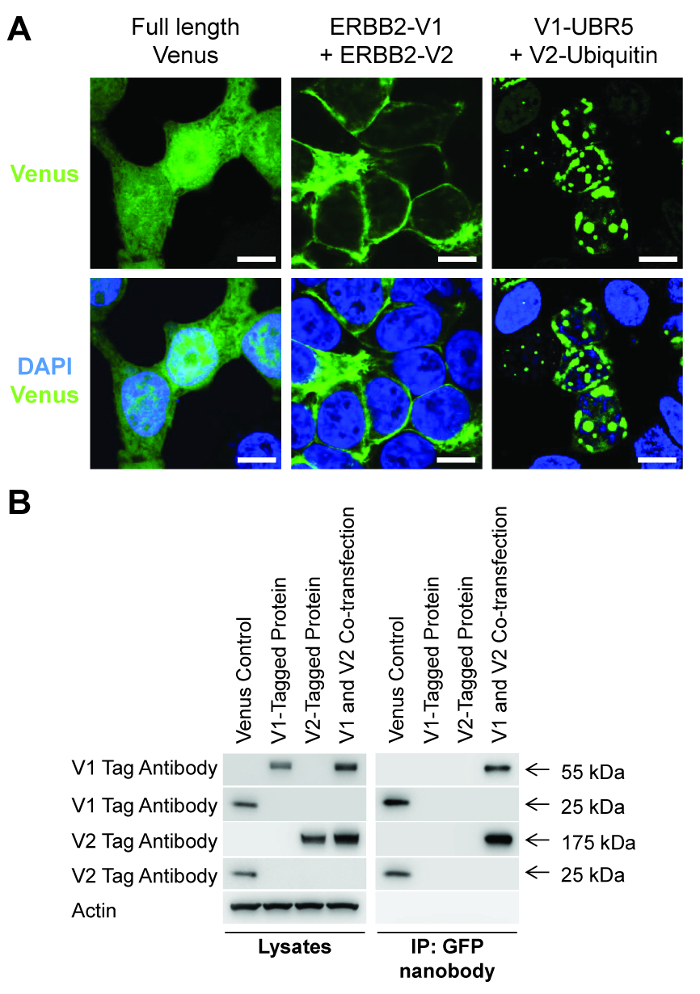
Figure 3: BiCAP allows visualization and isolation of interacting proteins. (A) Confocal fluorescence microscopy analysis of the fluorescent signal produced by the full-length Venus protein, the interaction between ERBB2-V1 and ERBB2-V2 and the interaction between V1-UBR5 and V2-Ubiquitin after transfection into HEK-293T cells. Scale bars, 10 µm. (B) HEK-293T cells were transfected with a control plasmid containing full-length Venus, ERBB2-V1, ERBB2-V2 or both ERBB2-V1 and ERBB2-V2, followed by immunoprecipitation with the GFP nanobody and Western blotting with the indicated antibodies. Please click here to view a larger version of this figure.
This specific purification afforded by the BiCAP protocol can be readily observed following the transfection of a control plasmid containing the full-length Venus protein, individual plasmids containing representative V1 or V2 tagged proteins or the co-transfection of these two plasmids (Figure 3B). After transfection of these plasmids into HEK-293T cells, and a 16 h incubation, the preparation of crude lysates for western blotting with V1 and V2 tag antibodies shows that each tagged protein is present within the appropriate sample. However, following affinity purification with the GFP nanobody, only the full-length Venus control protein and the interacting V1 and V2 tagged proteins within the co-transfection sample can be detected (Figure 3B).
This selective purification of interacting proteins can be followed by a number of downstream techniques. In our recent publication3 we utilized the BiCAP workflow to perform mass-spectrometry interactome analysis of the RTK ERBB2 as either a homodimer or a heterodimer with EGFR and ERBB3 (Figure 4). By following our data analysis pipeline (Figure 2) and visualizing the data using Cytoscape13 we identified distinct subsets of proteins that either interacted with all three ERBB2 dimers, or were specific to a pair of dimers, or an individual dimer. Closer analysis of these interaction profiles allowed us to identify novel patterns of adaptor protein binding regulating dimer-specific signal transduction events, which would not have been possible using standard co-immunoprecipitation approaches.
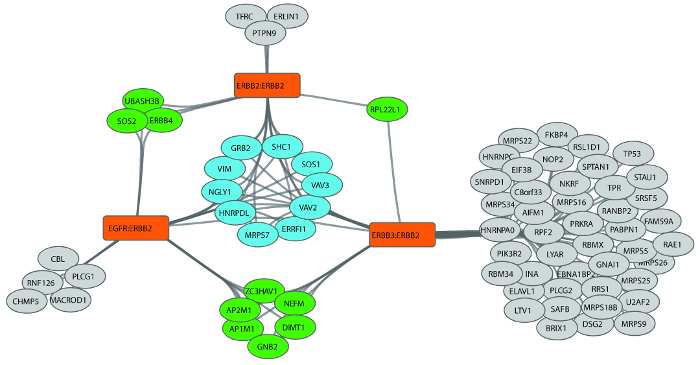
Figure 4: Analysis of the interactome diversity of ERBB2-containing dimers. The receptor baits used for BiCAP are shown in orange, the core group of 10 proteins common to the interactome of all three receptor dimers is shown in blue. Interacting proteins common to two receptor dimers are shown in green, and to one receptor in grey. This figure is republished fromCroucher et al.3 Reprinted with permission from AAAS. Please click here to view a larger version of this figure.
Discussion
BiCAP is a powerful method for isolating specific protein dimers while excluding the individual components and their competing binding partners3. BiCAP is based on adaptation of a fluorescence protein complementation assay called BiFC6. Existing methods, including BiFC and proximity ligation assays, have been used extensively to visualize and quantify protein interactions in live cells7, but did not provide an effective means of isolating and characterizing the dimers formed from these interactions.
Protein complementation based affinity purification techniques have also continued to evolve. Schopp et al. recently described a system for protein dimer proximity labeling using BioID14. By fusing the split fragments of the biotin ligase BirA to two interacting proteins, they directed enrichment of Ago2 containing protein complexes involved in miRNA-mediated gene silencing. Split-BioID complements BiCAP in allowing for selective enrichment of proteins within the proximity range of BioID, where BiCAP enriches direct complex interactors.
The success of the BiCAP technique depends on the efficient transfection of the BiFC vectors. While we routinely use HEK293T cells as a robust and easy-to-transfect cell line, the use of this cell line may preclude the identification of potentially cell-specific interaction partners. We would therefore recommend that the choice of cell line be carefully considered for each experiment, especially for studies with a specific disease focus. In addition to this, optimization of the transfection conditions should also be performed for each experimental setup. The values outlined in the protocol are aligned with our work on ERBB2 and were calibrated to approximate the levels of endogenous ERBB2 within breast cancer cell lines. It is prudent, when beginning the investigation of a specific protein-protein interaction, to perform several trials using varying quantities of both constructs in order to achieve expression levels that are consistent with those present in a physiological or patho-physiological setting.
Another critical element of this protocol is determining the correct time for the samples to be harvested following transfection. While the formation of protein complexes is generally a transient process, with the individual components combining and disassembling over time, the refolded Venus fragments have a very slow dissociation rate which may not reflect that of their fusion partners. In practical terms, this means that a reasonably strict timeframe following transfection is necessary to yield a realistic result and prevent the accumulation of highly over-expressed protein aggregates. Generally, once BiFC fluorescence is observable under a standard wide-field fluorescent microscope (e.g. ~16 hours for ErbB2 dimers), cells can be prepared for harvesting lysates or imaging, although this may need to be adjusted for each specific bait and prey protein.
Our recent publication demonstrated the utility of BiCAP for isolating RTK dimers3. This experimental setup had the advantage of a known orientation for the protein interaction, allowing the direct alignment of the V1 and V2 tags at the intracellular, C-terminal end of each receptor. The close alignment of these tags is necessary for their re-folding, and when investigating proteins interactions without a known orientation it is crucial to generate all variants of N-terminal and C-terminal tag fusions in order to identify a combination that will result in a positive BiFC signal. Even with the inclusion of this optimization step there is still the possibility of a false negative if the alignment of a specific protein interaction completely precludes any interaction between the tags.
Conversely, care should also be taken to prevent the occurrence of a false positive interaction. At longer time points (>36 h), we have previously observed the emergence of non-specific background fluorescence with some protein interaction pairs. Where possible, non-interacting controls should also be included to ensure the specificity of the observed protein interaction and inform a suitable time-frame for analysis following transfection.
BiCAP is a highly adaptable technique that is applicable to almost any pair of interacting proteins. To demonstrate the utility of this technique we have presented interactome data generated through the use of mass spectrometry. However, the purified complexes should be compatible with any desired downstream assay. For example, while the BiCAP protocol allows the purification of interacting proteins, it may also have genomic applications through the downstream use of CHIP-seq alongside proteomics. The application of BiCAP in this setting would provide increasing levels of detail for DNA binding motifs of specific transcription factors, which is also heavily regulated through complex dimerisation patterns.
BiCAP therefore represents a robust adaption of protein fragment complementation assays, which enables the interrogation of biochemical and genomic networks with greatly increased resolution. In the future, adaption and expansion of the BiCAP technique will increase our understanding of the complexity and plasticity of protein signaling networks, and their finely tuned role in the regulation of developmental, physiological and disease processes.
Disclosures
The authors have nothing to disclose
Acknowledgments
D.R.C is a Cancer Institute NSW Fellow and D.N.S was previously a Cancer Institute NSW Fellow. The research findings presented in this manuscript were funded by the Cancer Institute NSW (13/FRL/1-02 and 09/CDF/2-39), NHMRC (Project Grant GNT1052963), Science Foundation Ireland (11/SIRG/B2157), NSW Office of Science and Medical Research, Guest Family Fellowship and Mostyn Family Foundation. J.F.H. and R.S. were recipients of an Australian Postgraduate Award.
References
- Kolch W, Pitt A. Functional proteomics to dissect tyrosine kinase signalling pathways in cancer. Nat Rev Cancer. 2010;10(9):618–629. doi: 10.1038/nrc2900. [DOI] [PubMed] [Google Scholar]
- Pawson T, Kofler M. Kinome signaling through regulated protein-protein interactions in normal and cancer cells. Curr Opin Cell Biol. 2009;21(2):147–153. doi: 10.1016/j.ceb.2009.02.005. [DOI] [PubMed] [Google Scholar]
- Croucher DR, et al. Bimolecular complementation affinity purification (BiCAP) reveals dimer-specific protein interactions for ERBB2 dimers. Sci Signal. 2016;9(436):ra69. doi: 10.1126/scisignal.aaf0793. [DOI] [PubMed] [Google Scholar]
- Cassonnet P, et al. Benchmarking a luciferase complementation assay for detecting protein complexes. Nat Methods. 2011;8(12):990–992. doi: 10.1038/nmeth.1773. [DOI] [PubMed] [Google Scholar]
- Rossi F, Charlton CA, Blau HM. Monitoring protein-protein interactions in intact eukaryotic cells by beta-galactosidase complementation. Proc Natl Acad Sci U S A. 1997;94(16):8405–8410. doi: 10.1073/pnas.94.16.8405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magliery TJ, et al. Detecting protein-protein interactions with a green fluorescent protein fragment reassembly trap: scope and mechanism. J Am Chem Soc. 2005;127(1):146–157. doi: 10.1021/ja046699g. [DOI] [PubMed] [Google Scholar]
- Hu CD, Kerppola TK. Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis. Nat Biotechnol. 2003;21(5):539–545. doi: 10.1038/nbt816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubala MH, Kovtun O, Alexandrov K, Collins BM. Structural and thermodynamic analysis of the GFP:GFP-nanobody complex. Protein Sci. 2010;19(12):2389–2401. doi: 10.1002/pro.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fegan A, White B, Carlson JC, Wagner CR. Chemically controlled protein assembly: techniques and applications. Chem Rev. 2010;110(6):3315–3336. doi: 10.1021/cr8002888. [DOI] [PubMed] [Google Scholar]
- JoVE Science Education Database. Basic methods in cellular and molecular biology. plasmid purification. J Vis Exp. 2017.
- Eslami A, Lujan J. Western blotting: sample preparation to detection. J Vis Exp. 2010. [DOI] [PMC free article] [PubMed]
- Shearer RF, et al. The E3 ubiquitin ligase UBR5 regulates centriolar satellite stability and primary cilia formation via ubiquitylation of CSPP-L. Mol Biol Cell. 2018. [DOI] [PMC free article] [PubMed]
- Shannon P, et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schopp IM, et al. Split-BioID a conditional proteomics approach to monitor the composition of spatiotemporally defined protein complexes. Nat Commun. 2017;8:15690. doi: 10.1038/ncomms15690. [DOI] [PMC free article] [PubMed] [Google Scholar]