

Abstract
The maintenance of the genome and its faithful replication is paramount for conserving genetic information. To assess high fidelity replication, we have developed a simple non-labeled and non-radio-isotopic method using a matrix-assisted laser desorption ionization with time-of-flight (MALDI-TOF) mass spectrometry (MS) analysis for a proofreading study. Here, a DNA polymerase [e.g., the Klenow fragment (KF) of Escherichia coli DNA polymerase I (pol I) in this study] in the presence of all four dideoxyribonucleotide triphosphates is used to process a mismatched primer-template duplex. The mismatched primer is then proofread/extended and subjected to MALDI-TOF MS. The products are distinguished by the mass change of the primer down to single nucleotide variations. Importantly, a proofreading can also be determined for internal single mismatches, albeit at different efficiencies. Mismatches located at 2-4-nucleotides (nt) from the 3' end were efficiently proofread by pol I, and a mismatch at 5 nt from the primer terminus showed only a partial correction. No proofreading occurred for internal mismatches located at 6 - 9 nt from the primer 3' end. This method can also be applied to DNA repair assays (e.g., assessing a base-lesion repair of substrates for the endo V repair pathway). Primers containing 3' penultimate deoxyinosine (dI) lesions could be corrected by pol I. Indeed, penultimate T-I, G-I, and A-I substrates had their last 2 dI-containing nucleotides excised by pol I before adding a correct ddN 5'-monophosphate (ddNMP) while penultimate C-I mismatches were tolerated by pol I, allowing the primer to be extended without repair, demonstrating the sensitivity and resolution of the MS assay to measure DNA repair.
Keywords: Biochemistry, Issue 136, DNA polymerase proofreading, DNA repair, MALDI-TOF mass spectrometry, single nucleotide extension, dideoxyribonucleotide triphosphate, DNA mismatch, DNA replication error
Introduction
The proofreading functions of DNA polymerases during DNA replication are essential to ensure the high fidelity of genetic information that needs to be transferred to progeny1,2,3,4,5,6,7. Being able to assess the contributions of polymerase proofreading exonucleases would clarify the mechanisms safeguarding genetic stability.
Radioisotope labeling and gel-based assays in combination with densitometric analyses of the autoradiograms or phosphor imaging8,9,10 have traditionally been used to detect proofreading activity of DNA polymerases. While functional, these assays are laborious, expensive, and not amenable to high-throughput formats. In addition, radioisotopes suffer safety issues including waste disposal. Alternatively, proofreading activities have been analyzed by fluorometric techniques. For example, 2-aminopurine (2-AP) can be incorporated into extension products during in vitro polymerase proofreading assays to produce a fluorescent signal11,12. Unfortunately, these approaches suffer from a low specificity, since 2-AP can pair with both thymine and cytosine. More recent approaches include a sensitive G-quadruplex-based luminescent switch-on probe for a polymerase 3'-5' exonuclease assay13 as well as a singly-labeled fluorescent probe for a polymerase proofreading assay that overcomes some of the aforementioned drawbacks14. Enthusiasm for these fluorometric methods is diminished due to the need for the specific labeling of DNA substrates.
In contrast, a MALDI-TOF MS for DNA analysis has been employed in the PinPoint assay, where the primer extension reactions with unlabeled 4 ddNTPs can be used to identify polymorphisms at a given locus15,16,17 and has been widely adopted in clinical applications for mutation detections and cancer diagnoses18. Using these basic principles, we have created a label-free assay for the in vitro determination of DNA polymerase proofreading activity exploiting the high resolution, high specificity, and high-throughput potential of MALDI-TOF MS. Using the E. coli DNA polymerase I Klenow fragment as a model enzyme, dideoxyribonucleotide triphosphates (ddNTPs) as substrates can take a "snapshot" of proofreading products after a single nucleotide extension via MALDI-TOF MS (Figure 1).
Likewise, this method was also developed for a DNA repair assay where primers containing 3' penultimate dI lesions are subjected to a pol I repair assay which mimics endo V nicked repair intermediates. While not fully understood, the endo V repair pathway is the only repair system known to employ the pol I proofreading exonuclease activity for lesion excision19,20. Using MALDI-TOF MS, we show a clearly defined repair patch where dI can be excised by pol I when occurring in the last 2 nt of the primer before adding the correct complemented nucleotide.
For the study of proofreading and DNA repair, this method is faster and less laborious than previous methods and provides additional information towards mechanism and function.
Protocol
1. Primer/template Preparation
Design primers/templates with a balanced G+C content between 40% and 60% as in a sequencing or PCR primer design. Use primers of 18 to 21 nt for an appropriate annealing and better MS signals.
Design the template by setting 50 °C as the minimum melting temperature for the duplex region with at least 7 nt of 5'-overhang to separate the signals between the primer and the template. NOTE: For example, for the substrate P21/T28 in Table 1, the 21-nt primer is paired with a 28-nt template. Option: the use of alternative nucleic acids such as nuclease-resistant phosphorothioate bonds to replace the last 4 phosphodiester bonds at the 3' end of the template can prevent possible interference by a non-specific 3' end degradation of the templates (e.g., T28S4 in Table 1), although this will add some cost for the template preparation.
Using standard desalted oligonucleotides without any further purification is satisfactory for this study. Use oligonucleotide primers/templates at a concentration of 100 pmol/µL in H2O as a stock and store it at -20 °C. Check the quality and purity of the primers and templates by running the oligonucleotides on MALDI-TOF MS (steps 4 - 7) to ensure unique peak signals and a good signal to noise ratio.
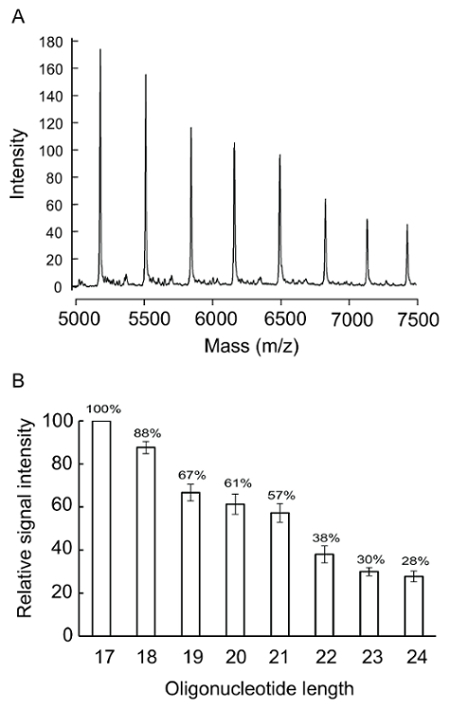
Determine the relative MALDI-TOF MS signal intensities (step 7) of the equal molar primers of the relevant sequences in the reaction for calibration and concentration normalization (Figure 2).
2. Proofreading Reactions
Note: The same proofreading reaction condition and protocol applies to a DNA repair of A-I, G-I, C-I, and T-I.
Using a T-G mismatch of substrate P21/T28 for the proofreading in Table 1 as an example, dilute primer P21 and template T28 to 12.5 pmol/µL with H2O; then transfer 12 µL of the diluted primer and 12 µL of the diluted template to a 1.5-mL sterilized microcentrifuge tube. NOTE: The amount of template/primer mix is sufficient for 2 reactions; proportionately increase the volume of the reagents accordingly for multiple reactions.
Close the tube tightly. Incubate it for 30 min in a covered 65-°C water bath, followed by 30 min in a 37-°C water bath, and finally on ice to ensure a proper annealing.
To a 1.5-mL sterilized microcentrifuge tube, add 8 µL of primer and template mix (50 pmol), 2 µL of 10x proofreading reaction buffer, 4 µL of 4 ddNTPs mix (2 mM of each 4 dNTPs), and H2O up to 18 µL. Flick the tube to mix. NOTE: 1x proofreading reaction buffer contains 50 mM NaCl, 10 mM MgCl2, 1 mM dithiothreitol, and 10 mM Tris-HCl (pH 7.9 at 25 °C). See the Table of Materials for the commercial source of 10x proofreading reaction buffer. The final concentration of each ddNTP in the reaction is 0.1 mM.
Dilute DNA polymerase in ice-cold 1x proofreading reaction buffer to the desired concentration [e.g., to a 1.5-mL microcentrifuge tube, add 4 µL of 1x proofreading reaction buffer and 1 µL of 5-U Klenow polymerase from a commercial source (Table of Materials)]. Store the diluted enzyme on ice at all times. NOTE: The amount of diluted DNA polymerase is sufficient for 2 reactions; proportionately increase the volume of the enzymes accordingly for multiple reactions. A volume of 2.0 µL, containing 2.0 units of Klenow polymerase, can proofread more than half of 50 pmol of T-G terminal mismatched P21/T28 in 5 min.
Prewarm the microcentrifuge tube with the substrate mix from step 2.3 to the desired reaction temperature (e.g., typically 37 °C for Klenow polymerase and 25 °C for T4 DNA polymerase). Then transfer 2.0 µL of the diluted DNA polymerase from step 2.4 to the prewarmed substrate mixture and flick the tube to mix its contents.
Centrifuge the tubes with enzyme and substrate for a few seconds at 3,200 x g at ambient temperature to spin down the components of the reactions. Immediately transfer the reaction to a heating block or a water bath at the desired incubation temperature as in step 2.5.
- Terminate the reaction
- Add an equal volume of buffered phenol, mix it by vortexing for a few seconds, and centrifuge it in a microcentrifuge at 3,200 x g at ambient temperature for 5 min.
- Transfer the aqueous phase to a clean microcentrifuge tube and add an equal volume of chloroform, mix it by vortexing for a few seconds and centrifuge it in a microfuge at 3,200 x g at ambient temperature for 3 min. Transfer the aqueous phase to a clean microfuge tube and incubate it at 95 °C for 5 min; then place it on ice. CAUTION: Phenol and chloroform are hazardous chemicals. Handling and waste disposal should follow institutional regulations.
- Alternatively, use an acid quenching protocol. For this, add 2 µL of 1 M HCl to stop the reaction on ice for 6 min, then add 0.64 µL of 1 M diethanolamine (DEA) to neutralize the pH of the reaction mixture and incubate it at 95 °C for 5 min; then place it on ice.
3. Resin Addition to Eliminate Salt Contamination
Using the resin scoop from the commercial desalting kit (see the Table of Materials), take sufficient resin to cover the dimples of the 384-dimple, 6-mg resin dimple plate.
Distribute the resin across all the dimples evenly (6 mg/dimple) by scraping across the top of the plate. Recover any excess resin and replace it in the bottle.
Transfer the final reaction products from step 2.7 from the microfuge tubes to a 384-well plate. Dispense 16 µL of H2O to dilute the final reaction products in each well and then seal the plate and centrifuge it to remove any bubbles.
Remove the seal, turn the 384-well plate with the reaction products upside down to cover the resin-filled dimple plate.
Turn over the well-to-dimple plates sandwich and carefully let the resin drop into the 384-well plate (be sure the 384-well plate is flush against the metal peg on the resin-filled dimple plate).
Remove the dimple plate on top, seal the 384-well plate containing the mixtures of the reaction products and resin, briefly centrifuge it at 3,200 x g to remove any bubbles and place it on a rotator for at least 15 min at room temperature for desalting its contents.
After the resin cleanup, centrifuge the 384-well plate at 3,200 x g for 5 min to compact the resin to the bottom of the wells.
4. Transfer Reaction Products to a Matrix Chip
Set the sample plates in the nanoliter dispenser (nanodispenser, see the Table of Materials).
Put the matrix chip (see the Table of Materials) and the 384-well plate from section 3.7 in the corresponding tray.
Operate the nanodispenser to spot the reaction products to the chip; push the RUN button on the touch screen of the nanodispenser to start.
Check the automated captured image of the sample spots containing saturation information on the screen to ensure the overall spotted volume on the chip is around 5 - 10 nL. Repeat the sample spotting if the volume is insufficient.
5. Setup the Assay Information on the Mass Spectrometry
Prepare a file in .xls format containing the anticipated signal information for importing. For example, use the setting of "P21_T28.xls" (Table 2) for the proofreading reaction of P21/T28 in steps 2, 3, and 4.
Create and define a new assay in the application program (see the Table of Materials) by right-clicking Import Assay Group in Designer Format and select the .xls file (e.g., "P21_T28.xls" from step 5.1).
Establish the target assay plate via the Customer:Project:Plate option tree on the left side of the screen by right-clicking the top of the tree and giving it a file name (e.g., "CTT20171201" represents the assay code and date).
Select the 384-well plate type and press OK; a blank plate will appear.
Select the appropriate assay (e.g., P21_T28) on the left side of the screen.
Assign the selected assay (e.g., P21_T28) for each sample spotted sample position on the chip by highlighting the well and right-clicking to select Add Plex.
Prepare a working list of all the tests on the chip in .xlsx format with no header (e.g., 1201.xlsx of Table 3). Import the working list via the Add New Sample Project option.
Assign the test in the working list (e.g., from 1201.xlsx) to each position of the P21_T28 assay and choose by right-clicking.
6. MALDI-TOF MS Operation
Link the mass spectrometer to the chip using the application program (see the Table of Materials).
Select the default setting at the right side of the screen.
Fill in the assay name from step 5.3 (e.g., CTT20171201), chip ID at the corner of the chip, and save the settings.
Start the MS control program (see the Table of Materials).
Press the In/Out button and take out the scout plate. Place the spotted chip from step 4.4 onto the scout plate and press the In/Out button for the spotted chip to enter the mass spectrometer.
Click the Acquire button of the application program (see the Table of Materials) to start the mass spectrometry and acquire data.
7. Data Analysis
Open the program for the data analysis (see Table of Materials).
Browse the tree of the database at the upper left corner and select the chip ID from step 6.3. Open the data file on the right side of the screen.
Click the spectrum icon to display the mass spectrum. To crop a specific range of m/z, right-click to select Customization Dialog and in the new window click on X-Axis and set the desired upper and lower limit and press OK to show the specified range of the spectrum.
- Export the spectrum for record keeping by right-clicking Export. NOTE: Using a scale of 1,600 width/1,200 unit is a reasonable size for data inspection on a computer screen.
- Select the JPEG file type, click on Destination, select Browse Disc (e.g., flash disc E:), type the file name (e.g., 1201-1.jpg), and then click on Export.
- For data quantitation, use the cursor and click on the peak to show the peak height at the upper left-hand corner.
- Alternatively, print out an exported JPEG file and measure the peak height with a ruler manually.
Representative Results
Templates and primers:
Using the procedure presented here, equal molar synthetic oligonucleotide templates and primers of relevant sequences obtained from commercial sources were checked for their purity and quality (Figure 3A; note the signals matched the designated mass and the low background) as well as for the relationship between the peak intensity and the analyte mass (Figure 2A). The peak heights were measured and calculated (Figure 2B) for the MS data normalization.
Reaction condition and mass spectra:
The use of single base primer extension reactions coupled with MALDI-TOF MS for genotyping has previously been shown to be feasible using standard ddNTPs terminators17. The proofreading assay using standard ddNTPs is similar to a singleplex reaction of genotyping but is much simpler and very easy to perform to obtain clean and reliable results.
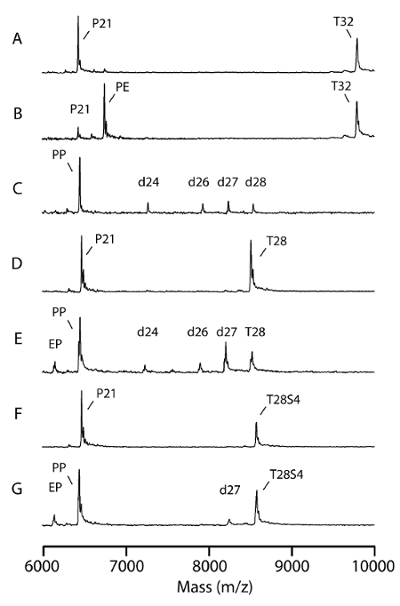
The MS spectra covered all the signals generated from both primers and templates (Figure 3). The proper design of the primers and templates generated a well-separated signal profile for both the templates and the primers (Figure 3). According to the m/z value, signals from templates and their non-specific partial degradation products (m/z > 7,000) were well separated from the mismatched primers and proofread products (m/z < 7,000 in Figure 3). If necessary, introducing nuclease resistant phosphorothioate bonds to replace the last 4 phosphodiester bonds at the 3' end of the template (Table 1; T28S4/P21) can reduce non-specific template hydrolysis (Figure 3G; d27).
In the absence of dNTPs, KF has a robust 3'-exonuclease and is capable of degrading both the primer and the template in vitro21. Like the native 4 dNTPs, the addition of the 4 ddNTPs prevents a 3' end degradation by KF (Figure 3C and 3E). Likewise, a one nucleotide extension of the 3'-ends adds increased stability to the primer. Using pol I proofreading conditions as previously described20 with 10 U (46 pmol) KF, more than 80% of the 50-pmol mismatched substrates were corrected in 5 min (Figure 3C, 3E, and 3G).
Intermediates and mechanism analysis:
MALDI-TOF MS resolution can be as fine as 1 dalton; indeed, all substrates, products, and intermediates with mass changes in the reaction can be resolved in the same MS spectrum of the selected range. For example, in a terminal T-G mismatch, the mismatched G at the primer terminus was corrected with the incorporation of ddA with an m/z difference of only 32. Such an m/z difference can be easily identified on the MS spectrum in an m/z range from 5,000 to 7,000 (Figure 4A). The change of each individual signal can be calculated by summing up all signals from the mismatched primer, excision intermediates, and proofread products as 100%. For example, in the T-G proofreading reaction, the corrected product relative to the excision product was 23% versus 17% at 5 min, 31% versus 18% at 10 min, and 69% versus 14% at 20 min (Figure 4A).
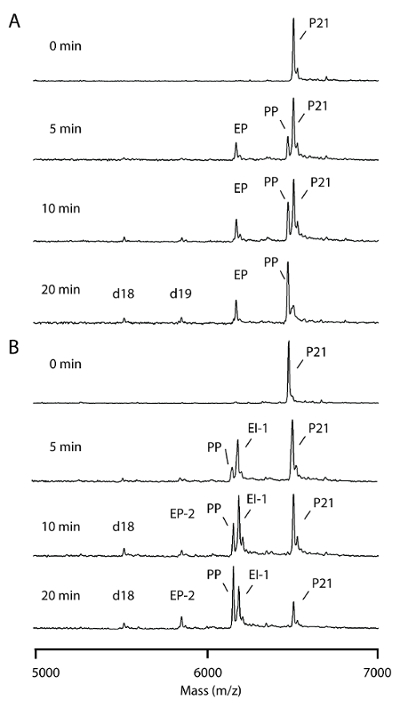
When a penultimate T-G mismatch is present in the primer, the last 2 nt should be excised followed by a ddA addition to complete the correction. Indeed, the correct products increased with time from 12% at 5 min, 28% at 10 min, to 45% at 20 min of the reaction. In addition, two forms of excision products for a penultimate T-G proofreading, the 1-nt excision intermediate and 2-nt excision products, were easily resolvable (Figure 4B).
Proofreading kinetics with single dNTP:
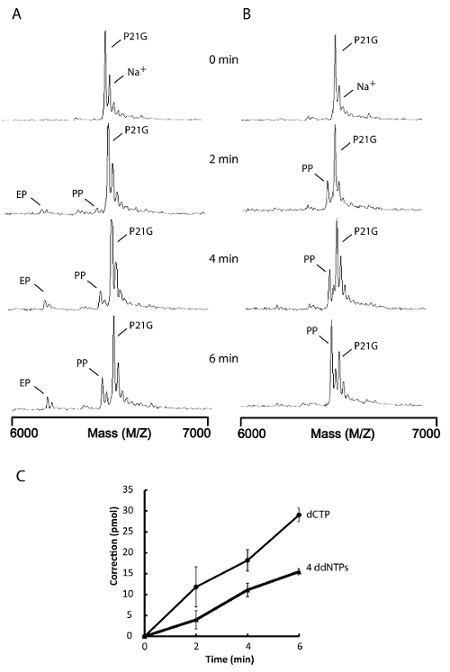
For E. coli DNA pol I, ddNTPs are not suitable substrates and should be regarded as inhibitors22. Alternatively, using a single 'correct' dNTP in the proofreading reaction instead will also suppress non-specific 3'-5' exonuclease activity and the generation of single nucleotide extension products suitable for MS analysis (Figure 5B). The proofreading rate obtained from reactions containing a single dCTP is about two times higher than reactions containing the 4 ddNTPs (Figure 5A, 5B, and 5C). Thus, the use of a single 'correct' dNTP in the proofreading assay yields a better result without the inhibitory effects of ddNTPs. This approach should provide greater sensitivity for proofreading kinetic analyses.
Proofreading of internal mismatches:
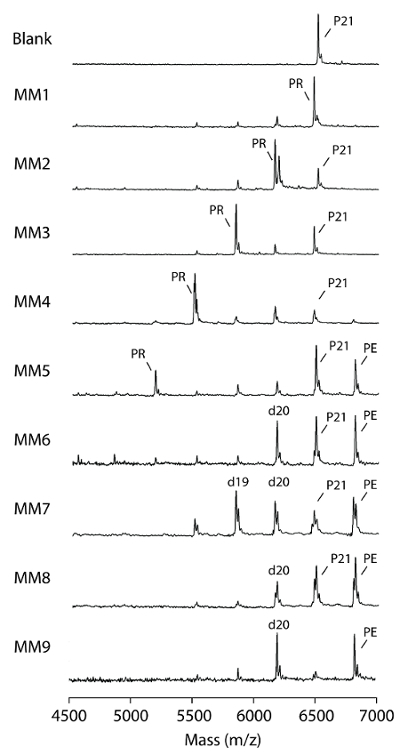
MS proofreading assays using KF for internal mismatches and the identification of proofread products are straightforward (i.e.,the proofreading exonuclease excises nucleotides from the 3' end to the internal mismatch of the primers), followed by the addition of matched ddNMP to the excision products via the polymerase function generating proofread products shorter than the primer substrates (Figure 6). A clear trend of proofreading efficiency was observed, where KF efficiently proofread mismatches at the last, penultimate, 3rd and 4th nucleotides from the 3' end of the primers (Figure 6; MM1, 2, 3, and 4). Mismatches at 5 nt from the 3' end of the primer were corrected partially with < 15% of mismatch correction, and > 15% of the primer showed an extension without proofreading (Figure6; MM5). KF failed to proofread mismatches positioned at 6, 7, 8, and 9 nt from the 3' end of the primer (Figure 6; MM6, 7, 8, and 9) but still extended a significant proportion of substrates (Figure 6; PE of MM6, 7, 8, and 9).
Repair of deoxyinosine lesion:
In E. coli, deoxyinosine in DNA is primarily processed by the endo V repair pathway20,23. Endo V initiates the reaction by an incision at the second phosphodiester bond 3' of the dI lesion. The strand break generated is believed to be used by pol I for a subsequent lesion excision and DNA resynthesis24. This procedure was applied to test pol I repair substrates containing dI at the 3' penultimate site (Table 1; A-I, C-I, G-I, and T-I) to validate this hypothesis. The repair of the 3' penultimate dI is analogous to the proofreading of the penultimate T-G mismatch (Figure 4B and 6; MM2 entry), in which the last 2 nt should be removed followed by an addition of a correct ddNMP to complete the repair. MS patterns clearly showed pol I corrected heteroduplexes of A-I, G-I, and T-I, albeit with different efficiencies (Figure 7; RP signals). However, the C-I substrate was refractory to the pol I repair (Table 1 and Figure 7D and 7E; very low RP) and demonstrated the primer extension to a great extent (Figure 7D; PE signal).
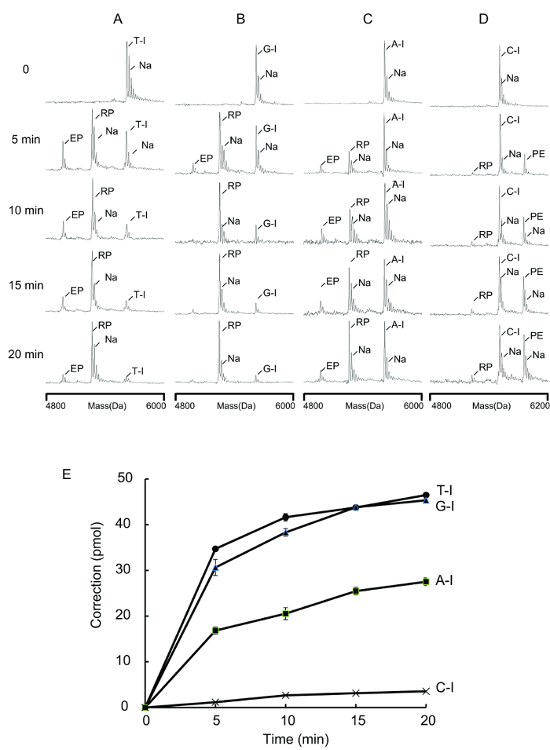
Figure 1. Model system for proofreading assay. A mismatched primer was annealed as indicated to a template forming a terminal mismatch (bold). Proofreading exonuclease of DNA polymerase would detect and remove the mismatched nucleotide forming an excision product. In the presence of a DNA polymerase and four ddNTPs, the excision product is extended by a single base complementary to the previous mismatched site. When subjected to MALDI-TOF, the difference in mass between the mismatched primer, the excision products, and the proofread products can be resolved. (This figure is adapted from Su et al.25 with permission.) Please click here to view a larger version of this figure.
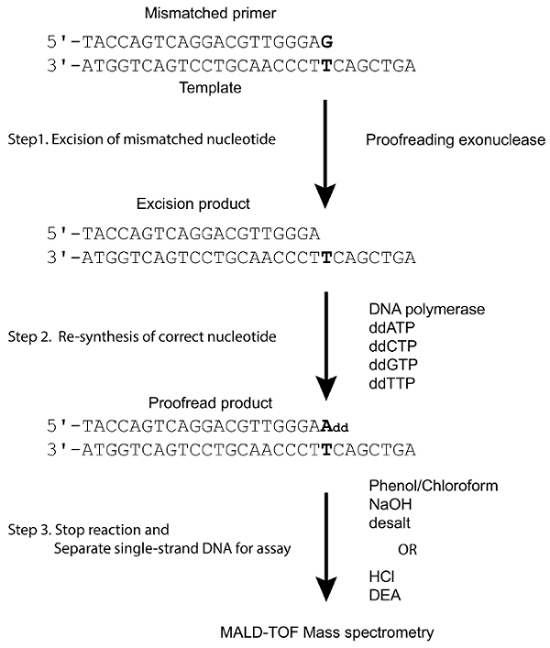
Figure 2. Peak intensity and oligonucleotide mass. Eight oligonucleotides from 17 to 24 nt with sequences complementary to the 3' end of T28 (Table 1) were tested. (A) A mixture of 50 pmol of each oligonucleotide in 40-µL solutions was subjected to an MS analysis. (B) The peak height of 17 nt was used as 100% for the calculation. The relative signal intensity for each oligonucleotide was the average of six determinations and the error bars represent 1 standard deviation. (This figure is adapted from Su et al.25 with permission.) Please click here to view a larger version of this figure.
Figure 3. Effect of various template design. Proofreading reactions were assessed with the P21 primer forming a terminal T-G mismatch with different templates. The proofreading assay was performed with 10 U (43 pmol) Klenow polymerase, 50 pmol substrate, and 4 ddNTPs at 37 °C for 5 min. (A) This panel shows a P21/T32 T-G substrate without enzyme. (B) This panel shows a primer extension of a P21/T32 T-A substrate by 3' exonuclease deficient Klenow. PE = primer extension product. (C) This panel shows a proofreading of the P21/T32 T-G substrate. PP = proofreading product; d26, d27, d28 = 3' end-degraded template. (D) This panel shows a P21/T28 T-G substrate without enzyme. (E) This panel shows a proofreading of the P21/T28 T-G substrate. PP = proofreading product; d26, d27 = 3' end-degraded template. (F) This panel shows a P21/T28S4 T-G substrate without enzyme. (G) This panel shows a proofreading of the P21/T28S4 T-G substrate. PP = proofreading product; EP = excision products; d27 = template degradation by-product. (This figure is adapted from Su et al.25 with permission). Please click here to view a larger version of this figure.
Figure 4. Time course analysis for excision products and proofreading products. Proofreading assays were performed with 2 U (8.6 pmol) KF and 50 pmol substrates as described in the protocol. The reactions were quenched at the indicated times. (A) This panel shows a proofreading of a 3' terminal T-G mismatch; EP = proofreading excision product; d18 & d19 = excess excision by-products; PP = proofreading products; P21 = mismatched primer. (B) This panel shows a proofreading of a 3'-penultimate T-G mismatch; EI-1 = excision intermediate; EP-2 = proofreading excision products; d18 = excess excision by-product; PP = proofreading products; P21 = mismatched primer; Na = sodium ion bound nucleotides. (This figure is adapted from Su et al.25 with permission.) Please click here to view a larger version of this figure.
Figure 5. Proofreading reaction with 4 ddNTPs versus a single dNTP. Proofreading assays were performed with 0.1 U (0.43 pmol) KF and 50 pmol substrates as described in the protocol. The reactions were quenched at the indicated times. (A) This panel shows a proofreading of a 3' terminal G-G mismatch with 4 ddNTPs; EP = proofreading excision product; PP = proofreading resynthesis products; P21G = mismatched primer. (B) This panel shows a proofreading of a 3' terminal G-G mismatch with dCTP; PP = proofreading resynthesis products; P21G = mismatched primer. (C) The correction levels are the average of three determinations and the error bars represent 1 standard deviation. Na+ = sodium ion bound nucleotides. (This figure is adapted from Su et al.25 with permission.) Please click here to view a larger version of this figure.
Figure 6. Proofreading of internal mismatches. Proofreading assays were performed with 2 U (8.6 pmol) Klenow polymerase and 50 pmol substrates in 37 °C for 20 min as described in the protocol. Blank = enzyme blank of an MM1 substrate. MM1 = a P21/T28 T-G substrate; MM2 to MM9 = substrates with internal mismatches, the numbers indicate the mismatch position relative to 3' end. P21 = mismatched primers; PR = proofread products; PE = extended primers; d19 and d20 = excess excision by-products, the numbers represent the size of nucleotides. (This figure is adapted from Su et al.25 with permission.) Please click here to view a larger version of this figure.
Figure 7. Repair of 3' penultimate deoxyinosine lesions. Repair assays were performed with 2 U Klenow polymerase and 50 pmol substrates in 37 °C as described in protocol step 2.3. The reactions were quenched at the indicated times. These panels show reactions with (A) T-I substrate; (B) G-I substrate; (C) A-I substrate; and (D) C-I substrate. RP = repair products; EP = excision products; PE = primer extension products, Na+ = sodium ion adducts. (E) The correction levels for T-I, G-I, and A-I were the average of three determinations and the error bars represent 1 standard deviation. The correction levels for C-I were the average of two determinations. (This figure is adapted from Su et al.25 with permission.) Please click here to view a larger version of this figure.
| Substrates | Sequences | Correction efficiency (pmol) |
| P21/T32 | 3'-TAATATGGTCAGTCCTGCAACCCTTCAGCTGA | 17.3 |
| 5'- TACCAGTCAGGACGTTGGGAG | ||
| P21/T28 | 3'-ATGGTCAGTCCTGCAACCCTTCAGCTGA | 18.4 |
| 5'-TACCAGTCAGGACGTTGGGAG | ||
| P21/T28S4 | 3'-ATGGTCAGTCCTGCAACCCTTCAGCTGA | 22.9 |
| MM1 | 5'-TACCAGTCAGGACGTTGGGAG | |
| MM2 | 3'-ATGGTCAGTCCTGCAACCCTTCAGCTGA | 13.5 |
| 5'-TACCAGTCAGGACGTTGGGGA | ||
| MM3 | 3'-ATGGTCAGTCCTGCAACCCTTCAGCTGA | 16.9 |
| 5'-TACCAGTCAGGACGTTGGAAA | ||
| MM4 | 3'-ATGGTCAGTCCTGCAACCCTTCAGCTGA | 15.1 |
| 5'-TACCAGTCAGGACGTTGAGAA | ||
| MM5 | 3'-ATGGTCAGTCCTGCAACCCTTCAGCTGA | 4.2 |
| 5'-TACCAGTCAGGACGTTAGGAA | ||
| MM6 | 3'-ATGGTCAGTCCTGCAACCCTTCAGCTGA | < 0.5* |
| 5'-TACCAGTCAGGACGTCGGGAA | ||
| MM7 | 3'-ATGGTCAGTCCTGCAACCCTTCAGCTGA | < 0.5 |
| 5'-TACCAGTCAGGACGCTGGGAA | ||
| MM8 | 3'-ATGGTCAGTCCTGCAACCCTTCAGCTGA | < 0.5 |
| 5'-TACCAGTCAGGACATTGGGAA | ||
| MM9 | 3'-ATGGTCAGTCCTGCAACCCTTCAGCTGA | < 0.5 |
| 5'-TACCAGTCAGGATGTTGGGAA | ||
| A-I | 3'-ACCAGCGACCCCTTATACAGTCAT | 8.2 |
| 5'-TGGTCGCTGGGGAATAIG | ||
| C-I | 3'-ACCAGCGACCCCTTATCCAGTCAT | 1.0 |
| 5'-TGGTCGCTGGGGAATAIG | ||
| G-I | 3'-ACCAGCGACCCCTTATGCAGTCAT | 15.4 |
| 5'-TGGTCGCTGGGGAATAIG | ||
| T-I | 3'-ACCAGCGACCCCTTATTCAGTCAT | 16.6 |
| 5'-TGGTCGCTGGGGAATAIG |
Table 1. Proofreading substrates and correction efficiency by Klenow fragment. This table shows the bold 4-nucleotides at the 3'-end of the template containing phosphorothioate modifications. The mismatched base pairs are in bold. The proofreading assays were performed with 2 U (8.6 pmol) Klenow polymerase, 50 pmol substrate, and 4 ddNTPs at 37 °C for 10 min. * These values are the lower limits for the proofreading level because the background noise prevents accurate estimates of < 1% of the total mass signals observed. (This figure is adapted from Su et al.25 with permission.)
| WELL | TERM | SNP_ID | 2nd-PCRP | 1st-PCRP | AMP_LEN | UP_CONF | MP_CONF | Tm | PcGC | PWARN | UEP_DIR | UEP_MASS | UEP_SEQ | EXT1_CALL | EXT1_MASS |
| W1 | iPLEX | T28 | NA | NA | NA | NA | NA | NA | NA | F | 8524.54 | ATGGTCAGTCCTGCAACCCTTCAGCTGA | |||
| W1 | iPLEX | P21 | NA | NA | NA | NA | NA | NA | NA | F | 6495.24 | TACCAGTCAGGACGTTGGGAA |
Table 2. T28_P21.xlsx file. The setting was used for Figure 3D, 3E, 4A, and 5A.
| 1201-1 |
| 1201-2 |
| 1201-3 |
| 1201-4 |
| 1201-5 |
| 1201-6 |
| 1201-7 |
Table 3. 1201.xlsx file. The setting was used for 7 reactions in Figure 4A.
Discussion
This study described a step-by-step proofreading activity assay analyzed by the chosen commercial instrument (see the Table of Materials) using MALDI-TOF MS. The major advantages include that the primer and template are label free and easy to perform, allowing for greater flexibility in designing experiments. A stream-lime complete processing of 30 proofreading tests would take 4 h, including 3 h for manually performing the proofreading reactions and their cleanup, while the MALDI-TOF MS analyses using the aforementioned protocol takes 1 h to complete. The primary drawback of this method is the amount of substrate required for the assay. To obtain strong signals and consistent results, we used 50 pmol of DNA substrate in a 20-µL reaction; thus, the amount of DNA is much higher than with radiolabeled DNA experiments8,9,10. Reducing the volumes and the sample size and fine-tuning the MS operation can lower the amount of substrate required.
Synthetic oligonucleotides obtained from commercial sources can be used for the proofreading study directly without any further purification or treatment. However, it is advisable to check the purity and quality of the DNA by MALDI-TOF MS. The phosphorothioate bonds were introduced to replace the last 4 phosphodiester bonds at the 3' end of the template (Table 1) to reduce the non-specific degradation of the template strand (Figure 3G), albeit at a higher cost per sample. Interestingly, the signals from the template and primer DNA can be well separated by MS by virtue of their differences in size; thus, this phosphorothioate modification is not required. In Figure 3E, for example, all the possible products of the primer P21 extension and the proofreading excision and resynthesis were less than 22 nt (m/z < 7,000), while all the degradation fragments from template T28 were greater than 24 nt (m/z > 7,000). The results in Figure 4A were from the same P21/T28 duplex set as those in Figure 3E. An enlarged window of m/z 6,000-7,000 shows only signals from the primer DNA without any confounding signals from the template.
The strategy of using primers with a ddNMP extension proved to be very stable, and, thus, ddNMP containing products are good indicators for the proofreading assay. The addition of ddNMP to excision products could 'freeze' the proofreading products in the state for analysis immediately post-excision. Alternatively, using a single correct dNTP (Figure 5B) instead of 4 ddNTPs to suppress the non-specific hydrolytic activity of the proofreading exonuclease is also feasible.
Stopping the reactions with a standard phenol/chloroform extraction procedure proved to be the best means for the reaction termination since, in one step, it enables the reactions to be stopped while also removing any protein interference. Alternatively, acid quenching, which is amenable to automation, then neutralized by non-metal alkaline without a phase separation, also demonstrated reliable results. Sodium ion adducts (Na+ in Figure 4, 5, and 7) are of concern since they can increase background and complicate the signal discrimination. A proper use of the clean resin can reduce such interference; yet, even when such adducts are present, they normally have much smaller peaks relative to their parent-signal, and this property can be used to distinguish between them (Figure 4, 5, and 7). Generally, the peak height of sodium adducts was proportional to the major peaks; the inclusion or exclusion of sodium adducts in the calculation did not significantly affect the quantification results. Therefore, for the quantification, we used the major peak height only for the calculation.
The proofreading experiments described here took advantage of internal mismatches to demonstrate the importance of the exonuclease domain partition assay of KF26 and the detailed DNA polymerase I capability to efficiently proofread the mismatch at up to 4 nt upstream from the primer 3'-end (Figure 5, MM1 - 4) possibly due to the base-pairing stability at the primer-template junctions27. In addition, this study also used primers containing penultimate deoxyinosine lesions to study a pol I repair of endo V-nicked repair intermediates. The results showed that T-I was repaired more efficiently than A-I and G-I; however, C-I was refractory to the pol I repair (Table 1 and Figure 7). These results confirm that a MALDI-TOF MS analysis can be very useful for proofreading and various short-patch DNA repair assays28 due to its high resolution.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank the NCFPB Integrated Core Facility for Functional Genomics (Taipei, Taiwan) and the NRPB Pharmacogenomics Lab (Taipei, Taiwan) for their technical support. This work was supported by research grants from the Taiwan Health Foundation (L-I.L.) and the Ministry of Science and Technology, Taipei, Taiwan, ROC [MOST 105-2320-B-002-047] for Woei-horng Fang, [MOST 105-2628-B-002 -051-MY3] for Kang-Yi Su, and [MOST-105-2320-B-002-051-MY3] for Liang-In Lin.
References
- Loeb LA, Kunkel TA. Fidelity of DNA synthesis. Annual Review of Biochemistry. 1982;51:429–457. doi: 10.1146/annurev.bi.51.070182.002241. [DOI] [PubMed] [Google Scholar]
- Kornberg A, Baker T. DNA replication. Oxford, UK: W.H. Freeman; 1992. [Google Scholar]
- Carroll SS, Benkovic SJ. Mechanistic aspects of DNA polymerases: Escherichia coli DNA polymerase I (Klenow fragment) as a paradigm. Chemical Reviews. 1990;90(7):1291–1307. [Google Scholar]
- Echols H, Goodman MF. Fidelity Mechanisms in DNA Replication. Annual Review of Biochemistry. 1991;60(1):477–511. doi: 10.1146/annurev.bi.60.070191.002401. [DOI] [PubMed] [Google Scholar]
- Johnson KA. The kinetic and chemical mechanism of high-fidelity DNA polymerases. Biochimica et Biophysica Acta. 2010;1804(5):1041–1048. doi: 10.1016/j.bbapap.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reha-Krantz LJ. DNA polymerase proofreading: Multiple roles maintain genome stability. Biochimica et Biophysica Acta. 2010;1804(5):1049–1063. doi: 10.1016/j.bbapap.2009.06.012. [DOI] [PubMed] [Google Scholar]
- Fidalgo da Silva E, Reha-Krantz LJ. DNA polymerase proofreading: active site switching catalyzed by the bacteriophage T4 DNA polymerase. Nucleic Acids Research. 2007;35(16):5452–5463. doi: 10.1093/nar/gkm591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petruska J, Goodman MF. Influence of neighboring bases on DNA polymerase insertion and proofreading fidelity. The Journal of Biological Chemistry. 1985;260(12):7533–7539. [PubMed] [Google Scholar]
- Mendelman LV, Petruska J, Goodman MF. Base mispair extension kinetics. Comparison of DNA polymerase α and reverse transcriptase. The Journal of Biological Chemistry. 1990;265(4):2338–2346. [PubMed] [Google Scholar]
- Lutz S, Burgstaller P, Benner SA. An in vitro screening technique for DNA polymerases that can incorporate modified nucleotides. Pseudothymidine as a substrate for thermostable polymerases. Nucleic Acids Research. 1999;27(13):2792–2798. doi: 10.1093/nar/27.13.2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Silva EF, Mandal SSS, Reha-Krantz LJ. Using 2-aminopurine fluorescence to measure incorporation of incorrect nucleotides by wild type and mutant bacteriophage T4 DNA polymerases. The Journal of Biological Chemistry. 2002;277(43):40640–40649. doi: 10.1074/jbc.M203315200. [DOI] [PubMed] [Google Scholar]
- Frey MW, Sowers LC, Millar DP, Benkovic SJ. The nucleotide analog 2-aminopurine as a spectroscopic probe of nucleotide incorporation by the Klenow fragment of Escherichia coli polymerase I and bacteriophage T4 DNA polymerase. Biochemistry. 1995;34(28):9185–9192. doi: 10.1021/bi00028a031. [DOI] [PubMed] [Google Scholar]
- Leung KH, et al. A highly sensitive G-quadruplex-based luminescent switch-on probe for the detection of polymerase 3'-5' proofreading activity. Methods. 2013;64(3):224–228. doi: 10.1016/j.ymeth.2013.05.017. [DOI] [PubMed] [Google Scholar]
- Song C, Zhang C, Zhao M. Rapid and sensitive detection of DNA polymerase fidelity by singly labeled smart fluorescent probes. Biosensors and Bioelectronics. 2011;26(5):2699–2702. doi: 10.1016/j.bios.2010.08.073. [DOI] [PubMed] [Google Scholar]
- Haff LA, Smirnov IP. Single-nucleotide polymorphism identification assays using a thermostable DNA polymerase and delayed extraction MALDI-TOF mass spectrometry. Genome Research. 1997;7(4):378–388. doi: 10.1101/gr.7.4.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haff LA, Smirnov IP. Multiplex genotyping of PCR products with MassTag-labeled primers. Nucleic Acids Research. 1997;25(18):3749–3750. doi: 10.1093/nar/25.18.3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondal T, et al. A novel MALDI-TOF based methodology for genotyping single nucleotide polymorphisms. Nucleic Acids Research. 2003;31(24):e155. doi: 10.1093/nar/gng156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su KY, et al. Pretreatment Epidermal Growth Factor Receptor (EGFR) T790M mutation predicts shorter EGFR tyrosine kinase inhibitor response duration in patients with non-small-cell lung cancer. Journal of Clinical Oncology. 2012;30(4):433–440. doi: 10.1200/JCO.2011.38.3224. [DOI] [PubMed] [Google Scholar]
- Lee CC, et al. Endonuclease V-mediated deoxyinosine excision repair in vitro. DNA Repair. 2010;9:1073–1079. doi: 10.1016/j.dnarep.2010.07.007. [DOI] [PubMed] [Google Scholar]
- Lee CC, et al. The excision of 3' penultimate errors by DNA polymerase I and its role in endonuclease V-mediated DNA repair. DNA Repair. 2013;12(11):899–911. doi: 10.1016/j.dnarep.2013.08.003. [DOI] [PubMed] [Google Scholar]
- Kucera RB, Nichols NM. DNA-Dependent DNA Polymerases. Current Protocols in Molecular Biology. 2008;84(1):3.5.1–3.5.19. doi: 10.1002/0471142727.mb0305s84. [DOI] [PubMed] [Google Scholar]
- Tabor S, Richardson CC. A single residue in DNA polymerases of the Escherichia coli DNA polymerase I family is critical for distinguishing between deoxy- and dideoxyribonucleotides. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(14):6339–6343. doi: 10.1073/pnas.92.14.6339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss B. Removal of deoxyinosine from the Escherichia coli chromosome as studied by oligonucleotide transformation. DNA Repair. 2008;7(2):205–212. doi: 10.1016/j.dnarep.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao W. Endonuclease V: an unusual enzyme for repair of DNA deamination. Cellular and Molecular Life Sciences. 2013;70(17):3145–3156. doi: 10.1007/s00018-012-1222-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su KY, et al. Application of single nucleotide extension and MALDI-TOF mass spectrometry in proofreading and DNA repair assay. DNA Repair. 2018;61:63–75. doi: 10.1016/j.dnarep.2017.11.011. [DOI] [PubMed] [Google Scholar]
- Carver TE, Hochstrasser RA, Millar DP. Proofreading DNA: recognition of aberrant DNA termini by the Klenow fragment of DNA polymerase I. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(22):10670–10674. doi: 10.1073/pnas.91.22.10670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santa Lucia J, Jr, Hicks D. The thermodynamics of DNA structural motifs. Annual Review of Biophysics and Biomolecular Structure. 2004;33:415–440. doi: 10.1146/annurev.biophys.32.110601.141800. [DOI] [PubMed] [Google Scholar]
- Su KY, et al. DNA polymerase I proofreading exonuclease activity is required for endonuclease V repair pathway both in vitro and in vivo. DNA Repair. 2018;64:59–67. doi: 10.1016/j.dnarep.2018.02.005. [DOI] [PubMed] [Google Scholar]