

Abstract
Polyamine-based Peptide Amphiphiles (PPAs) are a new class of self-assembling amphiphilic biomaterials-related to the peptide amphiphiles (PAs). Traditional PAs possess charged amino acids as solubilizing groups (lysine, arginine), which are directly connected to a lipid segment or can contain a linker region made of neutral amino acids. Tuning the peptide sequence of PAs can yield diverse morphologies. Similarly, PPAs possess a hydrophobic segment and neutral amino acids, but also contain polyamine molecules as water solubilizing (hydrophilic) groups. As is the case with PAs, PPAs can also self-assemble into diverse morphologies, including small rods, twisted nano-ribbons, and fused nano-sheets, when dissolved in water. However, the presence of both primary and secondary amines on a single polyamine molecule poses a significant challenge when synthesizing PPAs. In this paper, we show a simple protocol, based on literature precedents, to achieve a facile synthesis of PPAs using solid phase peptide synthesis (SPPS). This protocol can be extended to the synthesis of PAs and other similar systems. We also illustrate the steps that are needed for cleavage from the resin, identification, and purification.
Keywords: Chemistry, Issue 136, Peptide amphiphiles, Self-assembly, Biomaterials, Polyamine peptide amphiphiles, Peptide synthesis, Orthogonal protecting groups, nanofiber, nanoparticle
Introduction
Self-assembling peptide amphiphiles (PAs) are a class of biomaterials typically comprised of the following segments: (a) hydrophilic head, (b) linker region, and (c) hydrophobic tail. Most PAs described in the literature possess a hydrophilic head comprised of charged or polar amino acid residues1,2,3,4. PAs have found a wide range of applications in biomedicine including drug delivery, disease diagnostics, regenerative medicine, etc.5. Based on their amino acid sequence, PAs can form a wide variety of nanostructures including spherical micelles, and nano-filaments. We have recently reported a class of hybrid polyamine-based peptide amphiphiles, termed PPAs6. The morphologies, self-assembling kinetics, and metabolic degradation, of these biomaterials, were found to be related to their solubilizing head group. Moreover, the PPA nanostructures did not show toxicity towards mammalian cells (MiaPaCa2 and HeLa cell lines) at the tested concentrations. PPA-based nanocarriers are attractive drug-delivery vehicles because: (1) polyamine uptake and metabolism has been shown to be increased in cancerous cells, (2) cationic nanostructures can achieve endosomal escape7,8, which leads to higher circulation and residence within a cell, and (3) they should have a distinct metabolic profile when compared with PA; for example, they will be more stable towards proteases found in the human body (although they maybe sensitive to other enzymes, such as amine oxidases)9,10. Also, PPAs have been found to have diverse morphologies, physicochemical properties, nanoparticle stiffness, and assembly kinetics depending on the length and charge of individual PPA molecule6. Herein, we describe a detailed protocol for the synthesis, identification, and purification of PPAs that can also be applied to the preparation of PAs or similar hybrid peptide molecules.
Because polyamines are not commonly commercially available in their protected forms, and because protecting the primary and secondary amines of polyamines is of utmost importance for conjugating them with amino acids and other molecules, we have outlined the synthetic steps to achieve their protection. The overall goal of this protocol is to provide a simple method for conjugating polyamines to amino acids. Polyamines lack a carboxylic group; thus, they cannot be coupled to Rink Amide or Wang resins. Instead, resins such as 2-chlorotrityl chloride are recommended for the synthetic protocol. The main challenge for PPA synthesis is the presence of primary and secondary amine functional groups. For our purposes, we protected all the secondary amines in the polyamine while keeping the primary amino group on the polyamine free to allow the coupling reaction. The reaction was done on a solid support following the principles of solid phase peptide synthesis (SPPS) to facilitate the work-up after each coupling and deprotection step. The following protocol is for both the manual and automated synthesis of PPAs (although the verification of some steps will be challenging in an automated system). The synthesis of these molecules can also be carried out on an automated synthesizer or with the aid of a microwave reactor (automated or semi-automated). The reaction scheme has been summarized in Figure 1.
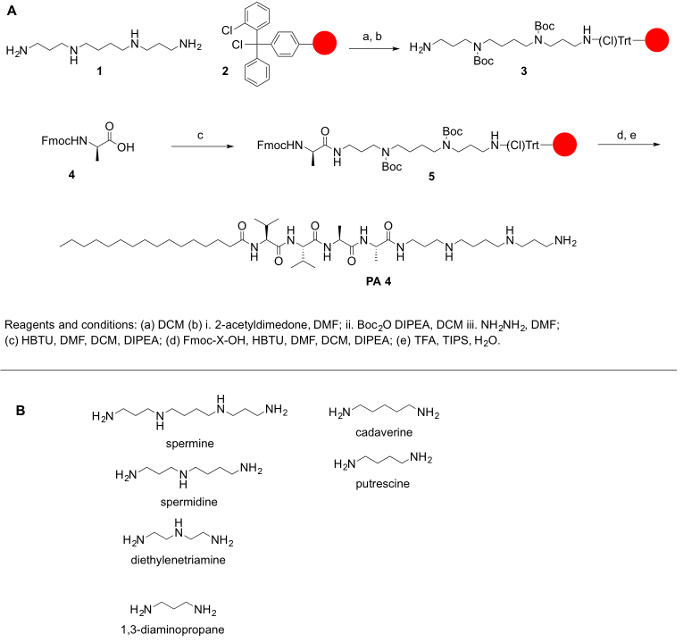
Figure 1: (A) A general reaction scheme for the synthesis of PPAs. (B) Representative polyamines that can be used to synthesized PPAs described here. Please click here to view a larger version of this figure.
Protocol
1. General Protocol for Synthesis of PPAs
Calculate the scale of synthesis (usually mmol). This scale is based on the mass of the target PPA amount. Keep in mind that the reaction efficiency of SPPS gradually decreases with an increase of amino acid sequence. Therefore, the exact reaction efficiency is difficult to calculate.
Calculate the weight of the resin to be used according to the loading of the resin. The loading is found on the container or the analysis protocol of the resin and expressed in mmol/g. The following formula can be used for calculating the weight of the resin:
Carefully weigh out 2-chlorotrityl chloride resin (in our case the loading is ~0.85 mmol)
Place the resin in a fritted synthesis vessel with the following specifications: Capacity – 50 mL, Fritted Disc – 25 mm, Porosity – Medium.
Add 15 mL of dichloromethane (DCM) to the resin.
Affix the synthesis vessel to a variable speed mechanical shaker (flask shaker/stirrer), and turn the vessels to a 45° angle (or horizontally) to maximize agitation.
Allow the resin beads to swell for 15 min. Swelling enhances the yield of reaction because it facilitates molecular diffusion and accessibility to the active site, enhancing coupling efficiency.
Add 8 equivalents (2 mmol for a 0.25 mmol scale) of the desired polyamine (Spermine, Spermidine, Diethyelenetriamine, 1,3-diaminopropane, etc.) to the resin and allow it to react for 5 h. Note: A lesser reaction period would reduce yield.
Do a Kaiser test (see Table of Materials) to confirm successful coupling of the polyamine to the resin. Successful coupling of the primary amines yields a violet/blue color, which corresponds to the free amine.
Protect the primary amine group using 4 equivalents of 1-(4,4-dimethyl-2,6-dioxocyclohex-1-ylidene)ethyl (Dde), dissolved in anhydrous methanol (15 mL), by shaking the reaction mixture overnight11. Note: A lesser reaction time reduces yield.
Perform a Kaiser test. An absence of blue color from the resin bead confirms successful protection. In case of unsuccessful protection of the primary amine, repeat the previous step.
Drain the DCM and wash the resins twice with a mixture of DCM and DMF (2:1, 15 mL).
Dissolve 20 equivalents (5 mmol) of di-tert butyl di-carbonate (Boc) in DCM (20 mL) and allow the reaction to proceed for 3 h.
Do a chloranil test (see Table of Materials) to confirm complete protection of the secondary amine. A positive test (protection) yields a colorless or light yellow color. Free secondary amines give a dark green/blue color, while primary amines give a red color.
Drain the solvent mixture and wash twice with a mixture of DCM and DMF (2:1, 15 mL).
Add a 2% solution of hydrazine in DMF (10 mL) and shake for 1 h.
Do a Kaiser test to confirm successful deprotection of the primary amine.
- Add the desired amino acids in the reverse sequence in which you would write/draw them. Peptides are drawn from the N termini to the C termini but are synthesized in the opposite direction, C to N. For example, for a PPA requiring the following peptide core -GLFD-, add the Aspartic acid (D), followed by Phenylalanine (F), Leucine (L), and finally Glycine (G). NOTE: For most amino acids, the following coupling cocktail is appropriate for attachment to the polyamine loaded resins.
- Mix 4 equivalents (1 mmol) of the Fmoc-protected amino acid, 3.95 equivalents of 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU), and 15 equivalents of diisopropylethyl amine (DIPEA).
- Dissolve them in a mixture of DCM and DMF (1:1, 15 mL) and sonicate the cocktail for 2 min (until complete dissolution).
- Wait an additional 3–5 min to ensure activation of the carboxylic acid on the amino acid.
- Add the reaction mixture to the vessel. Carry out the reaction for 2-4 h at ambient temperature. NOTE: We have found the following as efficient alternatives to HBTU (usually requiring lower amounts of the amino acids and the coupling agent): COMU, PyBOP, HATU, DIC/Oxima.
- Do a Kaiser test to confirm successful coupling.
- De-protect the Fmoc-group from the amino acid by adding a 20% solution of 4-methyl piperidine (10 mL) in DMF. Do it twice, each time shaking the reaction mixture for 15 min. NOTE: An efficient alternative for Fmoc removal is piperazine/DBU12.
- Perform a DCM (15 mL) wash between additions.
- Do a Kaiser test to confirm successful de-protection of the amino acid.
- Wash the resin thoroughly to remove all traces of 4-methyl. Wash the resin twice with DMF (10 mL), each wash lasting for 5 min, and finally with DCM (10 mL) for 10 min.
- Add all the remaining amino acids by successively repeating the coupling and de-protection steps.
- Finally, after coupling all the required amino acids, conjugate the hydrophobic tail to the last amino acid of the polyamine-peptide construct:
- Add 10 equivalents of the desired carboxylic acid functionality to 9.5 equivalents of HBTU and 12 equivalents of DIPEA dissolved in DCM and DMF mixture (1:1, 20 mL). Note: Bear in mind than some tails may need a different proportion of solvent (usually a larger% of DCM) or the addition or another solvent such as N-methylpyrrolidone (NMP).
- Sonicate the cocktail for 5–10 min until dissolution (we have seen it takes longer for the tail to completely dissolve) and add to the vessel. Note: For tails containing multiple reactive sites, they will need to be protected before coupling them.
- Carry out this reaction (coupling the hydrophobic tail) for at least 5 h, although it is advisable to carry it out overnight for the highest yield.
2. PPA Cleavage from Solid Support
The purpose of this step is the cleavage of the PPA from the resin, and to remove the Boc protecting groups from the amino acids and polyamine residues.
Wash the resin with DMF (8 mL for 2 min) and twice with DCM (8 mL, each time for 5 min). Before each addition, drain the solvent from the vessel. Once the last wash is performed, dry the resin under vacuum for 15 min.
Prepare the cleavage solution using the following mixture ratio: 28:1:1 trifluoroacetic acid (TFA): H2O : Triisopropyl silane (TIPS). To make 15 mL of the cleavage cocktail add 14 mL of TFA, to 0.5 mL of H2O and 0.5 mL of TIPS.
Add this cleavage solution to the resin and shake for 2–4 h at room temperature.
Collect the solution in a 25 or 50 mL round bottom flask.
Concentrate the TFA in vacuo to 1–2 mL using a rotary evaporator at reduced pressure while heating the mixture at 40 °C (do not surpass 55 °C to avoid decomposition of the PPA).
After evaporation, add the obtained TFA solution (dropwise) to a round bottom flask containing anhydrous cold ether (15 mL). This will immediately precipitate the PPA.
In addition, add anhydrous cold ether (5 mL) to the original flask where the TFA was concentrated.
Sonicate this flask to recover additional solids and combine with the ether solution from step 2.6. Note: The transferring pipette can be washed with a small volume of DCM. Avoid introducing DMF during the process because it tends to form a gel-like substance.
Place the flask (covered) inside the refrigerator and let it stand overnight to maximize precipitation.
Collect the precipitated material by vacuum filtration using a sintered disc filter funnel. The ideal pore sizes are fine or medium.
Wash the precipitate twice with cold ether (5–10 mL) to remove any residual organics.
3. Identification of the Crude Product Using the MALDI Dried-drop Method
- Matrix preparation for analysis in positive mode:
- To start molecular weight analysis using Matrix Assisted Laser Desorption/Ionization (MALDI) mass spectrometry, add 10 to 20 mg of α-cyano-4-hydroxycinnamic acid to a microcentrifuge tube.
- Add a solution of water/acetonitrile (1:1, 1.0 mL) with 0.1% Trifluoroacetic acid (TFA). Mix thoroughly by vortexing. Note: This matrix should be used for positively charged peptides. MALDI negative mode is similar but requires a matrix containing 9-aminoacridine with 0.1% ammonia as solvent additive.
- Add 1–2 µL of the sample to the stainless steel target plate for MALDI and dry the sample in air. Add 1–2 µL of the matrix and dry it again. Note: The plates have circular markings gridded along the target plate to help locate the particular spot.
Analyze the samples in MALDI instrument to verify their identity. Electrospray ionization (ESI) is a valid alternative to confirm the identity of the PPAs.
4. Purification of PPAs Using Preparative Reverse-phase High-performance Liquid Chromatography (HPLC) Purification
- Dissolve the PPA precipitate in a minimum amount of acetonitrile and water.
- As a rule of thumb, dissolve 100 mg of dry crude PPA in less than 5 mL of acetonitrile and water. More hydrophobic PPAs might require a greater percentage of acetonitrile to dissolve and might also require a larger volume of solvent in general, depending on the net charge of the PPAs (Table 1).
- If complete dissolution does not take place, add 1% TFA (ACN or H2O). It is also possible to add trace amounts of other compatible solvents including dimethylsulfoxide, methanol, or isopropanol (limit their content to 5%).
| Solvent | Positively Charged PPAs | Negatively Charged PPAs |
| 0.1% TFA in Water | 0.1% NH3 in Water | |
| 0.1% TFA in ACN | 0.1% NH3 in ACN |
Table 1: Solvent Systems. Proposed solvent system for positively and negatively charged PPAs.
Sonicate the PPA using a horn sonicator for 20 min at 10 s pulses with Amp1 of 48% or for 2-3 h in an ultrasonic bath.
- Then, filter using a 0.45 μm syringe filter followed by filtration using a 0.20 μm Polytetrafluoroethylene (PTFE) syringe filter. The PPA solution should be clear and free of all particulate materials.
- If filtration is too difficult, sonicate for an extended period of time. It is strongly advised that the PPA solution is purified immediately after syringe filtration, as prolonged storage might cause it to aggregate or gelate, which will necessitate re-sonication and, perhaps, re-filtration.
During and after purification, use either HPLC grade solvents or ones that are filtered through a 0.25 μm syringe filter/membrane. NOTE: The most common solvent conditions to purify PPAs consist in a gradient of H2O and ACN.
Use a standard reverse-phase HPLC instrument for this protocol. It should include an elution gradient programmer, a binary solvent delivery system, a UV detector capable of detecting at 220 and 254 nm, and a programmable fraction collector. The maximum flow rate should be 50 mL/min.
Use a C-18 reversed phase column for separation. Columns of the following dimensions can be used depending on the mass of solid being purified and the net charge of the PPA (Table 2).
| PPA Charge | Particle Size | Column Size | Mass of crude PPA |
| + ve Charged | 5 μm | 150 x 30 mm | 170 mg |
| - ve Charged | 5 μm | 150 x 30 mm | 170 mg |
| + ve Charged | 5 μm | 150 x 21.2 mm | 90 mg |
| - ve Charged | 5 μm | 150 x 21.2 mm | 90 mg |
Table 2: Suggested columns: Column dimensions, particle sizes and the maximum load capacity per injection for C18 reverse phase HPLC columns
Inject PPA with a volume determined by both the capacity of the column and by the volume of the injection loop of the HPLC. Run the HPLC gradient method according to the settings seen in Table 3 (elution time of 40 min).
| Time | Solvent A (Acetonitrile) | Solvent B (Water) | Flow Rate (mL/min) |
| 0 | 5% | 95% | Flow rate depends on the column packing and its size. |
| 2 | 5% | 95% | |
| 35 | 95% | 5% | |
| 38 | 100% | 0% | |
| 40 | 5% | 95% |
Table 3: Suggested gradient: Suggested reverse phase gradient showing the relative composition of water vs acetonitrile over a period of time. The flow rate will depend on the column specifications.
- Analyze the fractions collected on the various test tubes using MALDI and determine which tube (or tubes) contains the PPA. This is assessed by studying the molecular weight found in each tube.
- Check purity of the fractions on an analytical HPLC. Use an HPLC-gradient system equipped with a UV detector set at 220 nm.
- If there are impurities, do an additional cycle of purification by HPLC. The above gradient used for preparative HPLC can be scaled down to use with the analytical HPLC using the column converter online software. Each fraction much be ≥95% pure for physical characterization or biological evaluation.
Collect the fractions in a large round-bottomed flask or evaporation flask and remove all the acetonitrile and most of the water. The final PPA solution should be no more than 10 mL.
Transfer this concentrate to a 50 mL centrifuge tube. Be advised that PPAs are detergent-like substances; thus, bubbles will be generated during evaporation of the solvent. We have found that addition of ethyl alcohol diminishes bubble formation.
Vacuum concentrate the concentrate. During the vacuum evaporation, a large amount of the PPA might adhere to the walls of the container. Recover this by repeatedly rinsing the flask wall with HPLC water. The combined volume of the concentrated PPA and the rinsate should be no more than 30 mL.
Place the aqueous solution inside of a 50 mL tube.
- Flash freeze this PPA solution in liquid nitrogen: NOTE: Flash freezing results in smaller ice-crystals which will sublime faster in the lyophilizer. Moreover, slower freezing may allow the formation of self-assembled supramolecular structures. Another alternative (but less desirable) is to gradually freeze in a -80 °C freezer or to freeze using a dry ice + acetone mixture
- Before placing the centrifuge tube in the lyophilizer, perforate or loosen the cap of the tube to allow moisture to escape the sample and get collected in the refrigeration unit. Set the lyophilization unit set to -54 °C and 0.016 mbar. NOTE: Another option is to remove the cap and place a wipe (with a rubber band) on top of the opening. Also, we have found that freezing samples at an angle or breaking the ice into small portions allows for a faster and better lyophilization due to an increase in surface area.
- If the solid starts to melt, this may indicate traces of an organic solvent (usually acetonitrile) are present. Repeat the freezing step and assess the condition of lyophilization.
- If the melting persists, there are two potential solutions:
- Split the sample in two portions (to be placed in tubes), dilute with water, and freeze again. Do not use this solution often because large amounts of organic solvents may damage the equipment.
- Alternatively, place the sample in a flask and further evaporate the organic solvent under vacuum. Lyophilizing a sample that is in the liquid form usually leads to bumping and loss of the PPA compound.
5. Storage of PPAs
Store the PPAs in a -20 °C with the caps tightened and sealed with Parafilm on a tube with a proper label.
Before use, bring the PPA back to ambient temperature in a vacuum chamber to avoid condensation of water vapor on the PPAs.
Representative Results
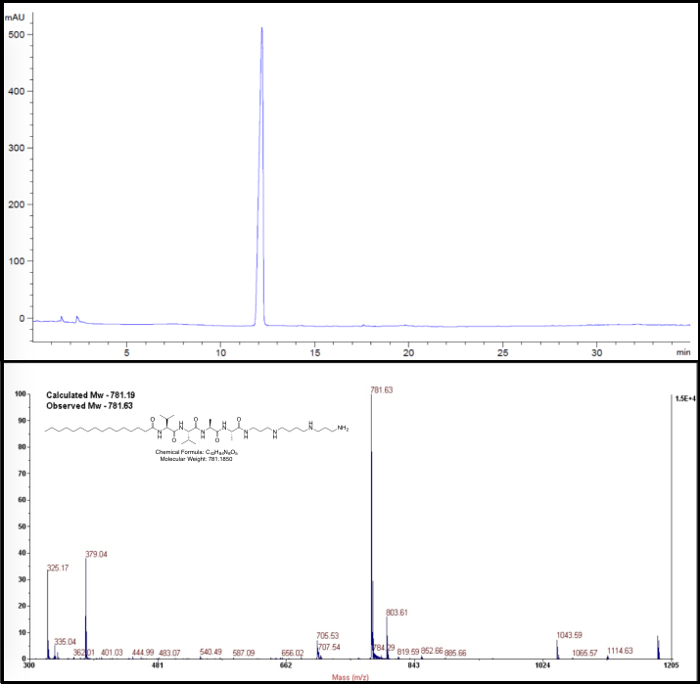
After the synthesis and purification and before physicochemical or biological evaluation, it is recommended the masses of the PPAs are re-checked and the purity ascertained using analytical HPLC. For material characterization or biological evaluation, PPAs need to have a purity of >95%. Figure 2 shows the HPLC trace (Top) and MALDI spectra (Bottom) confirming the presence of the product. HPLC analytical systems will integrate the area under the curve (AUC) and an AUC >95% can be related to product purity. In UV-based HPLC systems, expect to see a single, sharp peak. MALDI spectra should correspond to that of the calculated molecular weight of the PPA within ±1 Da (depending on the mode of analysis by MALDI).
The self-assembly of the PPAs can be visualized and analyzed using myriad techniques, including Transmission Electron Microscopy (TEM) (Figure 3A, B), Atomic Force Microscopy (AFM) (Figure 3C, D), Small Angle X-ray Scattering (SAXS), Scanning Electron Microscopy (SEM), and Dynamic Light Scattering (DLS). Successful self-assembly will result in well-defined nanostructures in both AFM and TEM. Failure to self-assemble will either result in the formation of irregular aggregates that are several hundred nanometers in size.
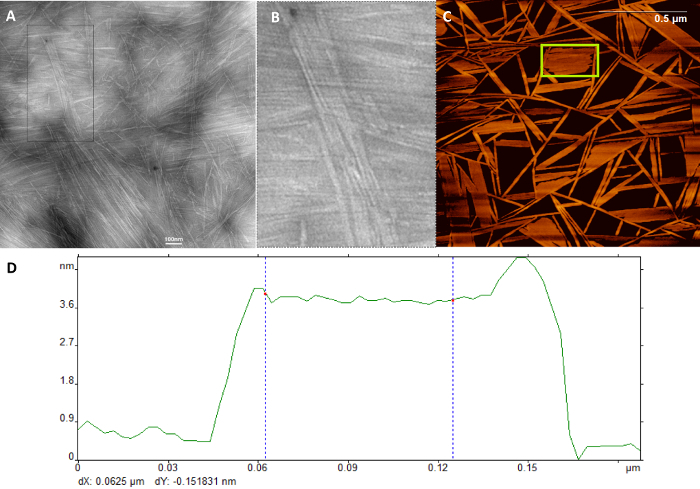
Figure 2: Representative Analytical HPLC chromatogram (Top) and MALDI spectrum (bottom) of the PPA C16V2A2Spermine. This figure has been modified from Samad et al. 20176; reused with permission of John Wiley & Sons, 2018. Please click here to view a larger version of this figure.
Figure 3: Representative Transmission electron microscope (TEM) micrograph. (A) TEM image showing the formation of nanofiber and fused nanosheets, (B) Magnified inset showing the formation of bundled nano-sheets, (C) Atomic force microscopy (AFM) micrograph, (D) and height map derived from the micrograph for C16V2A2KDiethyelenetriamine. The X and Y axes of (D) represents nano-sheet width and nano-sheet height respectively. This figure has been modified from Samad et al. 20176; reused with permission of John Wiley & Sons, 2018. Please click here to view a larger version of this figure.
Discussion
The protocols described here can be used to synthesize PPAs as wells as PAs and related peptide-based molecules (such as hybrid PA-peptoids). Although the synthesis of peptides using SPPS is a straightforward procedure, the synthesis of peptides containing biological homing molecules can be particularly challenging. Polyamines like spermine, spermidine, diethyelenetriamine, etc., can function as homing molecules for targeting cancer cells13. The PPAs can self-assemble into nanostructures with diverse morphologies6. Their positive charge can also aid with longer circulation time (due to endosomal escape) and a different metabolic profile (when compared with traditional PA). However, synthesizing PPAs and their analogues can be a particular challenge because of the presence of primary and secondary amines. The presented synthetic strategy overcomes this challenge by the rational use of orthogonal protecting group. The Dde molecule selectively undergoes reaction only with primary amines11. Boc, on the other hand, reacts indiscriminately with both primary and secondary amines and, therefore, can only be added after the primary amine is protected. We would like to mention that other protecting groups can be used instead of Boc. For example, reaction with acetic anhydride will permanently acetylate the secondary amines (this group will not be removed during cleavage). Likewise, the use of trifluoacetic anhydride will provide a base-sensitive protection while the benzylcarbamate will give a group that can be removed by H2/Ni14. Once the polyamines are protected as needed, the synthesis of the PPA can proceed using standard SPPS. The coupling the hydrophobic tail to the rest of the polyamine-peptide construct not only takes a longer period of time to couple, but also requires twice the molar amount of amino acids. Some of the hydrophobic tails might not dissolve well at room temperature in some traditional solvents or may precipitate even after initial dissolution. Such reactions are best carried out under mild heating (40 °C) by transferring all the reactants to round bottom flask. Alternatively, a jacketed synthesizer vessel might be used to keep the reactants warm throughout the reaction. If available, a microwave synthesizer may aid with the coupling15.
Although many forms of mass spectroscopy are available to determine the masses of PPAs, we used MALDI spectroscopy for the purpose of our identification. We have found MALDI to be more effective in producing the molecular ion peaks, minimizing fragmentation and adduct formation. MALDI should be programmed to generate an ion that corresponds to the most likely ionization state of the PPA. Besides programming the instrument to produce ion of a particular charge, we also need to combine the samples with the appropriate matrix that is suitable for the mode we will use.
PAs often have a tendency to form salt adducts on the MALDI plate. This problem is seen more often with negatively charged molecules. The most common salts are those of sodium (Na+ = 23 Da) and potassium (K+ = 39 Da). These adducts may be suppressed by adding 1–2 µL of acetic acid. The PPAs usually provide the H+ adduct. It is recommended to only use nanopore water or HPLC water throughout the synthetic and purification process to avoid the introduction of additional ions. Borosilicate glass containers and reaction vessels may contain appreciable amounts of sodium ions which will leach into the final product. Rinse the glassware carefully using nanopurewater for the last wash.
Identifying the molecular ion peaks also becomes difficult if the matrix solution is not fully saturated with the solid matrix. To ensure saturation, there should always be a small amount of the solid matrix that has not solubilized.
As a final note, please consider that some PPAs or PAs might not form any precipitate upon addition of ether. This was mainly observed in the more hydrophilic PPAs. In such events, we will first evaporate all ether, neutralize the excess TFA with NH4OH and add Water and Acetonitrile (with 0.1% TFA or NH4OH) to fully solubilize it and then proceed to purify as usual.
The protocol described here can be used to synthesize highly pure variants of self-assembling polyamine based peptide amphiphiles (PPAs) and also PAs. The orthogonal protection/deprotection steps can be used in other situations that require selective masking of primary and secondary amine groups.
Disclosures
The authors have no conflict of interest to declare
Acknowledgments
This project was funded by the University of Nebraska Medical Center (Start-up funds, MC-S); NIH-COBRE, 5P20GM103480 (T. Bronich) and the American Chemical Society, PRF# 57434-DNI7(MC-S).
References
- Cui H, Pashuck ET, Velichko YS, Weigand SJ, Cheetham AG, Newcomb CJ, Stupp SI. Spontaneous and x-ray-triggered crystallization at long range in self-assembling filament networks. Science. 2010;327:555–559. doi: 10.1126/science.1182340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pashuck ET, Cui H, Stupp SI. Tuning supramolecular rigidity of peptide fibers through molecular structure. Journal of the American Chemical Society. 2010;132:6041–6046. doi: 10.1021/ja908560n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupp SI, Zha RH, Palmer LC, Cui H, Bitton R. Self-assembly of biomolecular soft matter. Faraday Discussions. 2013;166:9–30. doi: 10.1039/c3fd00120b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conda-Sheridan M, Lee SS, Preslar AT, Stupp SI. Esterase-activated release of naproxen from supramolecular nanofibres. Chemical Communications. 2014;50:13757–13760. doi: 10.1039/c4cc06340f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata A, Palmer L, Tejeda-Montes E, Stupp SI. Nanotechnology in Regenerative Medicine. Springer; 2012. Design of biomolecules for nanoengineered biomaterials for regenerative medicine; pp. 39–49. [DOI] [PubMed] [Google Scholar]
- Samad MB, Chhonker YS, Contreras JI, McCarthy A, McClanahan MM, Murry DJ, Conda-Sheridan M. Developing Polyamine-Based Peptide Amphiphiles with Tunable Morphology and Physicochemical Properties. Macromolecular bioscience. 2017;17 doi: 10.1002/mabi.201700096. [DOI] [PubMed] [Google Scholar]
- Nel AE, Mädler L, Velegol D, Xia T, Hoek EM, Somasundaran P, Klaessig F, Castranova V, Thompson M. Understanding biophysicochemical interactions at the nano-bio interface. Nature Materials. 2009;8:543. doi: 10.1038/nmat2442. [DOI] [PubMed] [Google Scholar]
- Gujrati M, Malamas A, Shin T, Jin E, Sun Y, Lu Z-R. Multifunctional cationic lipid-based nanoparticles facilitate endosomal escape and reduction-triggered cytosolic siRNA release. Molecular Pharmaceutics. 2014;11:2734–2744. doi: 10.1021/mp400787s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Li J, Kanvinde S, Lin Z, Hazeldine S, Singh RK, Oupický D. Self-immolative polycations as gene delivery vectors and prodrugs targeting polyamine metabolism in cancer. Molecular Pharmaceutics. 2014;12:332–341. doi: 10.1021/mp500469n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planas-Portell J, Gallart M, Tiburcio AF, Altabella T. Copper-containing amine oxidases contribute to terminal polyamine oxidation in peroxisomes and apoplast of Arabidopsis thaliana. BMC Plant Biology. 2013;13:109. doi: 10.1186/1471-2229-13-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash IA, Bycroft BW, Chan WC. Dde - A selective primary amine protecting group: A facile solid phase synthetic approach to polyamine conjugates. Tetrahedron Letters. 1996;37:2625–2628. [Google Scholar]
- Ralhan K, KrishnaKumar VG, Gupta S. Piperazine and DBU: a safer alternative for rapid and efficient Fmoc deprotection in solid phase peptide synthesis. RSC Advances. 2015;5:104417–104425. [Google Scholar]
- Casero RA, Jr, Marton LJ. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nature Reviews Drug Discovery. 2007;6:373. doi: 10.1038/nrd2243. [DOI] [PubMed] [Google Scholar]
- Wuts PGM, Greene TW. Protection for the Amino Group. In Greene's Protective Groups in Organic Synthesis. John Wiley &, Sons, Inc; 2006. pp. 696–926. [Google Scholar]
- Palasek SA, Cox ZJ, Collins JM. Limiting racemization and aspartimide formation in microwave-enhanced Fmoc solid phase peptide synthesis. Journal of Peptide Science. 2007;13:143–148. doi: 10.1002/psc.804. [DOI] [PubMed] [Google Scholar]