

Abstract
Glia Maturation Factor (GMF) has recently been established as a regulator of the actin cytoskeleton with a unique role in remodeling actin network architecture. Conserved from yeast to mammals, GMF is one of five members of the ADF-H family of actin regulatory proteins, which includes ADF/Cofilin, Abp1/Drebrin, Twinfilin, and Coactosin. GMF does not bind actin, but does bind the Arp2/3 complex with high affinity. Through this association, GMF catalyzes the debranching of actin filament networks and inhibits actin nucleation by Arp2/3 complex. Here, we discuss GMF’s emerging role in controlling actin filament spatial organization and dynamics underlying cell motility, endocytosis, and other biological processes. Further, we attempt to reconcile these functions with its earlier characterization as a cell differentiation factor.
Keywords: actin, ADF-H, Arp2/3 complex, cell motility, lamellipodia
GMF: a small protein with a big reach
Glia Maturation Factor (GMF) is a 17 kDa protein with clear orthologues across species as diverse as yeast, flies, zebrafish, and mammals (Figure 1A). GMF was first identified over 40 years ago as a factor in brain extracts that could induce the differentiation of cultured glioblasts, gliomas, and neuroblastoma cells [1–4]. Thus, GMF was studied for decades as a signaling factor controlling brain cell differentiation and function [1, 2]. Inconsistent with its early assignment as a cell differentiation factor, GMF is conserved from single-celled yeast to mammals. In fungal species and invertebrates, there is typically a single GMF gene/protein. However, vertebrates express two different GMF genes, GMFβ and GMFγ. These isoforms are 82% identical in sequence [5] and have similar 3-D structures [6] (Figure 1B & D). GMFβ and GMFγ show overlapping yet distinguishable expression patterns in different tissues [7–9]. Although GMFβ has been described in some studies as a brain-specific isoform, its expression has also been detected at the protein level in lung, spleen, colon, and thymus [8], and even more ubiquitously as a transcript [5, 7, 10].
Figure 1.

GMF sequence and structure are evolutionarily conserved.
(A) Alignment of amino acid sequences of GMF from 6 different organisms plus human Cofilin-1. Conserved residues are shaded in the two sites (Sites 1 and 2) in GMF that mediate binding to Arp2/3-actin branch sites and the analogous sites in cofilin that mediate binding to actin.
(B) Heat map showing evolutionary conservation of surface residues on the structure of GMFγ (PDB ID 3L50).
(C) Surface rendered view of the crystal structure of human Cofilin-1 (PDB ID 4BEX).
(D) Overlay of the solution NMR structures of mouse GMFβ (blue) and mouse GMFγ (green) (PDB IDs 1V6F and 1WFS, respectively).
(E) Overlay of the crystal structures of human GMFγ (green) and human cofilin-1 (orange) (PDB IDs 3L50 and 4BEX, respectively). Abbreviations: Arp2/3, actin-related protein 2/3; GMF, glia maturation factor; NMR, nuclear magnetic resonance; PDB, protein data bank.
In 2006, pioneering work from Ikeda and colleagues showed that GMF localizes to and regulates the actin cytoskeleton (see Glossary), and noted its sequence similarity to ADF/Cofilin [11]. In 2009, the structure of GMF [6] was solved, revealing it to be a bona fide member of the ADF-H (actin depolymerizing factor homology) family, which includes: ADF/Cofilin, Twinfilin, Abp1 (actin binding protein 1)/Drebrin, and Coactosin [6, 12]. All known ADF-H proteins bind to either actin and/or actin related proteins (Arps), and have conserved roles in actin cytoskeleton remodeling [12]. Below, we highlight recent work defining GMF’s biochemical and cellular roles in binding Arp2/3 complex to remodel branched actin filament networks and thereby control endocytosis and cell motility. Finally, we attempt to integrate these new functions with its earlier assignment as a cell signaling and differentiation factor.
GMF branches out as an actin regulator
GMFγ was first established as a component of the actin cytoskeleton when it was shown to be highly expressed in microvascular endothelial cells and found to colocalize with actin at the leading edge of migrating cells [11]. It was also established that GMFγ has 39% similarity in sequence to ADF/cofilin, a prominent actin filament severing protein, and that GMFγ coimmunoprecipitates with the actin-nucleating Arp2/3 complex. Shortly thereafter, the NMR structures of GMFβ and GMFγ were solved, and found to be remarkably similar to ADF/cofilin [6] (Figure 1D-E). These pioneering studies prompted other groups to further investigate the cytoskeletal functions of GMF in other model systems.
In 2010, two studies in yeast revealed exciting new functions for the S. cerevisiae and S. pombe homologs of GMF, both called Gmf1 (Figure 2) [13, 14]. These studies showed that purified Gmf1 lacks appreciable affinity for G-actin or F-actin, but directly regulates activities of the Arp2/3 complex. The Arp2/3 complex contains two actin-related proteins, Arp2 and Arp3, and upon binding to an activator such as WASP or WAVE, it binds to the side of an existing actin ‘mother filament’ and nucleates formation of a ‘daughter filament’ branch (see Box 1). In this manner, Arp2/3 complex forms arborized or ‘dendritic’ actin filament networks, including those found at the leading edge of migrating cells and at sites of endocytosis. Gmf1 was found to inhibit actin nucleation co-stimulated by Arp2/3 complex and the VCA domain of WASP, as demonstrated both in bulk actin assembly assays and single-filament total internal reflection fluorescence (TIRF) microscopy assays [13, 14]. Inhibition of Arp2/3 complex involved competition between Gmf1 and the WASP VCA domain for binding Arp2/3 complex, and strong inhibition required low micromolar concentrations of Gmf1. Gmf1 also catalyzed debranching, or ‘pruning’ of daughter filaments by dissociation at branch sites [13]. Earlier it had been shown that branch junctions generated by Arp2/3 complex in vitro are unexpectedly stable, failing to dissociate for tens of minutes in the absence of additional factors [15, 16]. In contrast, branches in vivo at the leading edge and at endocytic sites turn over in seconds [17, 18], suggesting the existence of cellular factors that might catalyze debranching. Low nanomolar concentrations of Gmf1 induced rapid ‘pruning’ of daughter filaments from mother filaments without severing elsewhere along the filaments [13]. The much higher concentration of GMF required for inhibition of nucleation by Arp2/3 complex versus debranching may reflect a requirement for GMF to occupy two separate binding sites on Arp2/3 complex to block nucleation, one high affinity and one low affinity site (see below).
Figure 2.
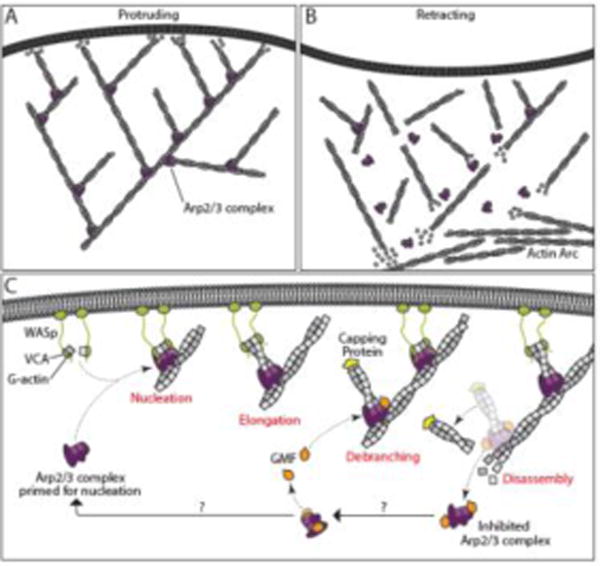
Model for GMF function in controlling actin organization and dynamics.
Illustration of a dendritic actin network being assembled by the Arp2/3 complex and driving membrane protrusion (A), and subsequently being debranched and disassembled to promote lamellipodial retraction (B). (C) Model showing steps in Arp2/3 complex cycle at the leading edge, with the roles of GMF highlighted. From the left, WASp VCA domains (green) bound to G-actin associate with Arp2/3 complex (purple) at the plasma membrane and stimulate nucleation of a daughter actin filament, forming a branch. WASp remains associated with the growing barbed end of the daughter filament until it is subsequently capped by Capping Protein (yellow). GMF (orange) binds Arp2/3 complex, possibly at two separate sites, to catalyze debranching, exposing pointed ends. Filaments are further disassembled by other factors (not shown). While GMF remains bound to the released Arp2/3 complex, it blocks nucleation. GMF dissociates from Arp2/3 complex, perhaps catalyzed by other Arp2/3 ligands or GMF phosphorylation, leaving Arp2/3 complex ‘primed’ for nucleation. Abbreviations: Arp2/3, actin-related protein 2/3; GMF, glia maturation factor; VCA, verprolin/cofilin/acidic domain; WASp, Wiskott-Aldrich syndrome protein.
Box 1. Arp2/3 complex and the formation of lamellipodia.
Lamellipodia are broad, flat, protrusions that form at the leading edge of cells that are migrating or spreading on a 2D surface. Their shape is determined by the branched actin networks assembled by Arp2/3 complex, and by a variety of other actin regulators that modulate lamellipodial protrusion and retraction dynamics. Lamellipodia form when upstream signals activate Rac1 GTPase, which in turn recruits to the membrane and activates the WAVE family of nucleation promoting factors (NPFs). The C-terminal VCA region of WAVEs induce conformational changes in Arp2/3 complex that increase its affinity for the sides of existing actin filaments and align the Arp2 and Arp3 subunits to mimic the barbed end of an actin filament. WAVEs also recruit the first actin monomers to Arp2/3 complex, and then dissociate, allowing the daughter filament to elongate [83]. Because WAVEs remain associated with the membrane, they are also proposed to continue tracking the growing barbed end of the daughter filament, controlling its growth rate and possibly protecting it from capping protein [84–86]. The branched actin networks found in lamellipodia are organized with the growing barbed ends of filaments oriented toward the membrane, such that network growth is directed toward the membrane and stimulates protrusion. As the network grows, with polymerization occurring near the leading edge to drive protrusion, older portions of the network move inward and are disassembled by cofilin and other proteins to recycle actin and actin-associated proteins. For a recent review on lamellipodia and cell migration, see [44].
These observations established GMF as a dedicated debranching factor and suggested that it may be involved in remodeling branched actin networks into unbranched arrays, as has been observed to occur only a short distance from the leading edge [19–21]. Other factors such as ADF/Cofilin and Coronin stimulate debranching in vitro [22, 23], but additionally promote severing of actin filaments along their lengths [13, 24], and may therefore lead to actin network disassembly rather than remodeling, i.e. they may promote the dissolution of filaments to yield actin monomers, as opposed to the rearrangement of filaments within a network. Subsequent studies showed that the two biochemical functions of GMF on Arp2/3 complex (inhibition of nucleation and stimulation of debranching) are conserved for the Drosophila homolog and both mammalian isoforms of GMF [14, 25–27]. Thus, GMF activities on Arp2/3 complex are conserved across great evolutionarily distances, consistent with the high degree of sequence conservation in GMF’s two key functional surfaces (Figure 1B).
Genetic observations from these studies suggest that GMF regulates Arp2/3 complex and actin dynamics in vivo [13, 14]. Budding yeast Gmf1 localizes to cortical actin patches, sites of endocytosis assembled by Arp2/3 [13, 14]. Furthermore, deletion of GMF1 exacerbates the growth defects of a cof1-22 mutant [13], and extends the lifetime of actin patches. In contrast, overexpression of Gmf1 reduces both the number of actin patches and the levels of Arp2/3 complex at actin patches, and rescues specific mutant alleles of Arp2/3 complex [14]. In addition, gmf1∆ mutants show synthetic defects in actin organization and cell growth with other actin turnover mutants [28]. Collectively, these genetic and biochemical observations provide strong evidence that Gmf1 promotes the remodeling and/or turnover of branched actin arrays in vivo. It is also noteworthy that gmf1Δ cells exhibit disorganized actin cables (actin structures polymerized by formins) [28], and defects in mitochondrial distribution likely caused by disorganized cables, as suggested by its alias name, Aim7, Altered inheritance of mitochondria 7 [29]. Importantly, the cable defects of gmf1∆ mutants may arise from reduced actin patch turnover, leading to an imbalance in the homeostatic distribution of actin subunits between patches and cables [30].
Mechanism of GMF-induced debranching
Mechanistic work has revealed that GMF debranches actin filaments by a mechanism related to cofilin severing of actin filaments, except that it is specialized for Arp2/3-actin junctions [13, 26, 31]. It has been reported that there are two binding sites for Gmf1 on Arp2/3 complex, one high affinity and one low affinity (with Kd = 10 nM and 1 μM, respectively) [26]. Furthermore, single particle electron microscopy showed that Gmf1 induces an open/inactive conformation in the Arp2/3 complex [41], related to Coronin’s effects on Arp2/3 structure [32, 33]. Employing fluorescence anisotropy, mutagenesis, and chemical cross-linking, it was found that Arp2/3 complex binding is mediated by a broad surface on Gmf1 (“Site 1”), analogous to the G/F (globular/filamentous) actin-binding surface on cofilin (Figure 1B-C) [26], and suggests that Site 2 (analogous to the F-site on cofilin) interacts with the first conventional actin subunit in the daughter filament. Both Site 1 and Site 2 were found to be important for Gmf1 functions in yeast cells, suggesting that debranching is critical for Gmf1 function in vivo. In parallel, when the crystal structure of GMFγ bound to mammalian Arp2/3 complex was solved [31], it validated one of the two proposed binding sites of GMF on the Arp2/3 complex, and provided an atomic level detail of the interaction. This structure reveals that GMF directly contacts the Arp2 and p40/ARPC1 subunits, which is presumed to represent GMF’s high affinity binding site on Arp2/3 complex, and suggests that surfaces on Arp2 may have co-evolved with GMF to maintain GMF interactions while excluding cofilin interactions [31]. A recent study using molecular dynamics simulations provides additional support for the existence of a second, lower affinity binding site for GMF on Arp2/3 complex, that involves interactions with the Arp3 subunit [34].
The above-mentioned studies also established that GMF catalyzes branch dissociation using its two distinct functional surfaces, with Site 1 binding to the Arp2 and p40/ARPC1 subunits and Site 2 binding to the first actin subunit in the daughter filament. Site 1 on GMF competes for binding with the nucleation promoting factor (NPF) WASP-VCA domain, explaining how GMF attenuates VCA-induced Arp2/3-mediated nucleation [31] (Figure 2C). Because GMF and cofilin are structural homologues with similarly-positioned functional surfaces/sites (Figure 1B), GMF may destabilize Arp2/3-actin branch junctions by a mechanism related to how cofilin destabilizes actin filaments to induce fragmentation. This argument is strengthened by GMF and cofilin both preferring the ADP-bound state of their ligands, Arp2/3 complex or actin, respectively. Given that cofilin induces a twist in F-actin conformation and alters actin-actin contacts [35, 36], it is possible that GMF exerts its debranching effects by similarly inducing conformational changes at Arp2/3-actin junctions.
GMF remodels actin networks at the leading edge
How are the conserved activities of GMF used in vivo to regulate branched actin networks, e.g., at sites of endocytosis and at the leading edge (Figure 2A-B)? In animal cells, GMF’s regulatory effects on actin networks appear to govern lamellipodial dynamics and cell motility (Box 1). In IA32 mouse embryonic fibroblasts, GMFβ localizes to mature rather than nascent lamellipodia, and overexpression of GMFβ demonstrates that it promotes lamellipodial retraction and ruffling, while depletion of GMFβ dramatically alters leading edge dynamics [10]. Consistent with observations made at yeast cortical actin patches [14], knockdown of GMFβ leads to increased Arp2/3 levels in lamellipodia. Conversely, GMFβ overexpression reduces Arp2/3 levels, suggesting that GMFβ catalyzes Arp2/3 turnover in actin networks [10]. Further, a point mutant was introduced into GMFβ to impair debranching, but not nucleation inhibition (designed based on the ‘site 2’ mutant in yeast Gmf1; [26]). Experiments using this mutant and a small molecule inhibitor of Arp2/3 complex demonstrated that GMFβ debranching activity is critical for proper leading edge dynamics [10]. Thus, GMF debranching activity is essential for its in vivo functions in promoting actin network remodeling and turnover. On the other hand, no tool has yet been generated to disrupt GMF inhibition of Arp2/3-mediated nucleation without also disrupting debranching, making it more difficult to assess the importance of GMF’s nucleation inhibition activity in vivo.
Additional support for GMF regulating lamellipodial dynamics comes from studies in Drosophila S2 cells and embryonic cells undergoing collective migration [25]. Here, GMF is enriched in lamellipodia undergoing retraction, a phase in which branched actin networks are rapidly disassembled and/or remodeled (debranched) into parallel bundles called ‘arcs’ (Figure 2B) [37]. GMF mutant flies also have reduced rates of border cell migration [25], demonstrating the importance of GMF function for directed cell migration in the physiological context of an intact animal. Similar roles may also explain the functions of the GMFγ isoform, which is highly expressed in immune tissues [8, 38]. GMFγ has important roles in hematopoietic lineage differentiation [9, 39–41], lamellipodial dynamics and chemotaxis in neutrophils and T-cells [42, 43] and vascular development in zebrafish [7].
The discovery that GMF drives lamellipodial retraction rather than protrusion lends important insights into how GMF promotes cell motility. Specifically, GMF may help remodel branched actin networks into unbranched structures such as actin arcs [37], which form parallel to the leading edge and treadmill rearwards [43]. Additionally, GMF may promote motility by governing the density of branches in actin networks at the leading edge. A number of studies, reviewed in more detail elsewhere [44], have explored the link between branch density, lamellipodial dynamics, and cell migration by altering expression levels of actin elongation factors (e.g., ENA/VASP, formins, and CARMIL), inhibitors of elongation (e.g., capping protein), branch stabilizers (e.g., cortactin), and branch destabilizers (e.g., coronin). Together, these studies suggest that densely branched networks comprised of shorter filaments lead to slow and steady lamellipodial protrusion, and there is an inverse correlation between protrusion rate and persistence [44]. Consistent with this view, depleting GMF, which should increase branch density, increases protrusion persistence [10]. In addition to these roles, GMF debranching is predicted to expose minus ends of filaments, thereby increasing the depolymerization flux and recycling of Arp2/3 complex, which in turn allows for continued network assembly at the leading edge [45].
Persistently protruding lamellipodia drive more continual directional cell migration [44, 46], and studies on GMF support this [10]. However, there are conflicting reports about the relationship between lamellipodial protrusion rates and cell speed. In one study, slow and steady protrusion correlated with faster cell speed [47], while in a different study, rapid protrusion correlated with faster cell speed [10]. It is possible that other parameters, such as the frequency and extent of lamellipodial retraction events, rather than protrusion rate, are the more relevant determinants of cell speed [46]. Finally, GMF may suppress unproductive lateral cell protrusions, leaving more Arp2/3 complex and actin monomers available for advancement of the leading edge of the cell [10, 25, 42]. This idea is similar to the proposed mechanism for Arpin, another direct inhibitor of the Arp2/3 complex that steers cell motility [48]. Indeed, in some cell types the Arp2/3 complex is dispensable for cell migration, yet required for steering cells toward haptotactic [49] or chemotactic signals [50].
Roles for GMF in endocytic traffic
In addition to its roles in cell migration, GMF regulates actin-dependent endocytosis in yeast and mammals [13, 14, 51], and promotes the turnover of focal adhesions in immune cells [43, 52]. Vinculin, a central component of focal adhesions that anchors cells to their substrate, binds to the Arp2/3 complex at focal adhesions [53]. This raises the possibility that GMF helps stimulate the turnover and recycling of Vinculin-Arp complexes, or possibly facilitates other Arp2/3-dependent steps in focal adhesion dynamics. GMFγ also has been shown to localize to early and late endosomes in macrophages, where it is required for proper internalization and trafficking of the TLR4 receptor [51]. Together, these studies suggest that GMF regulates Arp2/3 complex-dependent functions in endocytic trafficking.
Open questions about GMF’s activity and mechanism
To gain a more complete understanding of GMF’s regulatory effects on Arp2/3 complex, further studies are needed to examine GMF’s precise biochemical role in promoting debranching, and its activities in the context of other actin-associated proteins. GMF’s debranching activity is likely influenced by whether the Arp2 and Arp3 subunits of Arp2/3 complex are bound to ATP or ADP nucleotides. ATP hydrolysis and phosphate (Pi) release on Arp2 are thought to precede debranching [54], possibly leading to GMF targeting older branches while leaving younger branches intact. Consistent with this model, GMFγ preferentially binds ADP-Arp2/3 complex over ATP-Arp2/3 complex in solution [55]. GMF binding to Arp2/3 complex may also catalyze Pi release on Arp2 and/or Arp3, similar to cofilin’s stimulatory effects on Pi release from F-actin [56].
Additionally, it will be important to determine GMF’s functions in the presence of cofilin and coronin, both of which have been implicated in debranching. Cofilin has been observed to stimulate debranching in TIRF microscopy assays [23], yet recent structural work indicates that GMF-Arp2 contacts are evolutionarily divergent from cofilin-actin contacts [31], and there is minimal evidence to suggest cofilin directly binds Arp2/3 complex. Instead, binding of cofilin to actin at or near branch junctions may alter F-actin conformation [35] to weaken branch sites and catalyze daughter dissociation. Alternatively, cofilin debranching, as seen in TIRFM assays, may not reflect actual dissociation of the branch sites, but instead severing events located near branch junctions. Coronin also appears to have functions in debranching. Like GMF, coronin binds with high affinity to Arp2/3 complex and alters Arp2/3 conformation and activities [32, 33, 57–60]. Further, coronin was recently was shown to synergize with GMF in inhibiting Arp2/3-mediated actin nucleation [33]. Coronin also co-purifies with Arp2/3 complex on GMF affinity columns [13], and recent structural evidence suggests that coronin and GMF have non-overlapping binding sites on Arp2/3 complex [33]. Therefore, coronin and GMF may be capable of binding simultaneously to Arp2/3 complex, and it will be important to test how they affect debranching in combination. Some genetic evidence has supported a collaboration between GMF and coronin, e.g., yeast gmf1Δcrn1Δ double mutants having synthetic growth defects [28]. On the other hand, RNAi depletion of mammalian coronin-1B, which weakly stimulates debranching in vitro [22], shows opposite effects to GMFβ depletion [10], reducing instead of increasing lamellipodial persistence [22, 60]. This might be explained by GMF and coronin having distinct, and only partially overlapping mechanistic roles in regulating actin networks, although the interpretation of these results could also be confounded by coronin’s ability to promote cofilin-mediated severing [24, 61]. Last, it will be important to determine how the activities of GMF, cofilin, and coronin on actin network remodeling and turnover are affected by other binding partners of Arp2/3 complex, particularly cortactin and Abp1, which are suggested to stabilize branches.
Spatial and temporal control of GMF function in vivo
There is evidence to suggest that GMF activities are regulated in vivo by post-translational modification. indeed, there is strong precedent for ADF-homology family proteins being regulated in this manner. Cofilin is inhibited by phosphorylation of a serine residue at its N-terminus. This residue lies within the G/F-actin binding site of cofilin, and its phosphorylation by LIM kinase weakens F-actin binding and increases cofilin’s cooperativity of binding, leading to reduced mechanical discontinuities and reduced severing [62, 63]. Phosphorylation of GMFγ at its analogous Ser2 also has a regulatory role. Purified phosphomimetic GMFγ (S2E) has weakened affinity for Arp2/3 complex [55], and phosphorylated GMFγ in vivo accumulates more strongly on actin networks at the leading edge, possibly the result of failed network debranching [11]. Furthermore, signaling from GTPases Rac1 and Cdc42 promotes the phosphorylation of GMFγ at Ser2, as well as increased accumulation of GMFγ and F-actin at the leading edge and increased lamellipodial protrusion. Thus, it is possible that Ser2 phosphorylation of GMF inactivates its debranching activity to disrupt lamellipodial retraction and promote protrusion [10]. Although the kinase that phosphorylates Ser2 on GMF has not yet been identified, LIM kinase is an intriguing candidate, as this would provide a mechanism for coordinately regulating cofilin and GMF.
There are a number of additional putative phosphorylation sites on GMF (Figure 3). In GMFβ, four different Ser or Thr residues are predicted to be phosphorylated by one or more of the following kinases: PKA, PKC, CKII, and S6K [64]. These residues are conserved in GMFγ, which has two additional PKC and CKII kinase consensus sites not found in GMFβ [5]. Some of these phosphorylation sites have been implicated in GMF’s suggested role in MAP kinase signaling [65], as discussed below. Interestingly, all four sites shared by GMFβ and GMFγ reside in or near functional ‘Site 2’ (Figure 3; [26]), where GMF contacts the first actin subunit in the daughter filament, and therefore could regulate debranching. Additionally, phosphorylation on Tyr104 by c-abl kinase weakens GMFγ interactions with Arp2/3 complex in smooth muscle cells and alters F-actin levels [66]. Tyr104 is located near GMFγ “Site 1” (Figure 3), which contacts Arp2/3 complex, consistent with its effects on GMFγ-Arp2/3 interactions. However, the side chain of Tyr104 is not surface exposed on available structures of GMF, so it is not yet clear how c-abl gains access to this site. Continued investigation into how phosphorylation at each of these sites influences GMF activities and cellular functions is likely to reveal new mechanisms by which cells spatially and temporally tune actin network remodeling and turnover.
Figure 3.
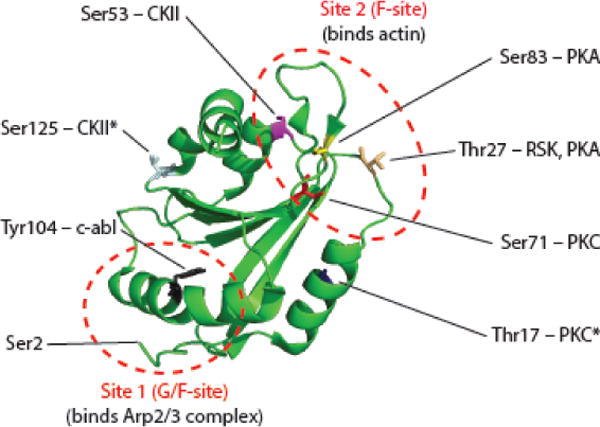
Suggested phosphorylation sites on human GMFγ.
Ribbon structure of human GMFγ (green), with labels indicating residues thought to be phosphorylated and the kinases that potentially target those residues based on consensus motifs (see main text for references). Asterisks (*) indicate residues in GMFγ not conserved in GMFβ. Abbreviations: Arp2/3, actin-related protein 2/3; GMF, glia maturation factor; Ser, serine; CKII, casein kinase II; c-Abl, abelson kinase 1; PKA, protein kinase A; PKC, protein kinase C; RSK, ribosomal s6 kinase.
Reconciling GMF’s actin functions with its roles in signaling and disease
GMF was initially identified as a factor present in brain extracts that induces differentiation of glioblast, glioma, and neuroblastoma cells in culture [1–4]. Subsequent studies indicated that GMFβ elicits these effects via (i) an extracellular route, without being internalized by the recipient cells, suggesting GMFβ is a secreted protein [67], and/or (ii) an intracellular signaling pathway [68]. Since GMFβ lacks a clear signal sequence for classical targeting to the secretory pathway, its release from cells would likely occur through non-classical “leaderless” secretion [69, 70]). In astrocytes, GMFβ is detected both in the cytosol and on the cell surface [68]. However, GMFβ activity has not been detected in astrocyte-conditioned media [71], suggesting healthy cells may not secrete GMFβ. GMFβ also has been identified as a surface-expressed protein on thymic epithelial cells that mature into CD4+ (helper) T-cells [72]; however, this may be due to autoimmune regulator (AIRE)-induced expression of non-thymic proteins used to educate developing T cells and prevent autoimmunity [73]. Thus, while it is intriguing to consider roles for GMF in functioning outside of the cell, this model requires further investigation. One possibility that warrants exploration is that GMFβ might be released specifically from sick or dying cells.
GMFβ was later localized to the cytosol of primary astrocytes and glioma cell lines [68], prompting investigations into its involvement in intracellular signaling. It was found that PKA phosphorylation of GMFβ inhibits the ERK1/2 branch of the MAP kinase pathway [74], yet activates the stress-responsive p38 pathway [65]. GMFβ-stimulated p38 signaling in turn induced NF-κB-mediated transcription, leading to secretion of: (i) growth factors such as BDNF and NGF to promote neurite outgrowth and neuroprotection, [75–78], and (ii) proinflammatory cytokines such as TNF-α and IFN-γ [79, 80]. These observations, combined with studies on GMFβ−/− knockout mice have suggested that GMFβ has neuroprotective roles in Alzheimer’s disease and Parkinson’s disease (see Box 2).
Box 2. Studies on GMF−/− mice and suggested roles in health and disease.
The generation of a GMFβ knockout mouse in 2004 revealed its importance in normal cognitive functions and that its loss promotes pathological neuroinflammatory disease progression [87]. GMFβ−/− mice displayed impaired motor skills and learning [87]. They also showed decreased susceptibility to a multiple sclerosis-like disease, which was attributed to a loss of GMFβ’s inflammation-inducing activity and might also explain a correlation between GMFβ levels and Alzheimer’s disease (AD) [80, 88–95]. Administration of an anti-GMFβ antibody to mice in a multiple sclerosis model greatly reduced the onset and severity of the disease [96]. However, since most cell types do not internalize antibodies, this observation returns us to the enigma of extracellular GMFβ (see main text), and the possibility that extracellular GMFβ is released by sick or dying cells. In support of this idea, GMFβ localizes to extracellular amyloid plaques and intracellular neurofibrillary tangles (NFTs) in a mouse model of AD and in post-mortem brains of AD patients [93, 94]. Further, a role for GMFβ in the progression of Parkinson’s disease has been suggested by the observation that dopaminergic neurons and astrocytes in GMFβ−/− mice have reduced sensitivity to oxidative stress [97, 98]. Collectively, these observations raise exciting possibilities for the involvement of GMF in disease states of the brain, and for GMF’s potential as a therapeutic target. In addition, recent research has suggested a role for GMFγ in promoting metastasis, which may be linked to GMF’s actin functions [99]. Overexpression of GMFγ in ovarian cancer cells promotes cell migration and invasion, and alters actin organization, and elevated GMFγ levels correlate with poor patient prognosis. Finally, transgenic mice overexpressing GMFβ have shorter lifespans and show accelerated aging particularly in the kidney, suggesting a role for GMF in aging [100].
How can we reconcile these observations for GMF with its well-defined biochemical and cellular functions in actin network remodeling? One possibility is that GMF signaling and cytoskeletal functions are mediated by distinct isoforms, GMFβ and GMFγ, respectively. However, the striking similarity in structure between GMFβ and GMFγ, (Figure 1) argues against this, as do studies showing that GMFβ and GMFγ both regulate actin [10, 11, 14, 25, 27, 81]. Further, at least one study has indicated that GMFγ, like GMFβ, functions in MAP kinase signaling [42]. Therefore, there is little evidence to support a ‘separate isoforms’ model. Alternatively, signaling and cytoskeletal functions of GMF could be integrated, such that association of GMF with actin networks spatially regulates MAP kinase signaling. This integration could lead to GMF availability for signaling being dependent on the local state of actin turnover, making MAP kinase signaling responsive to actin dynamics. Reciprocally, GMF modification by signaling pathways could affect its availability for actin remodeling. In fact, GMF inhibits ERK signaling [74], and ERK phosphorylates/activates components of the WAVE regulatory complex to promote Arp2/3-mediated actin assembly at the leading edge [82]. Therefore, GMF enrichment in retracting lamellipodia could not only promote debranching and direct Arp2/3 inhibition, but also indirectly block new actin assembly by inhibiting ERK signaling to WAVE (Figure 2).
Concluding Remarks and Future Directions
Over the past 40 years, our view of GMF has evolved from a secreted factor that promotes cellular differentiation, to an intracellular signaling protein, to a bona fide component of cellular actin networks with highly specific roles in controlling Arp2/3 complex activities and actin filament remodeling. This leaves it uncertain why GMF was initially identified in the extracellular space, or how it was able to induce cell differentiation, and whether this involved its internalization into cells. Future work is also needed to investigate the cross-relationships and interdependence of GMF functions in actin regulation and signaling. In addition, there are a number of questions yet to be answered about GMF mechanism and function in governing Arp2/3 complex activity, alone and in combination with other proteins (see Outstanding Questions). These mysteries deserve new attention using modern cell biological techniques, and taking advantage of separation-of-function mutations now available, to rigorously test the in vivo importance of these suggested functions. This will also be a crucial step in understanding which functions of GMF underlie its genetically demonstrated neuroprotective and anti-inflammatory functions.
Highlights.
GMF is structurally related to Cofilin, and is a conserved regulator of the actin cytoskeleton.
GMF triggers actin filament debranching by targeting Arp2/3 complex at branch sites.
GMF also inhibits free Arp2/3 complex from nucleating actin assembly.
GMF localizes to the leading edge, and its debranching activity is critical for lamellipodial retraction, a key step in directed cell migration.
Acknowledgments
We are grateful to Siyang (Sean) Guo, Adam Johnston, Luther Pollard, Shashank Shekhar, and Bengi Turegun for comments on the manuscript. This work was supported by a grant from NIH (R01-GM063691) to B.G., and was supported by Brandeis NSF MRSEC DMR-1420382.
Glossary
- Actin cytoskeleton
A cellular network of thin fibers found in all eukaryotic cells, wherein each filament is a linear polymer of globular actin subunits. The actin cytoskeleton drives many cellular processes, including endocytosis, cytokinesis, and cell crawling
- Actin patches
Branched actin filament networks at the cortex of cells, which represent individual sites of endocytosis and have rapid assembly and disassembly dynamics
- ADF-H
Actin depolymerizing factor homology, a domain shared by a super family of proteins that includes: ADF/Cofilin, Twinfilin, Abp1/Drebrin, GMF, and Coactosin. All five ADF-H family proteins govern actin cytoskeleton organization and/or dynamics, and bind to actin and/or actin-related proteins
- Arp2/3 complex
A conserved complex consisting of seven proteins (Arp2, Arp3, p40/ARPC1, p34/ARPC2, p21/ARPC3, p19/ARPC4, and p15/ARPC5) which nucleates new actin daughter filaments from the sides of existing mother filaments. Arp2/3 complex remains at the branch junction holding the mother and daughter filaments together a debranching event
- Cofilin
An essential protein that binds actin filaments and induces severing and depolymerization, and binds actin monomers to block nucleotide exchange (ATP for ADP). Founding member of ADF-H protein family
- Coronin
A conserved protein that binds Arp2/3 complex to inhibit actin nucleation and possibly promote debranching, and to actin filaments to enhance Cofilin-dependent severing and disassembly
- Daughter filament
An actin filament assembled by Arp2/3 complex off the side of a previously existing mother filament
- Debranching
The dissociation of an actin filament branch junction formed by Arp2/3 complex, which holds together a ‘mother’ and a ‘daughter’ filament
- Disassembly
The dissociation of actin subunits from filaments within a network
- F-actin
Filamentous form of actin; polymers of individual actin monomers.
- G-actin
Unpolymerized globular actin monomers, each 42 kDa
- Leaderless secretion
Mechanisms of protein secretion that bypass the canonical secretory pathway
- Leading edge
The ‘front’ of a polarized, migrating cell, i.e. the side facing the direction of migration. Typically consists of densely branched actin networks (lamellipodia) that generate protrusive force underlying cell crawling
- Mother filament
An existing actin filament, to which Arp2/3 complex associates and then nucleates a daughter filament at a 70 degree angle
- Phosphorylation
Post-translational modification of a protein by covalent attachment of a phosphoryl group to a specific residue. Phosphorylation is mediated by kinases, and can alter the function and/or conformation of the target protein
- Remodeling
The architectural reorganization of individual actin filaments within a larger polymer network
- Severing
Fragmentation of an actin filament into shorter pieces. Along with debranching and depolymerization, severing is critical for disassembly and remodeling of cellular actin networks
- Turnover
The disassembly of actin filaments into ADP-bound monomers, followed by recycling of monomers to an ATP-bound state, priming them for new rounds of assembly
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lim R, et al. Maturation-stimulating effect of brain extract and dibutyryl cyclic AMP on dissociated embryonic brain cells in culture. Exp Eye Res. 1973;79(1):243–6. [PubMed] [Google Scholar]
- 2.Pettmann B, et al. Isolation of a glial maturation factor from beef brain. FEBS Lett. 1980;118(2):195–9. doi: 10.1016/0014-5793(80)80217-1. [DOI] [PubMed] [Google Scholar]
- 3.Lim R, et al. Purification and characterization of glia maturation factor beta: a growth regulator for neurons and glia. Proc Natl Acad Sci U S A. 1989;86(10):3901–5. doi: 10.1073/pnas.86.10.3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaplan R, et al. Molecular cloning and expression of biologically active human glia maturation factor-beta. J Neurochem. 1991;57(2):483–90. doi: 10.1111/j.1471-4159.1991.tb03777.x. [DOI] [PubMed] [Google Scholar]
- 5.Asai K, et al. Isolation of novel human cDNA (hGMF-gamma) homologous to Glia Maturation Factor-beta gene. Biochim Biophys Acta. 1998;1396(3):242–4. doi: 10.1016/s0167-4781(97)00222-4. [DOI] [PubMed] [Google Scholar]
- 6.Goroncy AK, et al. NMR solution structures of actin depolymerizing factor homology domains. Protein Sci. 2009;18(11):2384–92. doi: 10.1002/pro.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zuo P, et al. The expression of glia maturation factors and the effect of glia maturation factor-gamma on angiogenic sprouting in zebrafish. Exp Cell Res. 2013;319(5):707–17. doi: 10.1016/j.yexcr.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 8.Inagaki M, et al. Sensitive immunoassays for human and rat GMFB and GMFG, tissue distribution and age-related changes. Biochim Biophys Acta. 2004;1670(3):208–16. doi: 10.1016/j.bbagen.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 9.Kawai Y, et al. Structure and promoter activity of the human glia maturation factor-gamma gene: a TATA-less, GC-rich and bidirectional promoter. Biochim Biophys Acta. 2003;1625(3):246–52. doi: 10.1016/s0167-4781(02)00627-9. [DOI] [PubMed] [Google Scholar]
- 10.Haynes EM, et al. GMFbeta controls branched actin content and lamellipodial retraction in fibroblasts. J Cell Biol. 2015;209(6):803–12. doi: 10.1083/jcb.201501094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ikeda K, et al. Glia maturation factor-gamma is preferentially expressed in microvascular endothelial and inflammatory cells and modulates actin cytoskeleton reorganization. Circ Res. 2006;99(4):424–33. doi: 10.1161/01.RES.0000237662.23539.0b. [DOI] [PubMed] [Google Scholar]
- 12.Poukkula M, et al. Actin-depolymerizing factor homology domain: A conserved fold performing diverse roles in cytoskeletal dynamics. Cytoskeleton (Hoboken) 2011;68(9):471–90. doi: 10.1002/cm.20530. [DOI] [PubMed] [Google Scholar]
- 13.Gandhi M, et al. GMF is a cofilin homolog that binds Arp2/3 complex to stimulate filament debranching and inhibit actin nucleation. Curr Biol. 2010;20(9):861–7. doi: 10.1016/j.cub.2010.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakano K, et al. GMF is an evolutionarily developed Adf/cofilin-super family protein involved in the Arp2/3 complex-mediated organization of the actin cytoskeleton. Cytoskeleton (Hoboken) 2010;67(6):373–82. doi: 10.1002/cm.20451. [DOI] [PubMed] [Google Scholar]
- 15.Amann KJ, Pollard TD. Direct real-time observation of actin filament branching mediated by Arp2/3 complex using total internal reflection fluorescence microscopy. Proc Natl Acad Sci U S A. 2001;98(26):15009–13. doi: 10.1073/pnas.211556398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mahaffy RE, Pollard TD. Kinetics of the formation and dissociation of actin filament branches mediated by Arp2/3 complex. Biophys J. 2006;91(9):3519–28. doi: 10.1529/biophysj.106.080937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003;112(4):453–65. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- 18.Smith MG, et al. The life cycle of actin patches in mating yeast. J Cell Sci. 2001;114(Pt 8):1505–13. doi: 10.1242/jcs.114.8.1505. [DOI] [PubMed] [Google Scholar]
- 19.Ydenberg CA, et al. Cease-fire at the leading edge: new perspectives on actin filament branching, debranching, and cross-linking. Cytoskeleton (Hoboken) 2011;68(11):596–602. doi: 10.1002/cm.20543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Urban E, et al. Electron tomography reveals unbranched networks of actin filaments in lamellipodia. Nat Cell Biol. 2010;12(5):429–35. doi: 10.1038/ncb2044. [DOI] [PubMed] [Google Scholar]
- 21.Yang C, Svitkina T. Visualizing branched actin filaments in lamellipodia by electron tomography. Nat Cell Biol. 2011;13(9):1012–3. doi: 10.1038/ncb2321. author reply 1013-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cai L, et al. Coronin 1B antagonizes cortactin and remodels Arp2/3-containing actin branches in lamellipodia. Cell. 2008;134(5):828–42. doi: 10.1016/j.cell.2008.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chan C, et al. Cofilin dissociates Arp2/3 complex and branches from actin filaments. Curr Biol. 2009;19(7):537–45. doi: 10.1016/j.cub.2009.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jansen S, et al. Single-molecule imaging of a three-component ordered actin disassembly mechanism. Nat Commun. 2015;6:7202. doi: 10.1038/ncomms8202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poukkula M, et al. GMF Promotes Leading-Edge Dynamics and Collective Cell Migration In Vivo. Curr Biol. 2014 doi: 10.1016/j.cub.2014.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ydenberg CA, et al. GMF severs actin-Arp2/3 complex branch junctions by a cofilin-like mechanism. Curr Biol. 2013;23(12):1037–45. doi: 10.1016/j.cub.2013.04.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sweeney MO. Tuning the architecture and growth rate of branched actin networks. ((UMI No. 3637249)).Brandeis University ProQuest Dissertations and Theses database. 2014 [Google Scholar]
- 28.Ydenberg CA, et al. Combinatorial genetic analysis of a network of actin disassembly-promoting factors. Cytoskeleton (Hoboken) 2015 doi: 10.1002/cm.21231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hess DC, et al. Computationally driven, quantitative experiments discover genes required for mitochondrial biogenesis. PLoS Genet. 2009;5(3):e1000407. doi: 10.1371/journal.pgen.1000407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burke TA, et al. Homeostatic actin cytoskeleton networks are regulated by assembly factor competition for monomers. Curr Biol. 2014;24(5):579–85. doi: 10.1016/j.cub.2014.01.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luan Q, Nolen BJ. Structural basis for regulation of Arp2/3 complex by GMF. Nat Struct Mol Biol. 2013;20(9):1062–8. doi: 10.1038/nsmb.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodal AA, et al. Conformational changes in the Arp2/3 complex leading to actin nucleation. Nat Struct Mol Biol. 2005;12(1):26–31. doi: 10.1038/nsmb870. [DOI] [PubMed] [Google Scholar]
- 33.Sokolova OS, et al. Structural Basis of Arp2/3 Complex Inhibition by GMF, Coronin, and Arpin. J Mol Biol. 2017;429(2):237–248. doi: 10.1016/j.jmb.2016.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Popinako A, et al. Analysis of the interactions between GMF and Arp2/3 complex in two binding sites by molecular dynamics simulation. Biochem Biophys Res Commun. 2018 doi: 10.1016/j.bbrc.2018.01.080. [DOI] [PubMed] [Google Scholar]
- 35.McGough A, et al. Cofilin changes the twist of F-actin: implications for actin filament dynamics and cellular function. J Cell Biol. 1997;138(4):771–81. doi: 10.1083/jcb.138.4.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Galkin VE, et al. Remodeling of actin filaments by ADF/cofilin proteins. Proc Natl Acad Sci U S A. 2011;108(51):20568–72. doi: 10.1073/pnas.1110109108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burnette DT, et al. A role for actin arcs in the leading-edge advance of migrating cells. Nat Cell Biol. 2011;13(4):371–81. doi: 10.1038/ncb2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsuiki H, et al. Cloning of a rat glia maturation factor-gamma (rGMFG) cDNA and expression of its mRNA and protein in rat organs. J Biochem. 2000;127(3):517–23. doi: 10.1093/oxfordjournals.jbchem.a022635. [DOI] [PubMed] [Google Scholar]
- 39.Shi Y, et al. Glia maturation factor gamma (GMFG): a cytokine-responsive protein during hematopoietic lineage development and its functional genomics analysis. Genomics Proteomics Bioinformatics. 2006;4(3):145–55. doi: 10.1016/S1672-0229(06)60027-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mao M, et al. Identification of genes expressed in human CD34(+) hematopoietic stem/progenitor cells by expressed sequence tags and efficient full-length cDNA cloning. Proc Natl Acad Sci U S A. 1998;95(14):8175–80. doi: 10.1073/pnas.95.14.8175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang QH, et al. Cloning and functional analysis of cDNAs with open reading frames for 300 previously undefined genes expressed in CD34+ hematopoietic stem/progenitor cells. Genome Res. 2000;10(10):1546–60. doi: 10.1101/gr.140200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aerbajinai W, et al. Glia maturation factor-{gamma} mediates neutrophil chemotaxis. J Leukoc Biol. 2011;90(3):529–38. doi: 10.1189/jlb.0710424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lippert DN, Wilkins JA. Glia maturation factor gamma regulates the migration and adherence of human T lymphocytes. BMC Immunol. 2012;13(1):21. doi: 10.1186/1471-2172-13-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krause M, Gautreau A. Steering cell migration: lamellipodium dynamics and the regulation of directional persistence. Nat Rev Mol Cell Biol. 2014;15(9):577–90. doi: 10.1038/nrm3861. [DOI] [PubMed] [Google Scholar]
- 45.Maiuri P, et al. Actin flows mediate a universal coupling between cell speed and cell persistence. Cell. 2015;161(2):374–86. doi: 10.1016/j.cell.2015.01.056. [DOI] [PubMed] [Google Scholar]
- 46.Harms BD, et al. Directional persistence of EGF-induced cell migration is associated with stabilization of lamellipodial protrusions. Biophys J. 2005;88(2):1479–88. doi: 10.1529/biophysj.104.047365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bear JE, et al. Antagonism between Ena/VASP proteins and actin filament capping regulates fibroblast motility. Cell. 2002;109(4):509–21. doi: 10.1016/s0092-8674(02)00731-6. [DOI] [PubMed] [Google Scholar]
- 48.Dang I, et al. Inhibitory signalling to the Arp2/3 complex steers cell migration. Nature. 2013;503(7475):281–4. doi: 10.1038/nature12611. [DOI] [PubMed] [Google Scholar]
- 49.Wu C, et al. Arp2/3 is critical for lamellipodia and response to extracellular matrix cues but is dispensable for chemotaxis. Cell. 2012;148(5):973–87. doi: 10.1016/j.cell.2011.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suraneni P, et al. The Arp2/3 complex is required for lamellipodia extension and directional fibroblast cell migration. J Cell Biol. 2012;197(2):239–51. doi: 10.1083/jcb.201112113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aerbajinai W, et al. Glia maturation factor-gamma negatively modulates TLR4 signaling by facilitating TLR4 endocytic trafficking in macrophages. J Immunol. 2013;190(12):6093–103. doi: 10.4049/jimmunol.1203048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aerbajinai W, et al. Glia Maturation Factor-gamma Regulates Monocyte Migration through Modulation of beta1-Integrin. J Biol Chem. 2016;291(16):8549–64. doi: 10.1074/jbc.M115.674200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chorev DS, et al. Regulation of focal adhesion formation by a vinculin-Arp2/3 hybrid complex. Nat Commun. 2014;5:3758. doi: 10.1038/ncomms4758. [DOI] [PubMed] [Google Scholar]
- 54.Le Clainche C, et al. ATP hydrolysis on actin-related protein 2/3 complex causes debranching of dendritic actin arrays. Proc Natl Acad Sci U S A. 2003;100(11):6337–42. doi: 10.1073/pnas.1130513100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boczkowska M, et al. Glia maturation factor (GMF) interacts with Arp2/3 complex in a nucleotide state-dependent manner. J Biol Chem. 2013;288(36):25683–8. doi: 10.1074/jbc.C113.493338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Blanchoin L, Pollard TD. Interaction of actin monomers with Acanthamoeba actophorin (ADF/cofilin) and profilin. J Biol Chem. 1998;273(39):25106–11. doi: 10.1074/jbc.273.39.25106. [DOI] [PubMed] [Google Scholar]
- 57.Chan KT, et al. Unraveling the enigma: progress towards understanding the coronin family of actin regulators. Trends Cell Biol. 2011;21(8):481–8. doi: 10.1016/j.tcb.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Humphries CL, et al. Direct regulation of Arp2/3 complex activity and function by the actin binding protein coronin. J Cell Biol. 2002;159(6):993–1004. doi: 10.1083/jcb.200206113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cai L, et al. Phosphorylation of coronin 1B by protein kinase C regulates interaction with Arp2/3 and cell motility. J Biol Chem. 2005;280(36):31913–23. doi: 10.1074/jbc.M504146200. [DOI] [PubMed] [Google Scholar]
- 60.Cai L, et al. Coronin 1B coordinates Arp2/3 complex and cofilin activities at the leading edge. Cell. 2007;128(5):915–29. doi: 10.1016/j.cell.2007.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brieher WM, et al. Rapid actin monomer-insensitive depolymerization of Listeria actin comet tails by cofilin, coronin, and Aip1. J Cell Biol. 2006;175(2):315–24. doi: 10.1083/jcb.200603149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang N, et al. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature. 1998;393(6687):809–12. doi: 10.1038/31735. [DOI] [PubMed] [Google Scholar]
- 63.Elam WA, et al. Phosphomimetic S3D cofilin binds but only weakly severs actin filaments. J Biol Chem. 2017;292(48):19565–19579. doi: 10.1074/jbc.M117.808378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lim R, Zaheer A. Phorbol ester stimulates rapid intracellular phosphorylation of glia maturation factor. Biochem Biophys Res Commun. 1995;211(3):928–34. doi: 10.1006/bbrc.1995.1901. [DOI] [PubMed] [Google Scholar]
- 65.Lim R, Zaheer A. In vitro enhancement of p38 mitogen-activated protein kinase activity by phosphorylated glia maturation factor. J Biol Chem. 1996;271(38):22953–6. doi: 10.1074/jbc.271.38.22953. [DOI] [PubMed] [Google Scholar]
- 66.Wang T, et al. GMF-gamma Phosphorylation at Tyr-104 Regulates Actin Dynamics and Contraction in Human Airway Smooth Muscle. Am J Respir Cell Mol Biol. 2014 doi: 10.1165/rcmb.2014-0125OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ito J, et al. Interaction of glia maturation factor with the glial cell membrane. Brain Res. 1982;243(2):309–14. doi: 10.1016/0006-8993(82)90254-2. [DOI] [PubMed] [Google Scholar]
- 68.Lim R, et al. Cell-surface expression of glia maturation factor beta in astrocytes. FASEB J. 1990;4(15):3360–3. doi: 10.1096/fasebj.4.15.2253851. [DOI] [PubMed] [Google Scholar]
- 69.Rubartelli A, et al. Secretion of thioredoxin by normal and neoplastic cells through a leaderless secretory pathway. J Biol Chem. 1992;267(34):24161–4. [PubMed] [Google Scholar]
- 70.Bendtsen JD, et al. Feature-based prediction of non-classical and leaderless protein secretion. Protein Eng Des Sel. 2004;17(4):349–56. doi: 10.1093/protein/gzh037. [DOI] [PubMed] [Google Scholar]
- 71.Lim R, et al. Distribution of immunoreactive glia maturation factor-like molecule in organs and tissues. Brain Res. 1987;430(1):93–100. doi: 10.1016/0165-3806(87)90179-9. [DOI] [PubMed] [Google Scholar]
- 72.Utsuyama M, et al. Glia maturation factor produced by thymic epithelial cells plays a role in T cell differentiation in the thymic microenvironment. Int Immunol. 2003;15(5):557–64. doi: 10.1093/intimm/dxg056. [DOI] [PubMed] [Google Scholar]
- 73.Perniola R, Musco G. The biophysical and biochemical properties of the autoimmune regulator (AIRE) protein. Biochim Biophys Acta. 2014;1842(2):326–37. doi: 10.1016/j.bbadis.2013.11.020. [DOI] [PubMed] [Google Scholar]
- 74.Zaheer A, Lim R. In vitro inhibition of MAP kinase (ERK1/ERK2) activity by phosphorylated glia maturation factor (GMF) Biochemistry. 1996;35(20):6283–8. doi: 10.1021/bi960034c. [DOI] [PubMed] [Google Scholar]
- 75.Zaheer A, et al. GMF-knockout mice are unable to induce brain-derived neurotrophic factor after exercise. Neurochem Res. 2006;31(4):579–84. doi: 10.1007/s11064-006-9049-3. [DOI] [PubMed] [Google Scholar]
- 76.Zaheer A, et al. Enhanced expression of neurotrophic factors by C6 rat glioma cells after transfection with glia maturation factor. Neurosci Lett. 1999;265(3):203–6. doi: 10.1016/s0304-3940(99)00253-0. [DOI] [PubMed] [Google Scholar]
- 77.Pantazis NJ, et al. Transfection of C6 glioma cells with glia maturation factor upregulates brain-derived neurotrophic factor and nerve growth factor: trophic effects and protection against ethanol toxicity in cerebellar granule cells. Brain Res. 2000;865(1):59–76. doi: 10.1016/s0006-8993(00)02194-6. [DOI] [PubMed] [Google Scholar]
- 78.Zaheer A, et al. Effects of glia maturation factor overexpression in primary astrocytes on MAP kinase activation, transcription factor activation, and neurotrophin secretion. Neurochem Res. 2001;26(12):1293–9. doi: 10.1023/a:1014241300179. [DOI] [PubMed] [Google Scholar]
- 79.Zaheer A, et al. A novel role of glia maturation factor: induction of granulocyte-macrophage colony-stimulating factor and pro-inflammatory cytokines. J Neurochem. 2007;101(2):364–76. doi: 10.1111/j.1471-4159.2006.04385.x. [DOI] [PubMed] [Google Scholar]
- 80.Kempuraj D, et al. Glia maturation factor induces interleukin-33 release from astrocytes: implications for neurodegenerative diseases. J Neuroimmune Pharmacol. 2013;8(3):643–50. doi: 10.1007/s11481-013-9439-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Aerbajinai W, et al. Glia maturation factor-gamma mediates neutrophil chemotaxis. J Leukoc Biol. 2011;90(3):529–38. doi: 10.1189/jlb.0710424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mendoza MC, et al. ERK-MAPK drives lamellipodia protrusion by activating the WAVE2 regulatory complex. Mol Cell. 2011;41(6):661–71. doi: 10.1016/j.molcel.2011.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Smith BA, et al. Three-color single molecule imaging shows WASP detachment from Arp2/3 complex triggers actin filament branch formation. Elife. 2013;2:e01008. doi: 10.7554/eLife.01008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Co C, et al. Mechanism of actin network attachment to moving membranes: barbed end capture by N-WASP WH2 domains. Cell. 2007;128(5):901–13. doi: 10.1016/j.cell.2006.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Khanduja N, Kuhn JR. Processive acceleration of actin barbed end assembly by N-WASP. Mol Biol Cell. 2013 doi: 10.1091/mbc.E12-11-0781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sweeney MO, et al. A novel role for WAVE1 in controlling actin network growth rate and architecture. Mol Biol Cell. 2015;26(3):495–505. doi: 10.1091/mbc.E14-10-1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lim R, et al. Impaired motor performance and learning in glia maturation factor-knockout mice. Brain Res. 2004;1024(1–2):225–32. doi: 10.1016/j.brainres.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 88.Zaheer A, et al. Reduced severity of experimental autoimmune encephalomyelitis in GMF-deficient mice. Neurochem Res. 2007;32(1):39–47. doi: 10.1007/s11064-006-9220-x. [DOI] [PubMed] [Google Scholar]
- 89.Zaheer A, et al. Diminished cytokine and chemokine expression in the central nervous system of GMF-deficient mice with experimental autoimmune encephalomyelitis. Brain Res. 2007;1144:239–47. doi: 10.1016/j.brainres.2007.01.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zaheer A, et al. Glia maturation factor modulates beta-amyloid-induced glial activation, inflammatory cytokine/chemokine production and neuronal damage. Brain Res. 2008;1208:192–203. doi: 10.1016/j.brainres.2008.02.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zaheer S, et al. Overexpression of glia maturation factor reinstates susceptibility to myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis in glia maturation factor deficient mice. Neurobiol Dis. 2010;40(3):593–8. doi: 10.1016/j.nbd.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zaheer S, et al. Augmented expression of glia maturation factor in Alzheimer’s disease. Neuroscience. 2011;194:227–33. doi: 10.1016/j.neuroscience.2011.07.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Thangavel R, et al. Glia maturation factor expression in entorhinal cortex of Alzheimer’s disease brain. Neurochem Res. 2013;38(9):1777–84. doi: 10.1007/s11064-013-1080-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Stolmeier D, et al. Glia maturation factor expression in hippocampus of human Alzheimer’s disease. Neurochem Res. 2013;38(8):1580–9. doi: 10.1007/s11064-013-1059-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zaheer S, et al. Enhanced expression of glia maturation factor correlates with glial activation in the brain of triple transgenic Alzheimer’s disease mice. Neurochem Res. 2013;38(1):218–25. doi: 10.1007/s11064-012-0913-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zaheer S, et al. Suppression of neuro inflammation in experimental autoimmune encephalomyelitis by glia maturation factor antibody. Brain Res. 2011;1373:230–9. doi: 10.1016/j.brainres.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Khan MM, et al. Suppression of glia maturation factor expression prevents 1-methyl-4-phenylpyridinium (MPP(+))-induced loss of mesencephalic dopaminergic neurons. Neuroscience. 2014;277:196–205. doi: 10.1016/j.neuroscience.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Khan MM, et al. Glia maturation factor deficiency suppresses 1-methyl-4-phenylpyridinium-induced oxidative stress in astrocytes. J Mol Neurosci. 2014;53(4):590–9. doi: 10.1007/s12031-013-0225-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zuo P, et al. High GMFG expression correlates with poor prognosis and promotes cell migration and invasion in epithelial ovarian cancer. Gynecol Oncol. 2014;132(3):745–51. doi: 10.1016/j.ygyno.2014.01.044. [DOI] [PubMed] [Google Scholar]
- 100.Imai R, et al. Transgenic mice overexpressing glia maturation factor-beta, an oxidative stress inducible gene, show premature aging due to Zmpste24 down-regulation. Aging (Albany NY) 2015;7(7):486–99. doi: 10.18632/aging.100779. [DOI] [PMC free article] [PubMed] [Google Scholar]