

Abstract
Vascular smooth muscle cells (SMC) play a major role in vascular diseases, such as atherosclerosis and hypertension. It has long been established in vitro that contractile SMC can phenotypically switch to function as proliferative and/or migratory cells in response to stimulation by oxidative stress, growth factors, and inflammatory cytokines. Reactive oxygen species (ROS) are oxidative stressors implicated in driving vascular diseases, shifting cell bioenergetics, and increasing SMC proliferation, migration, and apoptosis. In this review, we summarize our current knowledge of how disruptions to redox balance can functionally change SMC and how this may influence vascular disease pathogenesis. Specifically, we focus on our current understanding of the role of vascular nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOX) 1, 4, and 5 in SMC function. We also review the evidence implicating mitochondrial fission in SMC phenotypic transitions and mitochondrial fusion in maintenance of SMC homeostasis. Finally, we discuss the importance of the redox regulation of the soluble guanylate cyclase (sGC)-cyclic guanosine monophosphate (cGMP)-protein kinase G (PKG) pathway as a potential oxidative and therapeutic target for regulating SMC function.
Introduction
Vascular smooth muscle cells (SMC) regulate arterial vascular tone and provide structural stability for blood flow regulation needed for delivery of oxygen to tissues. SMC have classically been defined by expression of the following SMC contractile- or contraction-associated proteins: smooth muscle myosin heavy chain 11 (MYH11), smooth muscle α-actin (ACTA2), smooth muscle 22 α/transgelin (TAGLN), and H1-calponin (CNN1) [1]. It has been well-established that in response to growth factors, cytokines, and reactive oxygen species (ROS), cultured SMC can downregulate their contractile genes and phenotypically switch to a more “synthetic” state where cells have an increased proliferative and/or migratory capacity (reviewed in refs. [1–5]). Notably, the advent of rigorous SMC lineage tracing mouse models has provided in vivo evidence that SMC phenotypic switching can occur in the context of disease and has greatly expanded upon this classic two-state contractile-synthetic model of SMC function. Specifically, these studies provide evidence that: (1) in vascular disease, SMC contractile genes, such as ACTA2, can be expressed by other cell types making lineage tracing essential for accurate identification of SMC in vivo [6–9]; (2) SMCs undergo oligoclonal proliferation in diseases, such as atherosclerosis and pulmonary arterial hypertension [10–15]; (3) in atherosclerosis, SMCs can express marker genes and take on functions ascribed to other cell types to become, for example, macrophage and/or myofibroblast like cells [12, 14, 16]; and (4) SMCs can have both beneficial and detrimental roles in atherosclerotic disease progression [11, 12, 17].
An impaired SMC redox balance, exacerbated by the presence of excessive ROS, is a contributing factor to SMC phenotypic switching and has been associated with increased SMC proliferation, migration, apoptosis, and shifts in cell bioenergetics [3, 18–22]. Importantly, ROS levels are elevated in atherosclerosis, systemic hypertension, and pulmonary arterial hypertension, diseases in which SMC are major players [23–25]. It is thus likely that both extra- and intracellular sources of ROS acting on SMC contribute to vascular disease pathogenesis. Therefore, a greater understanding of the redox signaling pathways that influence SMC function and phenotypic state in the context vascular disease is of critical importance.
In this mini review, we have focused on our current understanding of how redox signaling can functionally change SMC and how this in turn may impact vascular disease development. Moving forward, we encourage more in-depth studies on the mechanisms of redox signaling on both SMC function and SMC phenotypic fate in vascular disease pathogenesis.
ROS and SMC function
Overproduction of ROS, leading to oxidative stress, has been implicated in numerous vascular pathologies [23–25]. ROS, such as superoxide (O2−) and hydrogen peroxide (H2O2), can induce SMC proliferation, migration, and apoptosis [3, 18–22]. While there are several producers of vascular ROS, studies in SMC biology have largely focused on nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOX) and mitochondrial mediated ROS generation. NOX proteins facilitate NADPH-mediated reduction of oxygen (O2) to superoxide (O2−) [26, 27]. Even the dismutated product of superoxide, namely H2O2, can acquire oxidative reactivity in the presence of peroxidase enzymes, Fenton chemistry, and low pKa of cysteine residues on proteins; [28] thereby taking on major mediator activity in NOX signaling. NOX1, NOX4, and NOX5 isoforms are present within SMC [29, 30]. Both NOX1 and NOX4 are bound to p22phox at cell membranes [29, 30]. In addition, the cytosolic side of NOX interacts with Rho GTPase RAC1 and p47phox/Nox organizer 1 (NOXO1) and NOX activator 1 (NOXA1). NOX4 binds with polymerase-δ activating protein 2 (POLDIP2) in the cytosol, which is thought to be involved in NOX4-mediated organization of the cell cytoskeleton [30]. Vascular NOX proteins also localize to different cellular compartments. NOX1 localizes to caveolae and endosomes on the plasma membrane; whereas, NOX4 localizes to the nucleus, endoplasmic reticulum (ER), and mitochondria [30]. NOX5 is the most unique vascular NOX in that it is expressed in human and porcine, but not mouse or rat, SMC [31–33]; localizes to the plasma and ER membrane [29, 30]; and contains EF hand motifs activated by intracellular calcium [29, 30].
Evidence suggests that NOX4 is involved in maintenance of SMC quiescence while NOX1 has a role in modulating SMC function. Rat aortic SMCs treated with platelet derived growth factor beta (PDGF-BB) or angiotensin II (Ang II), which induce SMC proliferation and hypertrophy, respectively, leads to downregulation of NOX4 expression and upregulation of NOX1 expression [34, 35]. PDGF-BB-induced increased NOX1 expression and H2O2 production in SMC has been shown to cause downstream activation of c-Jun N-terminal kinase (JNK), as well as cyclin D and extracellular signal-regulated kinase (ERK)1/2 signaling enhancing SMC migration and proliferation, respectively [29]. Ang II-induced SMC hypertrophy is regulated by NOX1 activation of Ras, p38 mitogen kinase activated protein kinase (MAPK)/protein kinase B (Akt), and epidermal growth factor (EGF) receptor pathways [29]. Interestingly, evidence suggests that basal production of H2O2 is the purview of NOX4, while both basal and Ang II-stimulated O2− production are the purview of NOX1 in rat aortic SMC [35]. Collectively, this indicates in all likelihood that NOX1 and NOX4 have highly specialized roles within SMC.
In cultured rat SMC, Nox1 mRNA expression was found to be enhanced in late passage SMC that express low levels of ACTA2, a hallmark of a phenotypically modulated SMC [36]. Cultured Nox1−/− mouse SMC exhibit decreased proliferative capacity and PDGF-BB-induced migratory capacity, while overexpression of NOX1 in SMC had the opposite effects [19]. Similarly, transgenic mice that over-express human NOX1 specifically in SMC had increased Ang II-induced vascular O2− production, hypertension, and vessel wall hypertrophy [37]. Apparently inconsistent findings were reported in studies with global Nox1 knockout mice which had decreased neointima formation and cell proliferation after femoral wire injury, reduced Ang II-induced vessel wall hypertrophy, and decreased susceptibility to aortic dissection [19, 38, 39]. However, the extent to which loss-of-SMC-produced NOX1 contributed to these observations is indeterminable. Overall, the data suggest NOX1 may be important in SMC function and potentially phenotypic plasticity in vascular disease.
In contrast to NOX1, NOX4 function has been associated with SMC contractile proteins in vitro. NOX4 colocalizes with ACTA2, MYH11, and CNN1 in differentiated rat aortic SMC [36]. In high-passage SMC with low levels of ACTA2 expression, NOX4 has been observed to relocate to focal adhesions [36]; siRNA knockdown of Nox4 in cultured rat aortic SMC led to decreased expression of ACTA2, MYH11, and CNN1, suggesting a relationship between NOX4 and SMC contractile protein expression [36]. siRNA knockdown of Nox4 in rat aortic SMC also results in decreased levels of serum response factor (SRF), a factor required for CArG box dependent expression of SMC contractile proteins [36]. In agreement with in vitro observations, NOX4 and CNN1 are coordinately expressed within the neointima and vessel wall 15 days post rat carotid balloon injury [36, 40]. Furthermore, studies by Tong et al.[41] in mice with TAGLN-driven overexpression of a human dominant negative mutant of NOX4 showed decreased carotid wire-injury-induced neointima formation, as compared to controls. These same mice also had reduced atherosclerotic lesion sizes after 12 weeks of Western diet feeding [42]. These data would support the hypothesis that SMC NOX4 expression may contribute to maintenance of SMC in a quiescent contractile state.
There is also evidence, however, that NOX4 may have stimuli-specific effects on SMC proliferation and apoptosis. Ismail et al.[43] showed that transforming growth factor beta 1 (TGF-β1), but not PDGF-BB or interferon gamma (IFNγ), increased NOX4 expression in cultured human pulmonary artery SMC. siRNA knockdown of NOX4 resulted in decreased baseline and TGF-β1-induced cell proliferation of human pulmonary artery SMC, as measured by MTT assay [44]; it should be noted this metabolic assay quantifies the reduction of a tetrazolium dye to formazan, indicating cell viability as a surrogate measure of cell proliferation. Moreover, this same group showed that NOX4 was increased in the media of human donor pulmonary arteries, hypoxia can induce NOX4 expression in cultured human pulmonary artery SMC, and that siRNA knockdown of NOX4 resulted in decreased hypoxia-induced proliferation [43, 44]. SMC NOX4 has also been associated with increased ROS production and decreased SMC viability. For example, overexpression of NOX4 in cultured mouse SMC increased both H2O2 production and SMC apoptosis [22]. Additionally, siRNA knockdown of NOX4 in human SMC decreased ROS levels and proliferation [45]. Taken together, this infers that NOX4 regulation of SMC function may therefore be dependent on the environmental stimuli and origin of the SMC being studied.
It is important to state that an inherent difficulty in studying NOX proteins is the diversity of activities these proteins can elicit depending on cell type, tissue, or disease state in which they are expressed (reviewed in refs. [23–26]). This is quite apparent in studies involving assessment of vascular NOX4 in vivo. Gray et al.[46] showed that global Nox4−/−ApoE−/− mice treated with streptozotocin (STZ) to induce diabetes and diabetes-induced atherosclerosis had no difference in lesion burden as compared to wild-type STZ-diabetic controls. However, Schürmann et al.[47] showed that inducible global Nox4−/−ApoE−/− mice subjected to an accelerated atherosclerosis model attained with partial carotid ligation and western diet feeding had increased aortic atherosclerotic lesion burden relative to similarly treated controls. It is reasonable to presume that the apparent contradictory results observed are due to the differences in disease model and experimental design used in the studies. While these global transgenic studies can inform as to the net effect of NOX4 in normal and disease development, their design does not yield information on cell-specific functions of NOX proteins. Therefore, the continued focused use of SMC-specific NOX mouse models in the context of vascular disease will be essential for understanding the specific contribution of SMC NOX proteins to SMC function, SMC phenotypic state, and vascular disease development.
Finally, in comparison to NOX1 and NOX4, not much is known about NOX5 function. As previously noted, NOX5 is present in human and porcine cells, but not in rat or mouse cells [31–33]. In a small sampling of non-diseased and diseased human coronary arteries, it was discovered that NOX5 expression was increased in diseased human coronary arteries and present within atherosclerotic lesions [48]. In culture, human aortic SMC treated with IFNγ upregulated NOX5 production [49]. With respect to the function of SMC-produced NOX5, siRNA knockdown of NOX5 in cultured human aortic SMC was found to impair PDGF-BB mediated proliferation and ROS production [50]. Likewise, work by Gole et al.[32] showed that adenoviral shRNA knockdown of NOX5 impaired basic fibroblast growth factor (bFGF)-induced porcine coronary SMC migration. Therefore, it is highly feasible that NOX5 is important in human-SMC responses to stress and SMC phenotype modulation.
Mitochondrial fission and SMC function
Mitochondria are the crucial energy producing organelles of the cell. In normal physiological conditions, SMC mitochondria ATP generation is accompanied by low levels of O2− and H2O2 production [51, 52]. Increased ROS levels and production in response to pathogenic stimuli causes mitochondrial damage, leading to a shift in mitochondria state from fusion to fission [51, 53]. Mitochondria fission is characterized by the fragmentation of mitochondria and mitochondrial networks in response to stress, facilitating the removal of severely stress-damaged mitochondria and the generation of new mitochondria [51]. Fission is a process regulated primarily by GTPase dynamin-related protein 1 (DRP1, gene name DNM1L) with assistance from other cofactor proteins (i.e., Mid49, Mid51, and Mff) [51, 54].
SMC phenotypic transitions, specifically in response to PDGF-BB treatment in vitro, have been shown to involve mitochondria shifts from fusion to fission. SMC DRP1 expression has been linked most closely to enhanced SMC proliferation and migration [55–57]. PDGF–BB treatment of human SMC results in increased phosphorylation of serine 616 of DRP1, which activates mitochondrial fission [55, 58]. Moreover, PDGF-BB treatment induces DRP1-dependent increases in SMC proliferation [55, 58, 59] and mitochondrial ROS production [55]. This stands in agreement with findings of increased DRP1 and serine 616 phosphorylated DRP1 levels, as well as increased mitochondrial fission and SMC proliferation rates in SMC derived from patients with pulmonary arterial hypertension [58]. Finally, adenoviral-mediated overexpression of a dominant negative mutant of DRP1 in a mouse femoral wire injury model showed decreased neointima formation and vessel wall ROS levels, suggesting DRP1-mediated mitochondrial fission may be important for vessel wall cell response to injury [55].
PDGF-BB mediates a shift in SMC metabolism from glycolysis to fatty-acid oxidation that is regulated in part by DRP1 [59, 60]. This is evidenced by impairment of the metabolic switch in SMC when PDGF-BB treatment has been combined with the selective DRP1 inhibitor Mdivi-1, 2-deoxyglucose, a non-hydrolyzable glucose that inhibits glycolysis, and/or the phosphoinositide 3-kinase inhibitor LY-294002 [59, 60]. Pharmacological stimulation of SMC glucose oxidation with trimetazidine has similarly been shown to reverse hypoxia-induced mitochondrial fission in human pulmonary artery SMC [61]. Interestingly, when SMC were treated with PDGF-BB, pharmacological inhibition of mitochondrial fission did not impact SMC autophagy and vacuole formation, a process important for the degradation and clearance of SMC contractile proteins [56, 60].
In addition to SMC proliferation, contractile, and osteogenic stimuli also induce mitochondrial fission. Liu et al. [62] have demonstrated ex vivo that rat mesenteric arteries contracted with a high-potassium solution (KPSS) have mitochondria fission occurring in vessel wall cells. Furthermore, SMC treated with KPSS in vitro showed increases in both cytosolic Ca2+ and mitochondrial ROS production, effects which were ameliorated by the addition of either Mdivi-1 or nitroglycerin [62]. Separately, work by Rogers et al.[63] has shown that treatment of human SMC with calcification-inducing osteogenic media increases DRP1 levels. Furthermore, Mdivi-1 inhibition of DRP1 impairs osteogenic media inducing cultured human SMC calcification [63]. Taken together, mitochondrial fission seems to be an essential process in SMC response to environmental stimuli and for SMC phenotypic switching. However, further studies are warranted to understand the direct mechanisms of connection between SMC response to environmental stimuli, mitochondrial fission, mitochondrial ROS generation, calcium signaling, and SMC function.
Mitochondrial fusion and SMC homeostasis
Mitochondrial fusion produces an interconnected mitochondrial network that is important in the maintenance of SMC homeostasis and quiescence [51, 64]. In response to cell energy demands and oxidative stress, mitofusion allows for complementation between healthy mitochondria and stress damaged mitochondria, diluting and mitigating damage for maintenance of cell homeostasis [51, 64]. Mitofusion occurs at both the inner and outer mitochondrial membranes with mitofusin 1 (MFN1) and mitofusin 2 (MFN2) acting at the outer mitochondrial membrane [51].
MFN2 is the most extensively studied in vascular SMC. Rat aortic SMC treated with PDGF-BB, bFGF, and endothelin-1 (ET-1) have decreased MFN2 expression levels [21, 65]. Overexpression of MFN2 in cultured rat or rabbit aortic SMC has been shown to impair SMC proliferation and arrest growth of SMC in cell cycle G0/G1 phase [21, 65–67]. Zhou et al.[67] discovered that MFN2 serine 442, which contains a protein kinase A (PKA) phosphorylation site, is an important residue for MFN2− mediated impairment of SMC proliferation [67]. MFN2 binding to Ras has been shown in aortic SMC over-expressing MFN2 [65]. Overexpression of MFN2 in SMC also decreases pAKT and pERK levels, implicating MFN2 as an inhibitor of the Ras-Raf-ERK1/2 signaling pathway [21, 65, 68].
MFN2 overexpression in cultured rat SMC has also been shown to increase mitochondrial mediated SMC apoptosis [21]. Rat SMC treated with H2O2 causes upregulation of MFN2 and caspases 3 and 9, markers of cell death [21]. Knockdown of Mfn2 by siRNA protects against H2O2 mediated apoptosis [21]. Overexpression of MFN2 in balloon injured rat carotid arteries led to a decrease in media and neointimal cell proliferation, increased medial cell death (TUNEL+), and reduced neointima formation as compared to controls [21]. Likewise, in hypercholesterolemic rabbits, overexpression of MFN2 has been shown to impair atherosclerotic lesion formation, though cell viability was not assessed [66]. Combined, these data suggest that MFN2 regulates SMC homeostasis and viability.
However, contradictory evidence exists that similar phenotypes occur when MFN2 levels are reduced. For example, work by Ding et al.[69] showed that shRNA knockdown of MFN2 in human SMC decreased SMC viability, as measured by NADPH reduction of tetrazolium to formazan. Similarly, Zhang et al.[70] found that siRNA knockdown of Mfn2 in rat pulmonary artery SMC decreases SMC proliferation; viability, as measured by MTT assay; and pAKT levels. Despite the apparent contradiction between studies, there is a general consensus that MFN2 is involved in cultured SMC viability, proliferation, and expression of pAKT and pERK1/2. We speculate that the quantity of MFN2 produced by SMC may therefore be a crucial determinant in regulation of SMC viability and proliferative capacity.
In addition to its role in mitochondrial fusion, MFN2 has been shown to link mitochondria with the endoplasmic reticulum (ER) [71, 72]. The transfer of calcium (Ca2+) between mitochondria and ER is important for regulating mitochondrial bioenergetics and cell viability [71–74]. The mitochondria-ER link promotes Ca2+ flux into the mitochondria, an activity that is associated with increased ATP generation and anti-proliferative activity in SMC [75, 76]. Li et al.[72] showed in cultured rat aortic SMC that there is increased mitochondrial fusion and mitochondria-ER links when SMC are in the G0/G1 versus S phase. Moreover, mitochondria-ER links were shown to be increased in rat aortic SMC when MFN2 was overexpressed and decreased with siRNA Mfn2 knockdown [72]. Separately, Morales et al.[76] showed that glucagon-like peptide-1 (GLP-1) enhancement of rat aortic SMC glucose uptake increased MFN2 levels. They also showed that MFN2 and PKA activity were important for GLP-1 mediated Ca2+ transfer from the ER to the mitochondria [76].
In both cardiac and skeletal muscles, NO and carbon monoxide have been shown to induce cGMP-PKG-dependent induction of PGC1α and mitochondrial fusion [77–79]. While there is no direct evidence that the sGC-cGMP-PKG pathway impacts mitochondrial bioenergetics, studies have shown that siRNA knockdown of PGC1α in human pulmonary artery SMC decreases MFN2 mRNA expression and vice versa [80]. Taken together, it is interesting to speculate on ROS regulation of sGC-cGMP-PKG as a master upstream regulator of SMC mitochondrial bioenergetics, as well as SMC function and phenotypic state.
The sGC-cGMP-PKG pathway
Excessive ROS generation can drive vascular pathogenesis and SMC phenotypic transitions by uncoupling the nitric oxide (NO)—soluble guanylate cyclase (sGC)—cyclic guanosine monophosphate (cGMP)—protein kinase G (PKG) pathway [81–83]. Oxidation of sGC heme iron (Fe2+ to Fe3+) impairs binding of NO to sGC [84–87]. H2O2 and peroxynitrite (ONOO−) are particularly strong oxidants that predispose the sGC enzyme to heme loss, insensitivity to NO signaling, and degradation via the ubiquitin protein degradation pathway [88, 89]. Uncoupling of the pathway, which is central to maintaining SMC in their quiescent state and mediating vessel dilation [90–93], has been implicated in several vascular diseases and has been specifically targeted in treatment of pulmonary arterial hypertension [94–96]. For example, in a rat carotid artery balloon injury model it was reported that sGC expression decreased by ~90% in the vessel wall, as compared to uninjured controls [97]. Likewise, inducible SMC-specific sGC knockout mice showed severely elevated blood pressure at baseline [98].
Physiological restoration of sGC to its NO-sensitive Fe2+ state requires redox regulation of the enzyme’s heme iron. Recently it was discovered that NADH cytochrome B5 reductase 3 (CYB5R3) in SMC functions as a regulator of sGC redox state and NO sensitivity; a mechanism previously unknown [99]. It was shown that CYB5R3 complexes with the oxidized sGC enzyme and is necessary for NO to signal cGMP production by the enzyme under oxidative conditions [99]. Circumvention of impaired NO-sGC signaling due to functional loss-of-sGC’s heme iron is the focus of a pipeline of pharmacological compounds that activate oxidized or heme-deficient sGC thus reactivating the sGC-cGMP signaling cascade. Indeed, Cinaciguat, or BAY 58–2667, is one of these pharmacological compounds and was recently shown to prevent neointima formation following wire injury and to decrease SMC migration and proliferation, as measured by in vitro scratch wound healing assay [100]. These sGC activators and the discovery of the sGC reductase CYB5R3 represent novel strategies and targets for modulating SMC function and treating vascular disease.
Concluding remarks
Oxidative stress and SMCs are integral contributors to various vascular diseases, including atherosclerosis, pulmonary arterial hypertension, and systemic hypertension [23–25]. As described herein, changes to SMC redox state can shift SMC from contractile to phenotypically diverse cells in vitro and drive changes in mitochondrial bioenergetics; this in turn can impact vascular disease. In addition, ROS can target and modulate sGC redox state in SMC, thus impacting the activation of downstream signaling proteins and kinases (i.e., PKG, PGC1α) that have been implicated in modulating SMC state, mitochondrial fission, and mitochondrial fusion. One possible connection between the pathways discussed here is that PDGF-BB-induced increases in NOX1 expression resulting in production of O2− and H2O2 could in turn oxidize sGC and thereby inhibit cGMP production and PKG activation (Fig. 1). It is also plausible that this PDGF-BB-mediated increase in SMC ROS production results in terminally damaged mitochondria, causing increased phosphorylation of DRP1 serine 616 and SMC mitochondrial fission (Fig. 1). Finally, it has been shown that the sGC-cGMP-PKG pathway regulates cytosolic Ca2+ [101] and that mitofusion protein MFN2 is a crucial mediator of Ca2+ flux between the mitochondria and ER [71–74]. It is therefore interesting to speculate that Ca2+ flux may be the downstream convergence point for these pathways and the currency for SMC phenotypic switching. However, the rate limiting factors and direct pathway integrations that factor into SMC responses to environmental cues and the balancing of SMC between quiescence, motility, proliferation, and viability remain incompletely understood and warrant further inquiry.
Fig. 1.
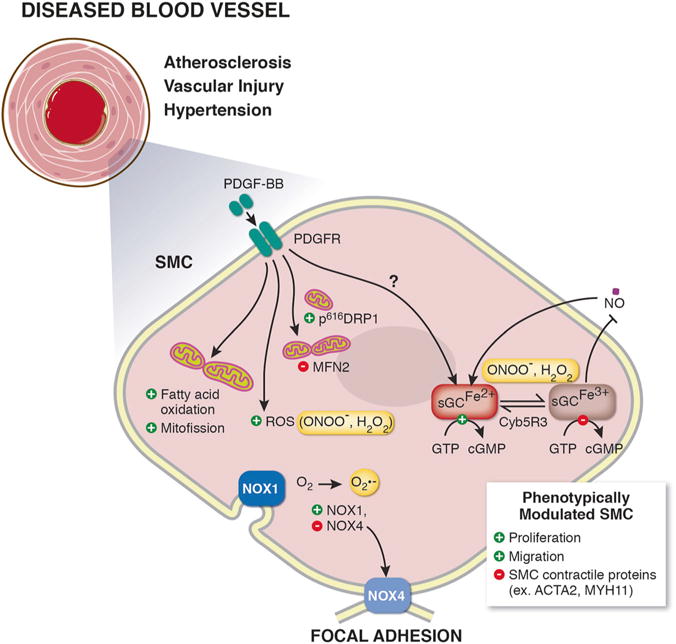
Schematic illustration showing mechanisms of smooth muscle cell (SMC) phenotypic switching. In response to vascular disease, SMC phenotypically switch from contractile cells to proliferative and migratory SMC characterized by downregulation of SMC contractile proteins (ex. smooth muscle α actin (ACTA2), smooth muscle myosin heavy chain 11 (MYH11)). SMC mitogen platelet derived growth factor beta (PDGF-BB) is known to induce SMC phenotypic switching. PDGF-BB can change cell bioenergetics, including shifting SMC metabolism to fatty-acid oxidation, increasing reactive oxygen species (ROS) production, and inducing mitochondrial fission, via increasing phosphorylation of dynamin-related protein 1 (DRP1) and decreasing mitofusin 2 (MFN2) levels. Vascular nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOX) NOX1 and NOX4 generate superoxide (O2−) and are known to drive SMC switching. The soluble guanylate cyclase (sGC)-cyclic guanosine monophosphate (cGMP)-protein kinase G (PKG) pathway signaling maintains SMC quiescence and mediates physiological vessel dilation. Excessive ROS generation results in oxidation of the sGC heme iron (Fe2+ to Fe3+) inhibiting binding of nitric oxide (NO) to sGC. Restoration of the sGC heme iron back to its NO-sensitive Fe2+ state is done endogenously by NADH cytochrome B5 reductase 3 (Cyb5R3)
Another critical next step in the field will be to move beyond a two-state view of SMC plasticity. Indeed, recent SMC lineage tracing studies have demonstrated that SMC can become foam cell, mesenchymal stem cell, or myofibroblast-like in atherosclerosis, with these phenotypic states likely having both beneficial and detrimental effects on disease progression [9, 11, 14, 17]. It is reasonable to predict that similar observations will be made in other vascular diseases. Therefore, the key challenges to the field moving forward will be to discern (1) if and how SMC produce and respond to ROS in vivo in a manner that directly contributes to SMC phenotypic state and function and; (2) to what extent, if any, this contributes to disease progression and/or resolution.
Acknowledgments
Financial support was provided by the National Institutes of Health Grants R01 HL 133864, R01 HL 128304, American Heart Association (AHA) Grant-in-Aid 16GRNT27250146 (ACS). This work was further supported by the Institute for Transfusion Medicine and the Hemophilia Center of Western Pennsylvania (ACS). We would like to acknowledge the contributions of Katherine C. Wood and Joseph C. Galley for their aid in critical analysis of the manuscript.
Footnotes
Compliance with Ethical Standards
Conflict of interest The authors declare that they have no conflict of interest.
References
- 1.Gomez D, Owens GK. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res. 2012;95:156–64. doi: 10.1093/cvr/cvs115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.San Martin Alejandra, Griendling KK. Redox control of vascular smooth muscle migration. Antioxid Redox Signal. 2010;12:625–40. doi: 10.1089/ars.2009.2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clempus Roza E, Griendling KK. Reactive oxygen species signaling in vascular smooth muscle cells. Cardiovasc Res. 2007;71:216–25. doi: 10.1016/j.cardiores.2006.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alexander MR, Owens GK. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu Rev Physiol. 2011;74:13–40. doi: 10.1146/annurev-physiol-012110-142315. [DOI] [PubMed] [Google Scholar]
- 5.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 6.Caplice NM, Bunch TJ, Stalboerger PG, Wang S, Simper D, Miller DV, et al. Smooth muscle cells in human coronary atherosclerosis can originate from cells administered at marrow transplantation. Proc Natl Acad Sci USA. 2003;100:4754–9. doi: 10.1073/pnas.0730743100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Allahverdian S, Chehroudi AC, McManus BM, Abraham T, Francis GA. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation. 2014;129:1551–9. doi: 10.1161/CIRCULATIONAHA.113.005015. [DOI] [PubMed] [Google Scholar]
- 8.Chen PY, Qin L, Baeyens N, Li G, Afolabi T, Budatha M, et al. Endothelial-to-mesenchymal transition drives atherosclerosis progression. J Clin Invest. 2015;125:4514–28. doi: 10.1172/JCI82719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gomez D, Shankman LS, Nguyen AT, Owens GK. Detection of histone modifications at specific gene loci in single cells in histological sections. Nat Methods. 2013;10:171–7. doi: 10.1038/nmeth.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sheikh AQ, Misra A, Rosas IO, Adams RH, Greif DM. Smooth muscle cell progenitors are primed to muscularize in pulmonary hypertension. Sci Transl Med. 2015;7:1–12. doi: 10.1126/scitranslmed.aaa9712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cherepanova OA, Gomez D, Shankman LS, Swiatlowska P, Williams J, Sarmento OF, et al. Activation of the pluripotency factor OCT4 in smooth muscle cells is atheroprotective. Nat Med. 2016;22:657–65. doi: 10.1038/nm.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015;21:628–37. doi: 10.1038/nm.3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chappell J, Harman JL, Narasimhan VM, Yu H, Foote K, Simons BD, et al. Extensive proliferation of a subset of differentiated, yet plastic, medial vascular smooth muscle cells contribute to neointimal formation in mouse injury and atherosclerosis models. Circ Res. 2016;119:1313–23. doi: 10.1161/CIRCRESAHA.116.309799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feil S, Fehrenbacher B, Lukowski R, Essmann F, Schulze-Osthoff K, Schaller M, et al. Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ Res. 2014;115:662–7. doi: 10.1161/CIRCRESAHA.115.304634. [DOI] [PubMed] [Google Scholar]
- 15.Gomez D, Owens GK. Reconciling smooth muscle cell oligoclonality and proliferative capacity in experimental atherosclerosis. Circ Res. 2016;119:1262–4. doi: 10.1161/CIRCRESAHA.116.310104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rong JX, Shapiro M, Trogan E, Fisher EA. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc Natl Acad Sci USA. 2003;100:13531–6. doi: 10.1073/pnas.1735526100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Durgin BG, Cherepanova OA, Gomez D, Karaoli T, Alencar GF, Butcher JT, et al. Smooth muscle cell-specific deletion of Col15a1 unexpectedly leads to impaired 1 development of advanced atherosclerotic lesions 2 3. Am J Physiol—Heart Circ Physiol. 2017;312:H943–58. doi: 10.1152/ajpheart.00029.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manea A, Tanase LI, Raicu M, Simionescu M. JAK/STAT signaling pathway regulates Nox1 and Nox4-based NADPH oxidase in human aortic smooth muscle cells. Arterioscler Thromb Vasc Biol. 2010;30:105–12. doi: 10.1161/ATVBAHA.109.193896. [DOI] [PubMed] [Google Scholar]
- 19.Lee MY, Martin AS, Mehta PK, Dikalova AE, Garrido AM, Datla SR, et al. Mechanisms of vascular smooth muscle NADPH oxidase 1 (Nox1) contribution to injury-induced neointimal formation. Arterioscler Thromb Vasc Biol. 2009;29:480–7. doi: 10.1161/ATVBAHA.108.181925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Waypa GB, Guzy R, Mungai PT, Mack MM, Marks JD, Roe MW, et al. Increases in mitochondrial reactive oxygen species trigger hypoxia-induced calcium responses in pulmonary artery smooth muscle cells. Circ Res. 2006;99:970–8. doi: 10.1161/01.RES.0000247068.75808.3f. [DOI] [PubMed] [Google Scholar]
- 21.Guo X, Chen K, Guo Y, Liao H, Tang J, Xiao R. Mitofusin 2 triggers vascular smooth muscle cell apoptosis via mitochondrial death pathway. Circ Res. 2007;101:1113–22. doi: 10.1161/CIRCRESAHA.107.157644. [DOI] [PubMed] [Google Scholar]
- 22.Xu S, Chamseddine AH, Carrell S, Miller FJ., Jr Nox4 NADPH oxidase contributes to smooth muscle cell phenotypes associated with unstable atherosclerotic plaques. Redox Biol. 2014;2:642–50. doi: 10.1016/j.redox.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee MY, Griendling KK. Redox signaling, vascular function, and hypertension. Antioxid Redox Signal. 2008;10:1045–59. doi: 10.1089/ars.2007.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nowak WN, Deng J, Ruan XZ, Xu Q. Reactive oxygen species generation and atherosclerosis. Arterioscler Thromb Vasc Biol. 2017;37:e41–53. doi: 10.1161/ATVBAHA.117.309228. [DOI] [PubMed] [Google Scholar]
- 25.Wong C-M, Bansal G, Pavlickova L, Marcocci L, Suzuki YJ. Reactive oxygen species and antioxidants in pulmonary hypertension. Antioxid Redox Signal. 2013;18:1789–96. doi: 10.1089/ars.2012.4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lassègue B, Griendling KK. NADPH oxidases: functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol. 2010;30:653–61. doi: 10.1161/ATVBAHA.108.181610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brandes RP, Kreuzer J. Vascular NADPH oxidases: molecular mechanisms of activation. Cardiovasc Res. 2005;65:16–27. doi: 10.1016/j.cardiores.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 28.Winterbourn C, Metodiewa D. Reactivity of biologically important thiol compunds with superoxide and hydrogen peroxide. Free Radic Biol Med. 1999;27:322–8. doi: 10.1016/s0891-5849(99)00051-9. [DOI] [PubMed] [Google Scholar]
- 29.Brown DI, Griendling KK. Nox proteins in signal transduction. Free Radic Biol Med. 2009;47:1239–53. doi: 10.1016/j.freeradbiomed.2009.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov. 2011;10:453–71. doi: 10.1038/nrd3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lassegue B, Clempus RE. Vascular NAD (P) H oxidases: specific features, expression, and regulation. Am J Physiol Integr Comp Physiol. 2003;285:R277–97. doi: 10.1152/ajpregu.00758.2002. [DOI] [PubMed] [Google Scholar]
- 32.Gole HKA, Tharp DL, Bowles DK. Upregulation of intermediate-conductance Ca2+-activated K+ channels (KCNN4) in porcine coronary smooth muscle requires NADPH oxidase 5 (NOX5) PLoS ONE. 2014;9:1–12. doi: 10.1371/journal.pone.0105337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fulton DJR. Nox5 and the regulation of cellular function. Antioxid Redox Signal. 2009;11:2443–52. doi: 10.1089/ars.2009.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lassègue B, Sorescu D, Szöcs K, Yin Q, Akers M, Zhang Y, et al. Novel gp91phox homologues in vascular smooth muscle cells: Nox1 mediates angiotensin II-induced superoide formation and redox-sensitive signaling pathways. Circ Res. 2001;88:888–95. doi: 10.1161/hh0901.090299. [DOI] [PubMed] [Google Scholar]
- 35.Dikalov SI, Dikalova AE, Bikineyeva AT, Schmidt HH, Harriso DG, Griendling KK. Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radic Biol Med. 2008;45:1340–51. doi: 10.1016/j.freeradbiomed.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clempus RE, Sorescu D, Dikalova AE, Pounkova L, Jo P, Sorescu GP, et al. Nox4 is required for maintenance of the differentiated vascular smooth muscle cell phenotype. Arterioscler Thromb Vasc Biol. 2007;27:42–8. doi: 10.1161/01.ATV.0000251500.94478.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dikalova A, Clempus R, Lassègue B, Cheng G, Mccoy J, Dikalov S, et al. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation. 2005;112:2668–76. doi: 10.1161/CIRCULATIONAHA.105.538934. [DOI] [PubMed] [Google Scholar]
- 38.Gavazzi G, Deffert C, Trocme C, Schappi M, Herrmann FR, Krause K-H. NOX1 deficiency protects from aortic dissection in response to angiotensin II. Hypertension. 2007;50:189–96. doi: 10.1161/HYPERTENSIONAHA.107.089706. [DOI] [PubMed] [Google Scholar]
- 39.Gavazzi G, Banfi B, Deffert C, Fiette L, Schappi M, Herrmann F, et al. Decreased blood pressure in NOX1-deficient mice. FEBS Lett. 2006;580:497–504. doi: 10.1016/j.febslet.2005.12.049. [DOI] [PubMed] [Google Scholar]
- 40.Szöcs K, Lassègue B, Sorescu D, Hilenski LL, Valppu L, Couse TL, et al. Upregulation of Nox-based NAD(P)H oxidases in restenosis after carotid injury. Arterioscler Thromb Vasc Biol. 2002;22:21–7. doi: 10.1161/hq0102.102189. [DOI] [PubMed] [Google Scholar]
- 41.Tong X, Khandelwal AR, Qin Z, Wu X, Chen L, Ago T, et al. Role of smooth muscle Nox4-based NADPH oxidase in neointimal hyperplasia. J Mol Cell Cardiol. 2015;89:185–94. doi: 10.1016/j.yjmcc.2015.11.013. [DOI] [PubMed] [Google Scholar]
- 42.Tong X, Khandelwal AR, Wu X, Xu Z, Yu W, Chen C, et al. Pro-atherogenic role of smooth muscle Nox4-based NADPH oxidase. J Mol Cell Cardiol. 2016;92:30–40. doi: 10.1016/j.yjmcc.2016.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ismail S, Sturrock A, Wu P, Cahill B, Norman K, Huecksteadt T, et al. NOX4 mediates hypoxia-induced proliferation of human pulmonary artery smooth muscle cells: the role of autocrine production of transforming growth factor-b 1 and insulin-like growth factor binding protein-3. Am J Physiol Lung Cell Mol Physiol. 2009;84132:489–99. doi: 10.1152/ajplung.90488.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sturrock A, Cahill B, Norman K, Huecksteadt TP, Hill K, Sanders K, et al. Transforming growth factor-beta1 induces Nox4 NAD(P)H oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2006;290:661–73. doi: 10.1152/ajplung.00269.2005. [DOI] [PubMed] [Google Scholar]
- 45.Przybylska D, Janiszewska D, Goździk A, Bielak-Zmijewska A, Sunderland P, Sikora E, et al. NOX4 downregulation leads to senescence of human vascular smooth muscle cells. Oncotarget. 2016;7:66429–43. doi: 10.18632/oncotarget.12079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gray SP, Di Marco E, Okabe J, Szyndralewiez C, Heitz F, Montezano AC, et al. NADPH oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation. 2013;127:1888–902. doi: 10.1161/CIRCULATIONAHA.112.132159. [DOI] [PubMed] [Google Scholar]
- 47.Schürmann C, Rezende F, Kruse C, Yasar Y, Löwe O, Fork C, et al. The NADPH oxidase Nox4 has anti-atherosclerotic functions. Eur Heart J. 2015;36:3447–56. doi: 10.1093/eurheartj/ehv460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guzik TJ, Chen W, Gongora MC, Guzik B, Lob HE, Mangalat D, et al. Calcium-dependent NOX5 nicotinamide adenine dinucleotide phosphate oxidase contributes to vascular oxidative stress in human coronary artery disease. J Am Coll Cardiol. 2008;52:1803–9. doi: 10.1016/j.jacc.2008.07.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Manea A, Manea SA, Florea IC, Luca CM, Raicu M. Positive regulation of NADPH oxidase 5 by proinflammatory-related mechanisms in human aortic smooth muscle cells. Free Radic Biol Med. 2012;52:1497–507. doi: 10.1016/j.freeradbiomed.2012.02.018. [DOI] [PubMed] [Google Scholar]
- 50.Jay DB, Papaharalambus CA, Seidel-Rogol B, Dikalova AE, Lassègue B, Griendling KK. Nox5 mediates PDGF-induced proliferation in human aortic smooth muscle cells. Free Radic Bio. 2009;45:329–35. doi: 10.1016/j.freeradbiomed.2008.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–5. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van der Bliek AM, Shen Q, Kawajiri S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb Perspect Biol. 2013;5:a011072–a011072. doi: 10.1101/cshperspect.a011072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–95. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 54.Loson OC, Song Z, Chen H, Chan DC. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell. 2013;24:659–67. doi: 10.1091/mbc.E12-10-0721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang L, Yu T, Lee H, Brien DKO, Sesaki H, Yoon Y. Decreasing mitochondrial fission diminishes vascular smooth muscle cell migration and ameliorates intimal hyperplasia. Cardiovasc Res. 2015;106:272–83. doi: 10.1093/cvr/cvv005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Salabei JK, Cummins TD, Singh M, Jones SP, Bhatnagar A, Hill BG. PDGF-mediated autophagy regulates vascular smooth muscle cell phenotype and resistance to oxidative stress. J Biochem. 2014;451:375–88. doi: 10.1042/BJ20121344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chalmers S, Saunter C, Wilson C, Coats P, Girkin JM, McCarron JG. Mitochondrial motility and vascular smooth muscle cell proliferation. Arter Thromb Vasc Biol. 2012;32:3000–11. doi: 10.1161/ATVBAHA.112.255174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marsboom G, Toth PT, Ryan JJ, Hong Z, Wu X, Fang YH, et al. Dynamin-related protein 1 (DRP1)-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ Res. 2012;110:1484–97. doi: 10.1161/CIRCRESAHA.111.263848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Salabei JK, Hill BG. Mitochondrial fission induced by platelet-derived growth factor regulates vascular smooth muscle cell bioenergetics. Redox Biol. 2013;1:542–51. doi: 10.1016/j.redox.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Perez J, Hill BG, Benavides GA, Dranka BP, Darley-Usmar VM. Role of cellular bioenergetics in smooth muscle cell proliferation induced by platelet-derived growth factor. J Biochem. 2013;428:255–67. doi: 10.1042/BJ20100090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Parra V, Bravo-Sagua R, Norambuena-Soto I, Hernandez-Fuentes Carolina P, Gomez-Contreras AG, et al. Inhibition of mitochondria fission prevents hypoxia-induced metabolic shift and cellular proliferation of pulmonary artery smooth muscle cells. Biochim Biophys Acta. 2017:S0925–4439. 30244–2. doi: 10.1016/j.bbadis.2017.07.018. [DOI] [PubMed] [Google Scholar]
- 62.Liu M, Jin J, Li S-L, Yan J, Zhen C-L, Gao J-L, et al. Mitochondrial fission of smooth muscle cells is involved in artery constriction. Hypertension. 2016;68:1245–54. doi: 10.1161/HYPERTENSIONAHA.116.07974. [DOI] [PubMed] [Google Scholar]
- 63.Rogers MA, Maldonado N, Hutcheson JD, Goettsch C, Goto S, Yamada I, et al. Dynamin-related protein 1 inhibition attenuates cardiovascular calcification in the presence of oxidative stress. Circ Res. 2017;121:1–12. doi: 10.1161/CIRCRESAHA.116.310293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Archer SL. Mitochondrial dynamics—mitochondrial fission and fusion in human diseases. N Engl J Med. 2013;369:2236–51. doi: 10.1056/NEJMra1215233. [DOI] [PubMed] [Google Scholar]
- 65.Chen K, Guo X, Ma D, Guo Y, Li Q, Yang D, et al. Dysregulation of HSG triggers vascular proliferative disorders. Nat Cell Biol. 2004;6:872–83. doi: 10.1038/ncb1161. [DOI] [PubMed] [Google Scholar]
- 66.Guo Y, Chen K, Gao W, Li Q, Chen L, Wang G, et al. Over-expression of Mitofusin 2 inhibited oxidized low-density lipoprotein induced vascular smooth muscle cell proliferation and reduced atherosclerotic lesion formation in rabbit. Biochem Biophys Res Commun. 2007;363:411–7. doi: 10.1016/j.bbrc.2007.08.191. [DOI] [PubMed] [Google Scholar]
- 67.Zhou W, Chen KH, Cao W, Zeng J, Liao H, Zhao L, et al. Mutation of the protein kinase A phosphorylation site influences the anti-proliferative activity of mitofusin 2. Atherosclerosis. 2010;211:216–23. doi: 10.1016/j.atherosclerosis.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 68.Fang X, Chen X, Zhong G, Chen Q, Hu C. Mitofusin 2 down-regulation triggers pulmonary artery smooth muscle cell proliferation and apoptosis imbalance in rats with hypoxic pulmonary hypertension Via the PI3K/Akt and mitochondrial apoptosis pathways. Int J Biochem Cell Biol. 2016;77:141–54. [Google Scholar]
- 69.Ding Y, Gao H, Zhao L, Wang X, Zheng M. Mitofusin 2-deficiency suppresses cell proliferation through disturbance of autophagy. PLoS One. 2015;10:e0121328. doi: 10.1371/journal.pone.0121328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang D, Ma C, Li S, Ran Y, Chen J, Lu P, et al. Effect of Mitofusin 2 on smooth muscle cells proliferation in hypoxic pulmonary hypertension. Microvasc Res. 2012;84:286–96. doi: 10.1016/j.mvr.2012.06.010. [DOI] [PubMed] [Google Scholar]
- 71.De Brito OM, Scorrano Mitofusin L. 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–11. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- 72.Li D, Li X, Guan Y, Guo X. Mitofusin-2-mediated tethering of mitochondria and endoplasmic reticulum promotes cell cycle arrest of vascular smooth muscle cells in G0/G1 phase. Acta Biochim Biophys. 2017;47:441–50. doi: 10.1093/abbs/gmv035. [DOI] [PubMed] [Google Scholar]
- 73.Bravo R, Vicencio JM, Parra V, Troncoso R, Munoz JP, Bui M, et al. Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J Cell Sci. 2011;124:2511–2511. doi: 10.1242/jcs.080762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bravo-Sagua R, Rodriguez AE, Kuzmicic J, Gutierrez T, Lopez-Crisosto C, Quiroga C, et al. Cell death and survival through the endoplasmic reticulum-mitochondrial axis. Curr Mol Med. 2013;13:317–29. doi: 10.2174/156652413804810781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chiong M, Cartes-Saavedra B, Norambuena-Soto I, Mondaca-Ruff D, Morales PE, García-Miguel M, et al. Mitochondrial metabolism and the control of vascular smooth muscle cell proliferation. Front Cell Dev Biol. 2014;2:1–9. doi: 10.3389/fcell.2014.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Morales PE, Torres G, Sotomayor-flores C, Peña-Oyarzún D, Rivera-Mejías P, Paredes F, et al. GLP-1 promotes mitochondrial metabolism in vascular smooth muscle cells by enhancing endoplasmic reticulum—mitochondria coupling. Biochem Biophys Res Commun. 2014;446:410–6. doi: 10.1016/j.bbrc.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 77.Nisoli E, Falcone S, Tonello C, Cozzi V, Palomba L, Fiorani M, et al. Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proc Natl Acad Sci USA. 2004;101:16507–12. doi: 10.1073/pnas.0405432101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Suliman HB, Carraway MS, Tatro LG, Piantadosi CA. A new activating role for CO in cardiac mitochondrial biogenesis. J Cell Sci. 2007;120:299–308. doi: 10.1242/jcs.03318. [DOI] [PubMed] [Google Scholar]
- 79.Mo L, Wang Y, Geary L, Corey C, Alef MJ, Beer-Stolz D, et al. Nitrite activates AMP kinase to stimulate mitochondrial biogenesis independent of soluble guanylate cyclase. Free Radic Biol Med. 2012;53:1440–50. doi: 10.1016/j.freeradbiomed.2012.07.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ryan JJ, Marsboom G, Fang YH, Toth PT, Morrow E, Luo N, et al. PGC1α-mediated mitofusin-2 deficiency in female rats and humans with pulmonary arterial hypertension. Am J Respir Crit Care Med. 2013;187:865–78. doi: 10.1164/rccm.201209-1687OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lee YC, Martin E, Murad F. Human recombinant soluble guanylyl cyclase: expression, purification, and regulation. Proc Natl Acad Sci USA. 2000;97:10763–8. doi: 10.1073/pnas.190333697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhao Y, Marletta MA. Localization of the heme binding region in soluble guanylate cyclase. Biochemistry. 1997;36:15959–64. doi: 10.1021/bi971825x. [DOI] [PubMed] [Google Scholar]
- 83.Karow DS, Pan D, Davis JH, Behrends S, Mathies RA, Marletta MA. Characterization of FUNCTIONAL HEME DOMAINS FOR SOLUBLE GUANYLATE Cyclase. Biochemistry. 2005;44:16266–74. doi: 10.1021/bi051601b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Panza Julia A, Quyyumi Arshed A, Brush John E, Epstein SE. Abnormal endothelium-dependent vascular relaxation in patients with essential hypertension. N Engl J Med. 1994;327:1141–52. doi: 10.1056/NEJM199007053230105. [DOI] [PubMed] [Google Scholar]
- 85.Shesely EG, Maeda N, Kim HS, Desai KM, Krege JH, Laubach VE, et al. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc Natl Acad Sci USA. 1996;93:13176–81. doi: 10.1073/pnas.93.23.13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Van Vliet BN, Chafe LL, Montani J-P. Characteristics of 24 h telemetered blood pressure in eNOS-knockout and C57Bl/6J control mice. J Physiol. 2003;549:313–25. doi: 10.1113/jphysiol.2003.041897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ohashi Y, Kawashima S, Hirata KI, Yamashita T, Ishida T, Inoue N, et al. Hypotension and reduced nitric oxide-elicited vasorelaxation in transgenic mice overexpressing endothelial nitric oxide synthase. J Clin Invest. 1998;102:2061–71. doi: 10.1172/JCI4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stasch J-P, Pacher P, Evgenov OV. Soluble guanylate cyclase as an emerging therapeutic target in cardiopulmonary disease. Circulation. 2011;123:2263–73. doi: 10.1161/CIRCULATIONAHA.110.981738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kalk P, Godes M, Relle K, Rothkegel C, Hucke A, Stasch J-P, et al. NO-independent activation of soluble guanylate cyclase prevents disease progression in rats with 5/6 nephrectomy. Br J Pharmacol. 2006;148:853–9. doi: 10.1038/sj.bjp.0706792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dey NB, Foley KF, Lincoln TM, Dostmann WR. Inhibition of cGMP-dependent protein kinase reverses phenotypic modulation of vascular smooth muscle cells. J Cardiovasc Pharmacol. 2005;45:404–13. doi: 10.1097/01.fjc.0000157455.38068.12. [DOI] [PubMed] [Google Scholar]
- 91.Anderson PG, Boerth NJ, Liu M, McNamara DB, Cornwell TL, Lincoln TM. Cyclic GMP-dependent protein kinase expression in coronary arterial smooth muscle in response to balloon catheter injury. Arterioscler Thromb Vasc Biol. 2000;20:2192–7. doi: 10.1161/01.atv.20.10.2192. [DOI] [PubMed] [Google Scholar]
- 92.Boerth NJ, Dey NB, Cornwell TL, Lincoln TM. Cyclic GMP-dependent protein kinase regulates vascular smooth muscle cell phenotype. J Vasc Res. 1997;34:245–59. doi: 10.1159/000159231. [DOI] [PubMed] [Google Scholar]
- 93.Lincoln TM, Dey NB, Boerth NJ, Cornwell TL, Soff GA. Nitric oxide–cyclic GMP pathway regulates vascular smooth muscle cell phenotypic modulation: implications in vascular diseases. Acta Physiol Scand. 1998;164:507–15. doi: 10.1111/j.1365-201x.1998.tb10700.x. [DOI] [PubMed] [Google Scholar]
- 94.Foerster J, Harteneck C, Malkewitz J, Schultz G, Koesling D. A functional heme-binding site of soluble guanylyl cyclase and pI subunits requires intact N-termini of alpha (1) and beta (1) subunits. Eur J Biochem. 1996;240:380–6. doi: 10.1111/j.1432-1033.1996.0380h.x. [DOI] [PubMed] [Google Scholar]
- 95.Arnold WP, Mittal CK, Katsuki S, Murad F. Nitric oxide activates guanylate cyclase and increases guanosine 3′:5′-cyclic monophosphate levels in various tissue preparations. Proc Natl Acad Sci USA. 1977;74:3203–7. doi: 10.1073/pnas.74.8.3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ignarro LJ, Adam JB, Horwitzq PM, Wood KS. Activation of soluble guanylate cyclase by NO-hemoproteins involves NO-heme exchange. J Biol Chem. 1986;261:4997–5002. [PubMed] [Google Scholar]
- 97.Sinnaeve P, Chiche JD, Nong Z, Varenne O, Van Pelt N, Gillijns H, et al. Soluble guanylate cyclase alpha(1) and beta(1) gene transfer increases NO responsiveness and reduces neointima formation after balloon injury in rats via antiproliferative and antimigratory effects. Circ Res. 2001;88:103–9. doi: 10.1161/01.res.88.1.103. [DOI] [PubMed] [Google Scholar]
- 98.Groneberg D, Konig P, Wirth A, Offermanns S, Koesling D, Friebe A. Smooth muscle—specific deletion of nitric oxide—sensitive guanylyl cyclase is sufficient to induce hypertension in mice. Circulation. 2010;121:401–9. doi: 10.1161/CIRCULATIONAHA.109.890962. [DOI] [PubMed] [Google Scholar]
- 99.Rahaman MM, Nguyen AT, Miller MP, Hahn SA, Sparacino-Watkins C, Jobbagy S, et al. Cytochrome b5 reductase 3 modulates soluble guanylate cyclase redox state and cGMP signaling. Circ Res. 2017;121:137–48. doi: 10.1161/CIRCRESAHA.117.310705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hirschberg K, Tarcea V, Páli S, Barnucz E, Gwanmesia PN, Korkmaz S, et al. Cinaciguat prevents neointima formation after arterial injury by decreasing vascular smooth muscle cell migration and proliferation. Int J Cardiol. 2013;167:470–7. doi: 10.1016/j.ijcard.2012.01.032. [DOI] [PubMed] [Google Scholar]
- 101.Tsai EJ, Kass DA. Cyclic GMP signaling in cardiovascular pathophysiology and therapeutics. Pharmacol Ther. 2010;122:216–38. doi: 10.1016/j.pharmthera.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]