

Abstract
The androgen receptor (AR) is a ligand-activated nuclear receptor that plays a critical role in normal prostate physiology, as well as in the development and progression of prostate cancer. In addition to the classical paradigm in which AR exerts its biological effects in the nucleus by orchestrating the expression of the androgen-regulated transcriptome, there is considerable evidence supporting a rapid, nongenomic activity mediated by membrane-associated AR. Although the genomic action of AR has been studied in depth, the molecular events governing AR transport to the plasma membrane and the downstream AR signaling cascades remain poorly understood. In this study, we report that AR membrane transport is microtubule-dependent. Disruption of the function of kinesin 5B (KIF5B), but not of kinesin C3 (KIFC3), interfered with AR membrane association and signaling. Co-immunoprecipitation and pulldown assays revealed that AR physically interacts with KIF5B and that androgen enhances this interaction. Furthermore, we show that heat shock protein 27 (HSP27) is activated by membrane-associated AR and that HSP27 plays an important role in mediating AR-mediated membrane-to-nuclear signal transduction. Together, these results indicate that AR membrane translocation is mediated by the microtubule cytoskeleton and the motor protein KIF5B. By activating HSP27, membrane-associated AR potentiates the transcriptional activity of nuclear AR. We conclude that disruption of AR membrane translocation may represent a potential strategy for targeting AR signaling therapeutically in prostate cancer.
Keywords: androgen receptor, microtubule, plasma membrane, kinesin, heat shock protein (HSP), HSP27, KIF5B, prostate cancer, steroid hormone receptor, type I nuclear receptor
Introduction
The androgen receptor (AR),3 along with the estrogen receptor (ER), glucocorticoid receptor (GR), progesterone receptor (PR), and the mineralocorticoid receptor (MR), is a member of the Type I nuclear receptor subfamily (1, 2). AR plays an important role in the development and maintenance of the male sexual phenotype (3, 4). Similar to other steroid hormone receptors, AR is located in the cytoplasm in the absence of androgens, forming a complex with chaperones and co-chaperones such as heat shock proteins and immunophilins (5). Upon androgen binding, AR undergoes conformational changes and translocates to the nucleus, where it binds to the androgen response elements (AREs) as a homodimer; activates the expression of AR target genes; and induces cell proliferation, differentiation, and survival (6, 7). This mode of action is the classical AR signaling pathway, also known as the genomic action. In addition to the classical pathway, many observations suggest androgens, as well as some other steroid hormones, can affect cellular processes in a nongenomic fashion (8). Cinar et al. found that within minutes of androgen stimulation, AR is localized to the membrane lipid rafts microdomain, interacts with AKT, and activates AKT signaling (9). Pedram et al. showed palmitoylation of a conserved motif in the ligand-binding domain is critical for membrane localization of estrogen receptor, progesterone receptor, and AR (10). Additionally, studies have shown AR interacts with caveolin-1 (Cav-1), a major component of the caveolae membrane structure (11, 12). Down-regulation of Cav-1 decreases AR membrane localization (11, 12). Overall, studies on membrane-associated AR are limited and very little is understood regarding the mechanisms underlying AR translocation to the plasma membrane.
In this study, we investigated the mechanism involved in AR plasma membrane trafficking. We found that the AR plasma membrane translocation depends on the microtubules and the KIF5B motor protein. We also identified membrane-associated AR potentiates the transcriptional activities of AR by enhancing AR nuclear import.
Results
Androgen induces AR plasma membrane translocation
We first employed an immunofluorescence (IF) assay to visualize androgen-induced AR membrane localization in LNCaP cells. As shown in Fig. 1A, when the cells were cultured in an androgen-depleted condition, AR was primarily in the cytoplasm. Ten min after the addition of R1881, a synthetic androgen, a fraction of AR could be observed at the periphery of cells, overlapping with the staining for ganglioside GM-1, a membrane lipid raft marker (13). At 20 min, as the majority of cytoplasmic AR had translocated to the nucleus, the membrane pool became more distinguishable. This fraction of AR appeared to peak at around 20 min after androgen stimulation, as the intensity declined after longer incubations. Similar observations were made in COS-7 cells transfected with AR (Fig. S1), suggesting AR membrane translocation is not unique to LNCaP cells.
Figure 1.

Androgen induces AR membrane translocation. A, intracellular localization of AR by immunofluorescence. LNCaP cells were cultured on chamber slides in phenol-red free RPMI 1640 containing 5% charcoal-stripped FBS and treated with 1 nm R1881 for the indicated times. Following fixation, the slides were stained for AR by an anti-AR antibody (green), for membrane marker GM-1 by Cholera Toxin Subunit B (red), and for nuclei by DAPI (blue). Co-localization of AR and GM-1 was visible after R1881 treatment for 10 and 20 min. Scale bar is 20 μm. B, top, Western blot analysis of AR in Triton X-100 soluble (TS) and Triton X-100 insoluble/octylglucoside soluble (TI) fractions isolated from LNCaP cells. Giα3 was used as the membrane marker and GAPDH as the cytoplasm marker. Bottom, AR levels in TI fractions were calculated as the AR/Giα3 ratios and expressed relative to that of the leftmost group. C, Western blot analysis of AR in LNCaP lysates fractionated by the sucrose gradient ultracentrifugation. Giα 3 was used as the membrane marker. D, AR levels in Giα3-positive fractions were quantitated by densitometry and expressed as % of total AR. The mean ± S.D. from three experiments are plotted. *, p < 0.05.
To confirm the IF results, we performed two membrane fractionation assays based on the low-density and Triton X-100–insoluble properties of membrane domains enriched with cytoplasmically oriented signaling molecules (9). LNCaP cells were lysed 20 min after the addition of R1881 and separated into Triton X-100–soluble (TS) and Triton X-100–insoluble/octylglucoside-soluble (TI) fractions. We first confirmed that there was no contamination from cytoplasmic and nuclear proteins in the TI fraction, which contains the membrane fraction (Fig. S2). As shown in Fig. 1B, AR level was low in the TI fraction under the androgen-depleted condition, but it was markedly induced by the addition of R1881. The induction was blocked by enzalutamide, a potent antiandrogen. Consistent with a previous report showing palmitoylation is critical for AR membrane translocation (10), treatment with 2-bromopalmitate (2-BP), a palmitoylation inhibitor, blocked the androgen-induced increase of AR in the TI fraction (Fig. S3).
Next, LNCaP lysates were fractionated by the sucrose gradient ultracentrifugation method and analyzed by Western blotting. As shown in Fig. 1C, AR was present at a very low level in Giα3-positive fractions under the androgen-depleted condition, and treatment with androgen significantly increased AR distribution in these fractions (Fig. 1C). When compared against AR in all fractions, we estimated the pool of membrane-associated AR accounted for 8–10% of total AR at 20 min after androgen stimulation (Fig. 1D).
The above analyses show AR undergoes a transient, ligand-induced membrane translocation, as previously reported by others (9). To determine the physiological relevance of this phenomenon, we performed the TS/TI fractionation assays with LNCaP cells treated with different concentrations of androgens. As shown in Fig. S4, AR membrane localization could be induced by dihydrotestosterone (DHT) as low as 1 nm, and by R1881 as low as 100 pm, suggesting AR membrane translocation occurs at both physiological and castrate levels of androgen (14, 15).
Microtubule cytoskeleton is involved in AR membrane transport
Studies have established that the microtubules play a critical role in facilitating the nuclear import of steroid receptors such as AR (16–18). However, little is known about the mechanism of AR membrane translocation. To test whether microtubules are involved in this process, LNCaP cells were treated with microtubule inhibitors docetaxel (stabilizes microtubules) and nocodazole (depolymerizes microtubules). As shown in Fig. 1C, treatment with docetaxel blocked the androgen-induced AR distribution in the membrane fractions. Similarly, both docetaxel and nocodazole abolished androgen induction of AR in the TI fraction (Fig. 2, A and B), suggesting microtubule dynamics are critical for AR trafficking to the membrane. The involvement of the microtubule cytoskeleton seems to be unique, as inhibition of actin cytoskeleton, which has been shown to be involved in protein transport (19), did not interfere with AR membrane translocation (Fig. 2C and Fig. S5).
Figure 2.

AR membrane translocation is microtubule-dependent. A–C, the effects of docetaxel (DTX), nocodazole (NCZ), and cytochalasin D (cytoD) on AR membrane translocation. LNCaP cells were pretreated with 10 nm docetaxel, 50 ng/ml nocodazole, or 0.5 μg/ml cytochalasin D for 16 h, then stimulated with 1 nm R1881 for 20 min. TS/TI fractions were extracted and analyzed by Western blotting. Left, representative blots are shown. Right, AR levels in TI fractions were calculated as the AR/Giα3 ratios and expressed relative to that of the leftmost group. The mean ± S.D. from three experiments are plotted. *, p < 0.05.
AR membrane transport is mediated by kinesin 5B
Protein movement along the microtubules is mediated by direction-specific motor proteins, which largely fall into two families, dyneins and kinesins. Whereas dyneins mediate cargo transport to the minus end of the microtubules, kinesins direct movement toward the plus end (20, 21). Among kinesin family proteins, KIF5B and KIFC3 have been shown to transport protein cargos to the plasma membrane (22, 23). To identify the specific motor protein mediating the transport of AR to the plasma membrane, we took advantage of the tail-domain mutants of KIF5B and KIFC3 (KIF5B-tail and KIFC3-tail, respectively). Because of the truncation of the motor domain, these mutants are capable of cargo binding but not transporting, thus acting as dominant-negative mutants (24). When they were co-expressed with AR, the KIFC3-tail had no effect on androgen-induced AR membrane translocation (Fig. 3A and Fig. S6A). In contrast, AR membrane translocation was abolished in the presence of the KIF5B-tail (Fig. 3B and Fig. S6B). A previous report has shown membrane-associated AR mediates rapid AKT activation stimulated by androgen (9, 25). Consistent with this study, we found androgen-induced AKT phosphorylation at 30 min was blocked in the presence of the KIF5B-tail, but not the KIFC3-tail (Fig. 3C). Additionally, KIF5B knockdown by an siRNA led to a significant reduction of androgen-induced AR membrane localization in LNCaP cells (Fig. 3, D and E). Collectively, these results show that AR transport to the membrane is mediated by the KIF5B motor protein.
Figure 3.

KIF5B mediates AR membrane transport. A and B, the effects of KIFC3-tail and KIF5B-tail on AR membrane translocation. LNCaP cells were transiently transfected with KIFC3-tail or KIF5B-tail for 48 h, and treated with 1 nm R1881 for 20 min. TS/TI fractions were extracted and analyzed by Western blotting. Membrane AR levels were calculated as AR/Giα3 ratios and expressed relative to that of the leftmost group. C, KIF5B-tail, but not KIFC3-tail, interferes with rapid activation of AKT. LNCaP cells were transfected with KIF5B-tail or KIFC3-tail, and treated with 1 nm R1881 treatment for 30 min. Whole cell lysates were collected and probed with antibodies against p-AKT and AKT. D, KIF5B knockdown. After LNCaP cells were transfected with siRNAs for 48 h, lysates were analyzed by Western blotting to confirm the knockdown of KIF5B. E, KIF5B knockdown interrupts AR membrane translocation. LNCaP cells were transfected siRNAs for 48 h and treated with 1 nm R1881 for 20 min. TS/TI fractions were extracted and analyzed for AR distribution. Left, representative blots are shown. Right, AR levels in TI fractions were calculated as the AR/Giα3 ratios and expressed relative to that of the leftmost group. The mean ± S.D. from three experiments are plotted. *, p < 0.05.
AR interacts with KIF5B through the N-terminal domain
If KIF5B is the motor protein responsible for the anterograde transport of AR, as suggested by the previous section, an interaction would be expected between AR and the KIF5B-tail domain, which is responsible for cargo recognition and binding. This appeared to be the case. In COS-7 cells co-transfected with AR and the KIF5B-tail or the KIFC3-tail plasmids (both are Myc-tagged), AR was co-precipitated with the KIF5B-tail, but not with the KIFC3-tail, by an anti-Myc antibody (Fig. 4A), suggesting there is a specific interaction between KIF5B and AR. A reciprocal co-immunoprecipitation (co-IP) assay performed with an antibody against AR in LNCaP cells demonstrated an interaction of endogenous AR and KIF5B (Fig. 4B). Additionally, we performed an in vitro Myc-pulldown assay. When protein G magnetic beads coated with the KIF5B-tail or KIFC3-tail were incubated with AR-expressing COS-7 lysates, AR was pulled down by the KIF5B-tail, but not by the KIFC3-tail (Fig. 4C), further supporting an interaction between AR and KIF5B.
Figure 4.

AR interacts with KIF5B. A, co-immunoprecipitation (co-IP) of KIF5B-tail or KIFC3-tail with AR. COS-7 cells were co-transfected with Myc-tagged KIF5B-tail or KIFC3-tail with AR. Co-IP assays were performed with an antibody against Myc-tag, followed by immunoblot with antibodies against AR and Myc-tag. Input, cell lysates were analyzed for the expression of AR and KIF5B-tail or KIFC3-tail. A control co-IP experiment was performed with a mouse IgG. B, the endogenous AR-KIF5B interaction. LNCaP cells were treated with EtOH or R1881 20 min, and a reciprocal co-IP assay was performed with an antibody against AR, followed by Western blot analyses for AR and KIF5B. C, In vitro pulldown assay for AR. Myc-tagged KIF5B-tail or KIFC3-tail was expressed in COS-7 cells, and enriched by protein G magnetic beads coated with an anti-Myc-tag antibody. The magnetic beads were subsequently incubated with lysates from COS-7 cells transfected with AR, and the eluted proteins were analyzed by immunoblotting. D, the KIF5B-tail–AR interaction is enhanced by androgen. COS-7 cells co-transfected with KIF5B-tail and AR were cultured in RPMI 1640 supplemented with 10% cs-FBS, and treated with EtOH or R1881 for 20 min. The lysates were analyzed by the co-IP assay.
We next investigated the effect of androgen on the AR–KIF5B association. As shown in Fig. 4D, a weak AR band co-precipitated with the KIF5B-tail under the androgen-deprived condition; however, a discernible stronger interaction was observed after the addition of androgen, suggesting that the AR–KIF5B interaction is ligand-dependent. A similar observation was made with the endogenous proteins (Fig. 4B).
To identify the domain(s) of AR mediating the interaction with KIF5B, a series of AR deletion constructs were generated (Fig. 5A) and used in the pulldown assay. As shown in Fig. 5, B and C, the full-length AR, as well as constructs containing the N-terminal domain (NTD) and the NTD plus the DNA-binding domain (DBD), were pulled down by the KIF5B-tail. In contrast, none of the constructs lacking the NTD were pulled down by the KIF5B-tail. These data suggest AR interacts with KIF5B through the NTD. Interestingly, the AR-NDH (NTD plus DBD and Hinge domain) construct was not pulled down by the KIF5B-tail, suggesting an inhibitory role of the hinge domain in the AR-KIF5B interaction.
Figure 5.

AR binds to KIF5B via the N-terminal domain. A, a series of AR deletion constructs were generated by deleting progressively from the C and N termini. B and C, these constructs were expressed in COS-7 cells and analyzed by the in vitro pulldown assay with KIF5B-tail–coated magnet beads. Western blots were probed with the N-20 (B) and C-19 (C) antibodies, which recognized the N and C termini of AR, respectively. Control pulldowns were performed with a mouse IgG. H, Hinge domain; LBD, ligand-binding domain; N, NTD only; ND, NTD and DBD; NDH, NTD plus DBD and H; DHL, DBD plus H and LBD; HL, H and LBD.
Membrane-associated AR cross-talks with nuclear-destined AR
Pedram et al. have previously identified a conserved motif in the ligand-binding domain of steroid hormone receptors that is critical for ligand-induced translocation to the plasma membrane (10). For AR, substituting the cysteine residue at position 807 with an alanine abolished AR palmitoylation, leading to reduced membrane localization (10). We believe this membrane-deficient mutant is suitable for understanding the functional relevance of membrane-associated AR. In our lab, we first confirmed that AR–C807A was defective in androgen-induced membrane localization by IF and TS/TI fractionation assays (Fig. S7, A and B). Subsequently, we performed the ligand-binding assay with AR–C807A, because the mutation is in the ligand-binding domain. Analysis of the data revealed similar binding curves and disassociation constants (Kd) for AR–C807A and AR–WT (Fig. S7, C and D), thus excluding the possibility that the observed deficiency in membrane localization of AR–C807A was because of decreased ligand-binding capacity. Furthermore, the cycloheximide chase assay showed that the protein stability was similar between AR–WT and AR–C807A (Fig. S7E).
When the transcriptional activity of AR–C807A was analyzed in COS-7 cells by the luciferase reporter assay, we found that although their activities were similar in the absence of androgen, the R1881-induced activity of AR–C807A was significantly less than that of the WT counterpart (Fig. 6A). This was confirmed by the expression of TMPRSS2, an AR-regulated gene, in DU145 cells ectopically expressing AR–WT or AR–C807A (Fig. 7C). Consistent with these results, treatment of LNCaP cells with the palmitoylation inhibitor 2-BP, which was shown to inhibit AR membrane localization, inhibited androgen-induced prostate-specific antigen (PSA) mRNA expression (Fig. 7D). These results suggest that abolishing AR membrane localization may ultimately impair the activity of AR as a transcriptional activator, supporting a cross-talk between membrane-associated and nuclear-destined AR.
Figure 6.

Loss of AR membrane association impairs its nuclear activity. A, luciferase reporter assay. COS-7 cells were co-transfected with the ARR3 luciferase reporter, pRL–TK, along with WT AR (AR–WT) or the AR–C807A mutant. Dual-Luciferase assays were performed 24 h after the cells were treated with the solvent (EtOH) or 1 nm R1881. The normalized luciferase activities were expressed relative to the leftmost group. B, IF analyses of COS-7 cells transfected with AR–WT or AR–C807A. Transfected cells were cultured on chamber slides for 48 h, and treated with 10 nm DHT for the indicated times. Following fixation, the slides were stained by an anti-AR, by CTB (for defining cell boundary), and by DAPI (for nuclei). The images were analyzed by the Image J software to quantitate the nuclear and cytoplasmic AR signals. At least 20 cells per group were analyzed. C, subcellular fractionation assay of COS-7 cells transfected with AR–WT or AR–C807A. Cells were stimulated by 1 nm R1881 for the indicated times and levels of AR were determined by Western blotting. The nuclear/cytoplasmic AR ratios were calculated relative to the 0 min time point. The mean ± S.D. from three experiments are plotted. *, p < 0.05.
Figure 7.

HSP27 mediates cross-talk between membrane-associated and nuclear-destined AR. A, KIF5B-tail blocks androgen-induced phosphorylation of HSP27. LNCaP cells transfected with KIF5B-tail or KIFC3-tail were treated with 1 nm R1881 for 15 and 25 min, and analyzed by Western blotting for total and phosphorylated HSP27. B, AR–WT, but not AR–C807A, induces HSP27 phosphorylation. COS-7 cells transfected with AR–WT or AR–C807A were treated with 1 nm R1881 for 0, 10, 20 min, and analyzed by Western blotting for total and phosphorylated HSP27. C, rescue of TMPRSS2 expression by HSP27D. DU145 cells were transfected with AR–WT/AR–C807A with vector/HSP27A/HSP27D, and treated with EtOH or 1 nm R1881 for 24 h. Following qRT-PCR, normalized TMPRSS2 mRNA levels were expressed relative to the leftmost group. D, inhibition of androgen-induced PSA expression by 2-BP is rescued by constitutively active HSP27. LNCaP cells were transfected with HSP27A/HSP27D or empty vector, treated with 10 μm 2-BP or DMSO for 16 h, followed by 1 nm R1881 or EtOH treatment for an additional 24 h. Following qRT-PCR, normalized PSA mRNA levels (by RPL30 housekeeping gene) were expressed relative to the leftmost group. E, HSP27D rescued cell growth. DU145 cells were transfected with AR/AR–C807A with vector/HSP27A/HSP27D, cultured in RPMI supplemented with 10% cs-FBS and treated with 1 nm R1881 for 1 to 3 days, and the SRB assay was performed. The data were expressed as -folds of the day 0 readings. The mean ± S.D. from at least three experiments are plotted. *, p < 0.05.
To understand how the nuclear function of AR is weakened by the lack of AR membrane localization, we looked into nuclear translocation by using an IF assay. In the absence of androgen, AR–WT and AR–C807A were both predominantly localized to the cytoplasm (Fig. S8A). Following androgen stimulation, AR–WT underwent a robust nuclear translocation, and became predominantly nuclear localized after 1 h. In comparison, the translocation of AR–C807A to the nucleus was much slower (Fig. S8A and Fig. 6B). Similar observations were made in COS-7 cells by using the subcellular fractionation assay (Fig. 6C and Fig. S8, B and C). These results suggest membrane-associated AR plays a critical role in potentiating the nuclear import of AR.
HSP27 mediates membrane-to-nuclear AR cross-talk
It has been previously reported that androgen induces phosphorylation of heat shock protein 27 (HSP27), which in turn facilitates AR nuclear translocation (26). However, it remains unclear whether HSP27 phosphorylation is mediated by membrane-associated AR. To address this question, we blocked AR membrane transport with the dominant-negative KIF5B-tail and examined HSP27 phosphorylation. As shown in Fig. 7A, androgen induced a rapid and robust induction of HSP27 phosphorylation in cells expressing the KIF3C-tail, which did not affect AR membrane localization (Fig. 3A). In contrast, androgen treatment failed to induce HSP27 phosphorylation in cells expressing the KIF5B-tail (Fig. 7A). Similarly, COS-7 cells expressing AR-C807A showed subdued HSP27 phosphorylation following androgen stimulation as opposed to those expressing AR-WT (Fig. 7B). These results suggest androgen-stimulated HSP27 phosphorylation is mediated by membrane-associated AR.
If HSP27 functions as a key mediator of AR signaling from the membrane to nucleus, it would be expected that expression of constitutive active HSP27 will at least in part rescue the defects of membrane-deficient AR. To this end, we performed rescue assays with HSP27-D (phosphorylation mimicking) and HSP27-A (phosphorylation deficient) mutants (27). As mentioned previously, androgen-induced TMRPSS2 expression was muted in AR–C807A–expressing DU145 cells. The co-transfection of HSP27-D, but not HSP27-A, rescued the expression of TMRPSS2 (Fig. 7C). Similarly, in LNCaP cells treated with 2-BP, the expression of PSA mRNA was rescued by HSP27-D, but not HSP27-A (Fig. 7D). Furthermore, although they grew similarly in an androgen-depleted condition (Fig. S9), AR–WT–expressing DU145 cells proliferated at a higher rate than those expressing AR–C807A in the presence of androgen (Fig. 7E), suggesting membrane localization is critical for the proliferative effects of AR. Co-expression of HSP27-D partially restored cell proliferation, whereas the co-transfection of HSP27-A failed to do so (Fig. 7E). Collectively, these results provide evidence supporting HSP27 as a key mediator of the AR membrane-to-nuclear signaling.
Discussion
As a member of the nuclear receptor superfamily, AR is best known for its activity in the nucleus as a ligand-dependent transcription factor, orchestrating the expression of androgen-regulated transcriptome which is critical for prostate development, homeostasis, and carcinogenesis (1, 15, 28). In recent years, AR's actions outside the nucleus, also known as the nongenomic function, have been increasingly recognized. A previous study has shown that upon androgen stimulation, AR appears in the membrane fraction within minutes, forms a complex with AKT, and induces the phosphorylation and activation of AKT (9, 25). However, the mechanism of AR transport to the plasma membrane and the downstream events regulated by membrane-associated AR are largely unknown. In this study, we are the first to demonstrate that AR membrane transport is microtubule-dependent, a similarity shared with its nuclear import. However, this anterograde transport of AR is mediated by the motor protein KIF5B, as opposed to dynein, which mediates the retrograde transport (17).
Previously, Lu et al. showed that caveolin-1 (Cav-1) knockdown by shRNAs decreased the amount of AR localized to the membrane, suggesting Cav-1 is involved in AR membrane trafficking (11). In our study, AR membrane translocation was observed in both Cav-1–positive COS-7 cells and Cav-1–negative LNCaP cells, indicating Cav-1 is not necessary for this process. Cav-1 may stabilize AR in the plasma membrane in Cav-1–expressing cells.
Our study also presents the first evidence that membrane-associated AR potentiates the classical AR actions by enhancing the nuclear import and activity of AR through the activation of HSP27. HSP27 is an ATP-independent molecular chaperone involved in cytoprotection in stress conditions (29). The activity of HSP27 is regulated by phosphorylation and phospho-HSP27 has been shown to suppress apoptosis, enhance invasion and survival of cancer cells (30). HSP27 is believed to drive prostate cancer cell metastasis through its regulation of MMP-2 (31). Particularly, HSP27 has been shown to be activated by androgen stimulation, and to enhance AR nuclear import and transcriptional activity through its interaction with AR (26). In our study, we found the phosphorylation of HSP27 is mediated by membrane-associated AR. However, the exact kinase(s) responsible for HSP27 phosphorylation remains to be elucidated. A possible candidate is ERK, which has been shown to phosphorylate HSP27 (32). Deng et al. showed testosterone induces rapid AR membrane association and phosphorylation of AKT and ERK (25).
As shown in Fig. 1A, androgen-induced AR membrane localization is transient. However, by using a novel androgen dendrimer conjugate (ADC) that selectively binds AR in the cytoplasm but prevents AR from being imported to the nucleus (33, 34), we found AR remained in the membrane fractions for longer periods (data not shown). This result suggests a feedback signal transmitted from nuclear AR to inhibit AR membrane association.
In addition to the canonical full-length AR, at least two AR isoforms have been found on the plasma membrane. AR8, a novel AR splice variant that contains the NTD and a unique 33 amino acid C terminus, is expressed in both clinical samples and prostate cancer cell lines (35). Localized primarily to the plasma membrane, AR8 promotes the association of Src and AR with EGFR in response to EGF stimulation, and is required for the optimal AR transcriptional activity induced by EGF or DHT (35). This is similar to our finding that membrane-associated AR potentiates the transcriptional activity of nuclear-destined AR. Another AR splice variant found on the plasma membrane is AR45, which lacks the NTD (36). A recent study identified AR45 in the plasma membrane lipid rafts of neuronal cells (37). Functionally, AR45 is postulated to play a role in the regulation of intracellular calcium signaling through its interactions with the membrane-associated Gαo and Gαq proteins (37). The molecular mechanisms of membrane transport of AR8 and AR45 have not been studied. However, based on our data that KIF5B interacts with the AR NTD, it is likely AR8 is transported by KIF5B.
Based on the current study and previous work from our lab and others, we propose a working model for role of the microtubule cytoskeleton in the regulation of AR intracellular localization and activity (Fig. 8). In the absence of androgen, AR is associated with the microtubules (16), which retains AR in the cytoplasm. Following androgen stimulation, AR undergoes conformational changes, which reduce AR binding to the microtubules but allow it to interact with KIF5B via the NTD. KIF5B then transports AR to the plasma membrane, where AR initiates a signaling cascade, possibly through interaction with AKT, leading to the phosphorylation and activation of ERK and HSP27, the latter of which further facilitates AR nuclear transport.
Figure 8.
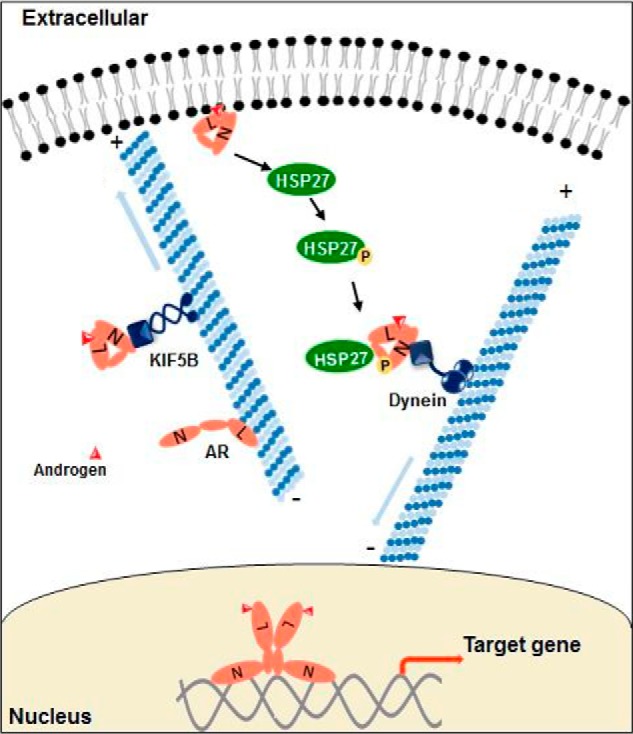
Schematic illustration of the anterograde AR transport and the cross-talk between membrane-associated and nuclear-destined AR.
It is now widely accepted that the AR signaling pathway remains active in castration-resistant prostate cancer (CRPC). Our study demonstrated that AR membrane transport can be induced by DHT concentrations found in CRPC tissues (38) (Fig. S4). Based on the existing evidence, we propose that membrane-associated AR plays a critical role in sustaining AR signaling in CRPC through at least two mechanisms. First, membrane AR potentiates the AR transcriptional activity in the presence of low levels of androgen by enhancing the nuclear translocation. Second, AR activates AKT, ERK, and Src pathways at the membrane, thereby promoting cell proliferation, invasion, and survival through these pathways (39). Therefore, targeting AR membrane transport represents a novel strategy to fully suppress AR signaling in CRPC.
Experimental procedures
Cell lines and reagents
LNCaP, DU-145, and COS-7 cells were obtained from American Type Culture Collection. The suppliers for the antibodies used in this study include Santa Cruz Technology (for AR N-20 and GAPDH antibodies), EMD Millipore (AR PG-21, Giα3, and Myc-tag), Cell Signaling Technology (AKT, phospho-AKT (Ser-473), HSP27, and phospho-HSP27 (Ser-82)), GeneTex (anti-LSD1), and Abcam (anti-KIF5B). Docetaxel was obtained from Selleck Chemicals (Houston, TX); nocodazole, protease inhibitor cocktails, and KIF5B siRNA were from Sigma Aldrich; and Alexa Fluor® 594 conjugated Cholera Toxin Subunit B (CTB) was from Thermo Fisher Scientific. For plasmids, pRK–Myc–KIFC3-tail was kindly provided by Dr. Kristen Verhey (University of Michigan), Myc–KIF5B-tail by Dr. Geri E. Kreitzer (Weill Cornell Medical College, Cornell University), and AR–C807A was a gift from Dr. Ellis Levin (University of California at Irvine). The pFLAG-CMV2-Hsp27-S15D/S78D/S82D and pFLAG-CMV2-Hsp27-S15A/S78A/S82A plasmids were obtained from Addgene (85188, 84997, supplied by Ugo Moens's lab).
Immunofluorescence
Cells were cultured on chamber slides at 37 °C and stained with CTB for 30 min at 4 °C. Following CTB staining, cells were washed with phosphate-buffered saline (PBS) once, fixed with 4% paraformaldehyde for 10 min at room temperature, and permeabilized with 0.2% Triton X-100 for 10 min at room temperature. Cells were then blocked with 5% BSA in PBST buffer (PBS with Tween 20) for 1 h, and incubated with anti-AR overnight at 4 °C, followed by staining with the Alexa Fluor® 488–labeled secondary antibody for 1 h at room temperature. The fluorescence signal was detected by using a Nikon A1 confocal microscope.
Membrane fractionation by serial detergent extraction
The membrane fractions were extracted by using the successive detergent method as previously described (40). Briefly, cells were washed with PBS and suspended in buffer A (25 mm morpholinoethanesulfonic acid, 150 mm NaCl, pH 6.5, 1% Triton X-100, 1 mm Na3VO4, 1 mm phenylmethylsulfonyl fluoride, and protease inhibitor cocktails) and incubated on ice for 30 min. The Triton-insoluble fractions were collected by centrifugation at 14,000 × g for 20 min at 4 °C. The supernatant (Triton soluble) was transferred to a new tube and the pellet (Triton insoluble) was resuspended in buffer B (10 mm Tris, pH 7.6, 1% Triton X-100, 500 mm NaCl, 1 mm Na3VO4, 60 mm n-octyl-β-d-glucoside, 1 mm phenylmethylsulfonyl fluoride, and protease inhibitor mixture) and incubated on ice for 30 min. The debris (octylglucoside insoluble) was cleared by centrifugation at 14,000 × g for 20 min at 4 °C.
Sucrose gradient ultracentrifugation
Low-density membrane fractions enriched with lipid rafts were isolated by using the sucrose gradient ultracentrifuge method as described previously (41). Briefly, cells were washed once with PBS and suspended in lysis buffer (50 mm HEPES, 300 mm KCl, 0.1 mm EDTA, 1 mm PMSF, 0.1% Nonidet P-40, and protease inhibitor cocktails). Lysates were scraped from the dishes and cleared by centrifugation at 3000 × g at 4 °C for 10 min. The supernatant was mixed with an equal volume of 80% sucrose (in HEPES buffer), placed at the bottom of an ultracentrifuge tube, and layered with 4 ml each of 35 and 5% sucrose. Samples were centrifuged at 120,000 × g at 4 °C for 18 h in a swing bucket rotor (Beckman SW41 Ti). After ultracentrifugation, the layers were carefully collected and analyzed by Western blotting.
Western blotting
Cells were washed with ice-cold PBS and lysed with 1× Cell Lysis Buffer (Cell Signaling Technology) containing a phosphatase inhibitor and the protease inhibitor mixture (Sigma). After incubating the cells on ice for 30 min, lysates were collected by centrifugation at 12,000 × g for 10 min. Protein concentrations were determined by the BCA Protein Assay kit (Pierce). The samples were separated on 10% SDS-polyacrylamide gels and transferred onto polyvinylidene fluoride (PVDF) membranes. After blocking in TBST buffer (150 mm NaCl, 10 mm Tris, pH 7.4, 0.1% Tween 20) containing 5% nonfat milk, the blots were incubated with a primary antibody overnight at 4 °C and a fluorescent-labeled secondary antibody for 1 h at room temperature. The fluorescent signals were obtained by the Odyssey IR Imaging System (LI-COR Biosciences).
Cell culture, transient transfection, and luciferase reporter assay
LNCaP and DU-145 cells were cultured in RPMI 1640 and COS-7 cells in Dulbecco's modified Eagle's medium (high glucose), supplemented with antibiotics (penicillin and streptomycin) and 10% fetal bovine serum. Transient transfection was performed by using TurboFect (Thermo Scientific) or Lipofectamine 3000 (Invitrogen) for plasmids, and X-tremeGENE (Roche) for siRNAs, following the manufacturers' instructions.
For reporter gene assay, cells were co-transfected with the ARR3-luc luciferase reporter construct and pRL-TK, along with a plasmid encoding for AR–WT or AR–C807A. After incubating with the transfection mixture for 4 h, cells were replated in culture medium containing 10% charcoal-stripped fetal bovine serum (cs-FBS) and treated with 0 or 1 nm R1881. Dual-Luciferase assay was performed at 24 h post treatment using the Dual-Luciferase Reporter Assay System (Promega). The Firefly luciferase activity was normalized by the Renilla luciferase.
Co-immunoprecipitation
Cells were prepared in lysis buffer (50 mm Tris-HCl, pH 8.0, 120 mm NaCl, 0.5% Nonidet P-40, and protease inhibitor cocktails), precleared with anti-mouse IgG antibody for 1 h at 4 °C, and immunoprecipitated with an anti–Myc-tag antibody at 4 °C overnight. The antibody and associated complex were collected by protein G beads (Thermo Fisher), washed three times with the lysis buffer, and analyzed by Western blotting.
In vitro pulldown assay
Myc-tagged KIF5B-tail was expressed in COS-7 cells, and enriched by protein G magnetic beads coated with an anti-Myc-tag antibody. The enriched kinesin was further incubated with lysates from COS-7 cells transfected with AR or AR deletion constructs, and the proteins pulled down by kinesin were analyzed by Western blotting.
Ligand-binding assay
COS-7 cells transfected with AR–WT and AR–C807A were cultured in a 6-well plate for 48 h. Cells were washed twice in serum-free medium and incubated with 0.25, 0.5, 1, 2.5, 5, and 7.5 nm [3H]R1881 (methyltrienolone, Perkin-Elmer) in the presence or absence of 200-fold cold R1881 at 37 °C in a CO2 incubator for 1 h. At the end of incubation, cells were washed three times with ice-cold PBS and the wash solution was completely removed after each wash. Seven hundred μl absolute ethanol was added to each well and the cells were incubated at room temperature with gentle rocking for 30 min. The ethanoic extracts were transferred to scintillation vials with 4 ml scintillation fluid for radioactivity counting.
Nuclear and cytoplasmic fractionation
Nuclear and cytoplasmic factions were extracted by NE-PERTM Nuclear and Cytoplasmic Extraction kit (Thermo Fischer Scientific) according to the manufacturer's instructions. GAPDH was used as the cytoplasmic marker, and LSD1 as the nuclear marker.
Statistical analysis
Statistical analysis was performed using GraphPad Prism and Microsoft Excel. The Student's two-tailed t test was used to determine the difference in means between two groups. p < 0.05 is considered significant. Data are presented as mean ± S.D. from at least three independent experiments.
Author contributions
Jianzhuo Li, X. F., H.-W. P., F. M.-J., and H. Z. conceptualization; Jianzhuo Li and S. C. data curation; Jianzhuo Li, Jing Li, S. X., D. L., and Y. D. formal analysis; Jianzhuo Li validation; Jianzhuo Li, S. C., and Y. D. methodology; Jianzhuo Li writing-original draft; Jianzhuo Li, D. L. C., and H. Z. writing-review and editing; H. Z. supervision; H. Z. funding acquisition.
Supplementary Material
Acknowledgments
We are indebted to Dr. Kristen Verhey for providing the pRK–Myc–KIFC3-tail plasmid, to Dr. Dr. Geri E. Kreitzer for the Myc–KIF5B-tail plasmid, and to Dr. Ellis Levin for the AR–C807A expression vectors. We thank Sudurika Mukhopadhyay for help with manuscript preparation.
This work was supported by American Cancer Society Grants RSG-14-016-01-CCE, DOD W81XWH-12-1-0275, and W81XWH-14-1-0480; Louisiana Cancer Research Consortium Fund; and the Tulane University Carol Lavin Bernick Faculty Grants (to H. Z.). This work was also supported by National Institutes of Health Grants R01 DK074970 and DK107444 (to F. M.-J.) and Department of Veterans Affairs Merit Review Award BX003725. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S9.
- AR
- androgen receptor
- IF
- immunofluorescence
- TS
- Triton X-100–soluble
- TI
- Triton X-100–insoluble/octylglucoside-soluble
- 2-BP
- 2-bromopalmitate
- co-IP
- co-immunoprecipitation
- NTD
- N-terminal domain
- CRPC
- castration-resistant prostate cancer
- CTB
- Cholera Toxin Subunit B
- cs-FBS
- charcoal-stripped fetal bovine serum
- DHT
- dihydrotestosterone
- PSA
- prostate-specific antigen.
References
- 1. Tan M. H. E., Li J., Xu H. E., Melcher K., and Yong E.-L. (2015) Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 36, 3–23 10.1038/aps.2014.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mangelsdorf D. J., Thummel C., Beato M., Herrlich P., Schütz G., Umesono K., Blumberg B., Kastner P., Mark M., Chambon P., and Evans R. M. (1995) The nuclear receptor superfamily: The second decade. Cell 83, 835–839 10.1016/0092-8674(95)90199-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yeh S., Tsai M.-Y., Xu Q., Mu X.-M., Lardy H., Huang K.-E., Lin H., Yeh S.-D., Altuwaijri S., Zhou X., Xing L., Boyce B. F., Hung M.-C., Zhang S., Gan L., and Chang C. (2002) Generation and characterization of androgen receptor knockout (ARKO) mice: An in vivo model for the study of androgen functions in selective tissues. Proc. Natl. Acad. Sci. U.S.A. 99, 13498–13503 10.1073/pnas.212474399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Notini A. J., Davey R. A., McManus J. F., Bate K. L., and Zajac J. D. (2005) Genomic actions of the androgen receptor are required for normal male sexual differentiation in a mouse model. J. Mol. Endocrinol. 35, 547–555 10.1677/jme.1.01884 [DOI] [PubMed] [Google Scholar]
- 5. Pratt W. B., and Toft D. O. (1997) Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr. Rev. 18, 306–360 10.1210/edrv.18.3.0303 [DOI] [PubMed] [Google Scholar]
- 6. Beato M. (1989) Gene regulation by steroid hormones. Cell 56, 335–344 10.1016/0092-8674(89)90237-7 [DOI] [PubMed] [Google Scholar]
- 7. Foradori C. D., Weiser M. J., and Handa R. J. (2008) Non-genomic actions of androgens. Front. Neuroendocrinol. 29, 169–181 10.1016/j.yfrne.2007.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Simoncini T., and Genazzani A. R. (2003) Non-genomic actions of sex steroid hormones. Eur. J. Endocrinol. 148, 281–292 10.1530/eje.0.1480281 [DOI] [PubMed] [Google Scholar]
- 9. Cinar B., Mukhopadhyay N. K., Meng G., and Freeman M. R. (2007) Phosphoinositide 3-kinase-independent non-genomic signals transit from the androgen receptor to Akt1 in membrane raft microdomains. J. Biol. Chem. 282, 29584–29593 10.1074/jbc.M703310200 [DOI] [PubMed] [Google Scholar]
- 10. Pedram A., Razandi M., Sainson R. C. A., Kim J. K., Hughes C. C., and Levin E. R. (2007) A conserved mechanism for steroid receptor translocation to the plasma membrane. J. Biol. Chem. 282, 22278–22288 10.1074/jbc.M611877200 [DOI] [PubMed] [Google Scholar]
- 11. Lu M. L., Schneider M. C., Zheng Y., Zhang X., and Richie J. P. (2001) Caveolin-1 interacts with androgen receptor. A positive modulator of androgen receptor mediated transactivation. J. Biol. Chem. 276, 13442–13451 10.1074/jbc.M006598200 [DOI] [PubMed] [Google Scholar]
- 12. Deng Q., Wu Y., Zhang Z., Wang Y., Li M., Liang H., and Gui Y. (2017) Androgen receptor localizes to plasma membrane by binding to caveolin-1 in mouse Sertoli cells. Int. J. Endocrinol. 2017, 3985916 10.1155/2017/3985916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moreno-Altamirano M. M. B., Aguilar-Carmona I., and Sánchez-García F. J. (2007) Expression of GM1, a marker of lipid rafts, defines two subsets of human monocytes with differential endocytic capacity and lipopolysaccharide responsiveness. Immunology 120, 536–543 10.1111/j.1365-2567.2006.02531.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nishiyama T. (2014) Serum testosterone levels after medical or surgical androgen deprivation: A comprehensive review of the literature. Urol. Oncol. 32, 38.e17–38.e28 10.1016/j.urolonc.2013.03.007 [DOI] [PubMed] [Google Scholar]
- 15. Heinlein C. A., and Chang C. (2004) Androgen receptor in prostate cancer. Endocr. Rev. 25, 276–308 10.1210/er.2002-0032 [DOI] [PubMed] [Google Scholar]
- 16. Zhang G., Liu X., Li J., Ledet E., Alvarez X., Qi Y., Fu X., Sartor O., Dong Y., and Zhang H. (2015) Androgen receptor splice variants circumvent AR blockade by microtubule-targeting agents. Oncotarget 6, 23358–23371 10.18632/oncotarget.4396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Thadani-Mulero M., Nanus D. M., and Giannakakou P. (2012) Androgen receptor on the move: Boarding the microtubule expressway to the nucleus. Cancer Res. 72, 4611–4615 10.1158/0008-5472.CAN-12-0783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Harrell J. M., Murphy P. J. M., Morishima Y., Chen H., Mansfield J. F., Galigniana M. D., and Pratt W. B. (2004) Evidence for glucocorticoid receptor transport on microtubules by dynein. J. Biol. Chem. 279, 54647–54654 10.1074/jbc.M406863200 [DOI] [PubMed] [Google Scholar]
- 19. Pollard T. D., and Cooper J. A. (2009) Actin, a central player in cell shape and movement. Science 326, 1208–1212 10.1126/science.1175862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gennerich A., and Vale R. D. (2009) Walking the walk: How kinesin and dynein coordinate their steps. Curr. Opin. Cell Biol. 21, 59–67 10.1016/j.ceb.2008.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Soldati T., and Schliwa M. (2006) Powering membrane traffic in endocytosis and recycling. Nat. Rev. Mol. Cell Biol. 7, 897–908 10.1038/nrm2060 [DOI] [PubMed] [Google Scholar]
- 22. Noda Y., Okada Y., Saito N., Setou M., Xu Y., Zhang Z., and Hirokawa N. (2001) KIFC3, a microtubule minus end–directed motor for the apical transport of annexin XIIIb–associated Triton-insoluble membranes. J. Cell Biol. 155, 77–88 10.1083/jcb.200108042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schmidt M. R., Maritzen T., Kukhtina V., Higman V. A., Doglio L., Barak N. N., Strauss H., Oschkinat H., Dotti C. G., and Haucke V. (2009) Regulation of endosomal membrane traffic by a Gadkin/AP-1/kinesin KIF5 complex. Proc. Natl. Acad. Sci. 106, 15344–15349 10.1073/pnas.0904268106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gelfand V. I., Le Bot N., Tuma M. C., and Vernos I. (2001) A dominant negative approach for functional studies of the kinesin II complex. in Kinesin Protocols (Vernos I., ed) pp. 191–204, part of the Methods in Molecular Biology series, Humana Press, New York: 10.1385/1592590691 [DOI] [PubMed] [Google Scholar]
- 25. Deng Q., Zhang Z., Wu Y., Yu W.-Y., Zhang J., Jiang Z.-M., Zhang Y., Liang H., and Gui Y.-T. (2017) Non-genomic action of androgens is mediated by rapid phosphorylation and regulation of androgen receptor trafficking. Cell. Physiol. Biochem. 43, 223–236 10.1159/000480343 [DOI] [PubMed] [Google Scholar]
- 26. Zoubeidi A., Zardan A., Beraldi E., Fazli L., Sowery R., Rennie P., Nelson C., and Gleave M. (2007) Cooperative interactions between androgen receptor (AR) and heat-shock protein 27 facilitate AR transcriptional activity. Cancer Res. 67, 10455–10465 10.1158/0008-5472.CAN-07-2057 [DOI] [PubMed] [Google Scholar]
- 27. Jovcevski B., Kelly M. A., Rote A. P., Berg T., Gastall H. Y., Benesch J. L. P., Aquilina J. A., and Ecroyd H. (2015) Phosphomimics destabilize Hsp27 oligomeric assemblies and enhance chaperone activity. Chem. Biol. 22, 186–195 10.1016/j.chembiol.2015.01.001 [DOI] [PubMed] [Google Scholar]
- 28. Pomerantz M. M., Li F., Takeda D. Y., Lenci R., Chonkar A., Chabot M., Cejas P., Vazquez F., Cook J., Shivdasani R. A., Bowden M., Lis R., Hahn W. C., Kantoff P. W., Brown M., Loda M., Long H. W., and Freedman M. L. (2015) The androgen receptor cistrome is extensively reprogrammed in human prostate tumorigenesis. Nat. Genet. 47, 1346–1351 10.1038/ng.3419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Konda J. D., Olivero M., Musiani D., Lamba S., and Di Renzo M. F. (2017) Heat-shock protein 27 (HSP27, HSPB1) is synthetic lethal to cells with oncogenic activation of MET, EGFR and BRAF. Mol. Oncol. 11, 599–611 10.1002/1878-0261.12042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Katsogiannou M., Andrieu C., and Rocchi P. (2014) Heat shock protein 27 phosphorylation state is associated with cancer progression. Front. Genet. 5, 346 10.3389/fgene.2014.00346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Voll E. A., Ogden I. M., Pavese J. M., Huang X., Xu L., Jovanovic B. D., and Bergan R. C. (2014) Heat shock protein 27 regulates human prostate cancer cell motility and metastatic progression. Oncotarget 5, 2648–2663 10.18632/oncotarget.1917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Robitaille H., Simard-Bisson C., Larouche D., Tanguay R. M., Blouin R., and Germain L. (2010) The small heat-shock protein Hsp27 undergoes ERK-dependent phosphorylation and redistribution to the cytoskeleton in response to dual leucine Zipper-bearing kinase expression. J. Invest. Dermatol. 130, 74–85 10.1038/jid.2009.185 [DOI] [PubMed] [Google Scholar]
- 33. Harrington W. R., Kim S. H., Funk C. C., Madak-Erdogan Z., Schiff R., Katzenellenbogen J. A., and Katzenellenbogen B. S. (2006) Estrogen dendrimer conjugates that preferentially activate extranuclear, nongenomic versus genomic pathways of estrogen action. Mol. Endocrinol. 20, 491–502 10.1210/me.2005-0186 [DOI] [PubMed] [Google Scholar]
- 34. Navarro G., Xu W., Jacobson D. A., Wicksteed B., Allard C., Zhang G., De Gendt K., Kim S. H., Wu H., Zhang H., Verhoeven G., Katzenellenbogen J. A., and Mauvais-Jarvis F. (2016) Extranuclear Actions of the androgen receptor enhance glucose-stimulated insulin secretion in the male. Cell Metab. 23, 837–851 10.1016/j.cmet.2016.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yang X., Guo Z., Sun F., Li W., Alfano A., Shimelis H., Chen M., Brodie A. M. H., Chen H., Xiao Z., Veenstra T. D., and Qiu Y. (2011) Novel membrane-associated androgen receptor splice variant potentiates proliferative and survival responses in prostate cancer cells. J. Biol. Chem. 286, 36152–36160 10.1074/jbc.M111.265124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ahrens-Fath I., Politz O., Geserick C., and Haendler B. (2005) Androgen receptor function is modulated by the tissue-specific AR45 variant. FEBS J. 272, 74–84 10.1111/j.1432-1033.2004.04395.x [DOI] [PubMed] [Google Scholar]
- 37. Garza-Contreras J., Duong P., Snyder B. D., Schreihofer D. A., and Cunningham R. L. (2017) Presence of androgen receptor variant in neuronal lipid rafts. eNeuro 4, ENEURO.0109–17.2017 10.1523/ENEURO.0109-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mohler J. L., Gregory C. W., Ford O. H. 3rd, Kim D., Weaver C. M., Petrusz P., Wilson E. M., and French F. S. (2004) The androgen axis in recurrent prostate cancer. Clin. Cancer Res. 10, 440–448 [DOI] [PubMed] [Google Scholar]
- 39. Zarif J. C., Lamb L. E., Schulz V. S., Nollet E. A., and Miranti C. K. (2015) Androgen receptor non-nuclear regulation of prostate cancer cell invasion mediated by Src and matriptase. Oncotarget 6, 6862–6876 10.18632/oncotarget.3119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhuang L., Lin J., Lu M. L., Solomon K. R., and Freeman M. R. (2002) Cholesterol-rich lipid rafts mediate Akt-regulated survival in prostate cancer cells. Cancer Res. 62, 2227–2231 10.18632/oncotarget.3119 [DOI] [PubMed] [Google Scholar]
- 41. Song K. S., Li S., Okamoto T., Quilliam L. A., Sargiacomo M., and Lisanti M. P. (1996) Co-purification and direct interaction of Ras with caveolin, an integral membrane protein of caveolae microdomains: Detergent-free purification of caveolae membranes. J. Biol. Chem. 271, 9690–9697 10.1074/jbc.271.16.9690 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.