

Abstract
Set7/9 (also known as Set7, Set9, Setd7, and Kmt7) is a lysine methyltransferase that catalyzes the methylation of multiple substrates, including histone H3 and non-histone proteins. Although not essential for normal development and physiology, Set7/9-mediated methylation events play important roles in regulating cellular pathways involved in various human diseases, making Set7/9 a promising therapeutic target. Multiple Set7/9 inhibitors have been developed, which exhibit varying degrees of potency and selectivity in vitro. However, validation of these compounds in vivo has been hampered by the lack of a reliable cellular biomarker for Set7/9 activity. Here, we report the identification of Rpl29, a ribosomal protein abundantly expressed in all cell types, as a major substrate of Set7/9. We show that Rpl29 lysine 5 (Rpl29K5) is methylated exclusively by Set7/9 and can be demethylated by Lsd1 (also known as Kdm1a). Rpl29 is not a core component of the ribosome translational machinery and plays a regulatory role in translation efficiency. Our results indicate that Rpl29 methylation has no effect on global protein synthesis but affects Rpl29 subcellular localization. Using an Rpl29 methylation-specific antibody, we demonstrate that Rpl29K5 methylation is present ubiquitously and validate that (R)-PFI-2, a Set7/9 inhibitor, efficiently reduces Rpl29K5 methylation in cell lines. Thus, Rpl29 methylation can serve as a specific cellular biomarker for measuring Set7/9 activity.
Keywords: biomarker, post-translational modification (PTM), epigenetics, histone methylation, ribosome function, Lsd1, Rpl29, Set7/9
Introduction
Covalent post-translational modifications (PTMs)7 of proteins, such as phosphorylation, acetylation, methylation, and ubiquitination, increase the functional diversity and complexity of the proteome. PTMs can alter the properties of proteins, including their folding, stability, targeting to specific subcellular locations, interactions with ligands or other proteins, and functional states. Lysine methylation, which involves the transfer of one, two, or three methyl groups to the ϵ-nitrogen of a lysine side chain, is a widespread PTM that contributes to essentially all aspects of cell physiology (1). Given their abundance, histone proteins were among the first characterized methyl-lysine proteins (2). Studies over the last two decades have established that histone lysine methylation plays critical roles in gene regulation and chromatin dynamics, with the sites (lysine residues) and degrees of methylation (mono-, di-, and trimethylation) often correlating with distinct chromatin states and functional outcomes. For example, mono- and trimethylation of histone H3 lysine 4 (H3K4me1 and H3K4me3) mark enhancer and active promoter regions, respectively, and H3K36me3 is usually associated with transcribed regions, whereas H3K27me3 and H3K9me3 are typically enriched in repressed and heterochromatin regions, respectively (3). The relative slow development of proteomic tools and the fact that most proteins are much less abundant than histones have hindered the identification of lysine methylation in other proteins. Nonetheless, numerous non-histone proteins have been shown to be methylated at lysine residues (4, 5), although the regulation and functional relevance of most of these methylation events remain to be explored.
Lysine methylation is generated by lysine methyltransferases (KMTs) and removed by lysine demethylases (KDMs). The majority of known KMTs contain a catalytic SET domain, named after the three founding members of the family: Su(var)3–9, Enhancer of zeste, and Trithorax (6). Another protein family known to methylate lysine is the seven β-strand methyltransferase family (7), including Dot1L, which methylates H3K79, and several other members that methylate lysines in non-histone proteins (5). There are also two families of KDMs. Lysine-specific demethylase 1 (Lsd1, also known as Kdm1a) and Lsd2 (Kdm1b) belong to the family of FAD-dependent monoamine oxidases. They remove mono- and dimethylation from H3K4 (8, 9), and Lsd1 has also been shown to demethylate non-histone proteins (4, 10, 11). The second family of KDMs is the Jumonji C (Jmjc) domain-containing proteins, which can remove mono-, di-, and trimethylation by an oxidative mechanism requiring Fe(II) and α-ketoglutarate as co-factors (12).
Set7/9 (also known as Set7, Set9, Setd7, and Kmt7), a SET domain-containing protein, was originally identified as a histone H3K4 monomethyltransferase associated with transcriptional activation (13, 14). However, Set7/9 exhibits little activity toward H3K4 in nucleosomes (15), and Set7/9-deficient (Set7/9−/−) mouse embryonic fibroblasts (MEFs) have normal levels of H3K4 methylation (16), suggesting that histone H3 is not the primary substrate of Set7/9. Indeed, multiple non-histone substrates of Set7/9 have been identified, including p53, Taf10, Dnmt1, estrogen receptor α, Stat3, E2F1, Rb, and Pdx1 (4, 15, 17–24). Structural and functional analysis revealed that the minimal consensus sequence for Set7/9 substrate recognition is K/R-S/T-K (in which the methylation site is underlined) (25, 26).
Genetic studies in mouse suggest that Set7/9 is not essential for mammalian development (16, 27, 28). However, Set7/9, via modulating the stability and activities of its substrates, has been implicated in regulating cellular pathways involved in various human diseases, including cancer, diabetes, pulmonary and renal fibrosis, and inflammation (4, 24, 29–32). Therefore, Set7/9 has emerged as a promising target for therapeutic interventions. Indeed, multiple Set7/9 inhibitors have been developed (33–39). Although these compounds exhibit certain degrees of potency in biochemical assays in vitro, it has been difficult to validate their inhibitory effects toward Set7/9 in vivo. Despite the identification of multiple Set7/9 substrates, a reliable biomarker that can be conveniently used to measure Set7/9 activity in cells is still unavailable. The challenges include potential functional redundancy of KMTs on Set7/9 substrates, low levels of expression and methylation of Set7/9 substrates, and lack of detection reagents.
In this study, we identified ribosomal protein L29 (Rpl29) as a major substrate of Set7/9. Rpl29, a component of the large (60S) ribosomal subunit abundantly expressed in all cell types, plays a regulatory role in translation efficiency but is not essential for protein synthesis (40). We showed that Rpl29 lysine 5 (Rpl29K5) is methylated exclusively by Set7/9. This methylation event had no effect on global protein synthesis but facilitated Rpl29 nuclear enrichment. We demonstrated that Rpl29K5 methylation is present ubiquitously in cell lines and validated that the Set7/9 inhibitor (R)-PFI-2 (36) efficiently reduces Rpl29K5 methylation in cells. Thus, Rpl29 methylation is a specific cellular biomarker for Set7/9 activity, which would be valuable for developing and testing therapeutics that target Set7/9.
Results
Set7/9 specifically methylates Rpl29 at Lys-5
In an attempt to generate a “pan” methyl-lysine antibody, New Zealand White rabbits were immunized with a peptide library that contained a single dimethyl lysine (Kme2) surrounded by six degenerate amino acids on each side (XXXXXX-Kme2-XXXXXX), and the polyclonal antibody, D3977, was purified by immunoaffinity. As Set7/9 is a promiscuous enzyme that methylates multiple substrates (4), we tested the reactivity of the D3977 antibody by Western blot analysis of the wildtype (WT) J1 mouse embryonic stem cell (mESC) line and a Set7/9−/− mESC line derived from a Set7/9-null blastocyst-stage embryo (27). The antibody recognized multiple proteins, with the vast majority of them showing no obvious differences in both cell lines. However, a prominent signal of ∼22 kDa detected in WT cells was absent in Set7/9−/− cells (Fig. 1A), suggesting that the protein could be a Set7/9-specific substrate or, alternatively, its expression could be silenced in Set7/9−/− cells. To determine the identity of the protein, we performed immunoaffinity enrichment with the D3977 antibody, followed by LC and tandem MS (LC-MS/MS) analysis. The ∼22-kDa protein turned out to be Rpl29, a component of the large (60S) ribosomal subunit. Of note, peptides containing dimethyl Lys-5 of Rpl29 were identified in WT, but not Set7/9−/−, samples, suggesting that Set7/9 catalyzes dimethylation of Rpl29 at Lys-5 (Rpl29K5me2) (Fig. 1B). Indeed, Lys-5 is present in an optimal sequence motif recognized by Set7/9, i.e. K-S-K (25, 26), and previous work has shown that human and rat Rpl29 can be methylated at Lys-5 (41, 42).
Figure 1.

Set7/9 methylates Rpl29 at Lys-5. A, Western blot analysis of WT (J1) and Set7/9−/− mESC lysates with a pan dimethyl-lysine antibody (D3977), which recognized an ∼22-kDa protein (arrow) in J1 cells, but not in Set7/9−/− cells. B, MS/MS spectrum of dimethyl Lys-5 of Rpl29. C, FLAG-tagged Set7/9 or an inactive Set7/9 mutant (H297A) was stably expressed in Set7/9−/− mESCs, and the cell lysates were immunoblotted with FLAG, D3977, and Rpl29 antibodies, respectively. Note that Rpl29 methylation was restored by Set7/9, but not by the inactive mutant. Untrans, untransfected. D, FLAG-tagged Rpl29 or a K5R mutant was transiently expressed in J1 mESCs (empty vector was transfected as a negative control), the cell lysates were immunoprecipitated (IP) with FLAG antibody (FLAG IP), and the samples were immunoblotted with D3977 and FLAG antibodies, respectively. Note that the K5R substitution abolished Rpl29 methylation. E, in vitro methylation assay using 3H-labeled SAM, GST-Set7/9 protein, and biotinylated Rpl29 peptide-(2–15) with Lys-5 being unmodified (me0), mono (me1)-, di (me2)-, or trimethylated (me3). Histone H3-(1–18) peptide was used as a positive control, and GST-Set7/9 alone served as a negative control. The presence of biotinylated peptides was verified by Western blotting probed with horseradish peroxidase (HRP)-conjugated streptavidin. Note that Set7/9 can methylate Rpl29K5me0 and me1 peptides, but not Rpl29K5me2 and me3 peptides.
To confirm that Set7/9 is responsible for Rpl29 methylation, we stably transfected Set7/9−/− mESCs with a plasmid expressing FLAG-tagged Set7/9 or a catalytically inactive variant that harbors a histidine-to-alanine mutation at amino acid 297 (H297A) in the SET domain (15). WT Set7/9 restored Rpl29 methylation, with methylation abundance correlating with Set7/9 levels in different stable clones, whereas the H297A mutant protein failed to rescue Rpl29 methylation (Fig. 1C). To verify the methylation site, we transiently transfected FLAG-tagged Rpl29 or an Rpl29 mutant with Lys-5 being substituted with arginine (K5R). The FLAG-tagged proteins were immunoprecipitated and immunoblotted with the D3977 antibody. As shown in Fig. 1D, Lys-5 substitution led to complete elimination of Rpl29 methylation, indicating that Lys-5 is the only site methylated by Set7/9.
Set7/9 is typically a monomethyltransferase, although it has also been shown to deposit dimethyl marks on specific substrates (26). To confirm that Set7/9 indeed catalyzes Rpl29K5 dimethylation, we performed an in vitro methylation assay using recombinant GST-Set7/9 protein and biotinylated Rpl29 peptides (amino acids 2–15) with Lys-5 being unmodified or methylated. As shown in Fig. 1E, Set7/9 efficiently methylated the unmodified and monomethyl peptides, but not di- and trimethyl peptides. By measuring the density of the methyl signals, as well as the biotinylated peptide signals (for normalization), using the NIH ImageJ software, we estimated that the methyl signal deposited on the monomethyl peptide was ∼70% of that deposited on the unmodified peptide (Fig. 1E). The data suggest that Set7/9 catalyzes both mono- and dimethylation of Rpl29 at Lys-5, with strong activity for producing the dimethyl mark.
Having established that Set7/9 specifically methylates Rpl29 at Lys-5, a rabbit mAb against an Rpl29 peptide with dimethyl Lys-5 was developed. Dot-blot analysis showed that the antibody, named dimethyl-RPL29 (Lys-5) (D8T9P) rabbit mAb, exhibited strong reactivity with the dimethyl peptide, slight cross-reactivity with the monomethyl peptide, and no reactivity with the trimethyl and unmodified peptides (Fig. 2A). Western blot analysis detected a single ∼22-kDa protein in WT mESCs, but not in Set7/9−/− and Rpl29−/− mESCs (Rpl29−/− cells were generated by CRISPR-Cas9 technology, Fig. S1), thus demonstrating that the antibody is specific for methylated Rpl29 (Fig. 2B). Taken together, our results show that Rpl29 is a major substrate of Set7/9.
Figure 2.
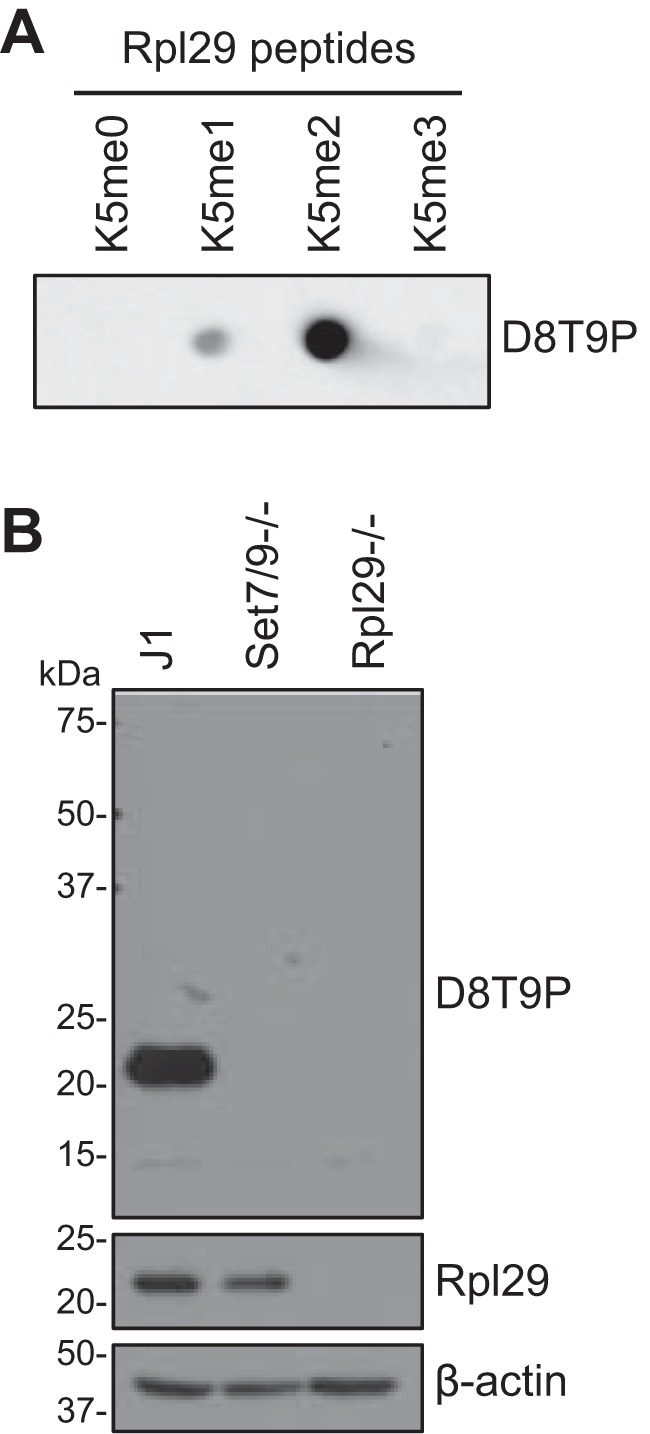
Characterization of the dimethyl-RPL29 (Lys-5) (D8T9P) rabbit mAb. A, dot-blot analysis using Rpl29 peptide with Lys-5 being unmodified (me0), mono (me1)-, di (me2)-, or trimethylated (me3) (10 ng/dot), which showed that D8T9P strongly recognized the Rpl29K5me2 peptide and exhibited weak cross-reactivity with the Rpl29K5me1 peptide and no reactivity with the Rpl29K5me0 and Rpl29K5me3 peptides. B, Western blot analysis of J1, Set7/9−/−, and Rpl29−/− mESCs with D8T9P and Rpl29 antibodies, respectively, which demonstrated that the D8T9P antibody was specific for methylated Rpl29. β-Actin was used as a loading control.
Rpl29 is demethylated by Lsd1
Several Set7/9 substrates, including p53, Dnmt1, E2F1, and Stat3, have been shown to be demethylated by the lysine demethylase Lsd1 (4, 10, 11, 19, 21, 22, 43). We therefore assessed whether Rpl29 methylation is regulated by Lsd1. In mESCs, deletion of Lsd1 (19) led to an increase in Rpl29K5 methylation (Fig. 3A) and, conversely, overexpression of FLAG-tagged Lsd1 resulted in a decrease in Rpl29K5 methylation (Fig. 3B). These results suggest that Lsd1 is a key enzyme that demethylates Rpl29 in mESCs.
Figure 3.

Lsd1 demethylates Rpl29. A, J1 and Lsd1−/− mESC lysates were immunoblotted with Lsd1, Set7/9, dimethyl-RPL29 (Lys-5) (D8T9P) rabbit mAb, Rpl29, and β-actin antibodies, respectively. Note that Rpl29 methylation was increased in Lsd1−/− cells. B, J1 mESCs were stably transfected with a plasmid encoding FLAG-Lsd1 or the empty vector, and Western blot analysis showed that Lsd1 overexpression reduced Rpl29 methylation. C and D, Western blot analysis of mouse cell lines (C) and human cancer cell lines (D) for the levels of Rpl29K5 methylation, Rpl29, Set7/9, and Lsd1.
To determine whether Rpl29K5 is also methylated in differentiated cells, we examined the murine fibroblast cell line NIH 3T3 by Western blot analysis. It turned out that the Rpl29K5 methylation level was substantially higher in NIH 3T3 cells compared with mESCs (Fig. 3C). Although comparable levels of Set7/9 were detected in NIH 3T3 cells and mESCs, Lsd1 expression was markedly different, with a much higher level in mESCs, which is likely the major determinant of the difference in Rpl29K5 methylation levels in these cell lines (Fig. 3C).
Human and murine Rpl29 are identical in their N-terminal 64 amino acids, and thus their methylated forms would be recognized equally well by the dimethyl-RPL29 (Lys-5) (D8T9P) rabbit mAb. We assessed the presence of RPL29K5 methylation in a panel of human cancer cell lines. As expected of a ribosomal protein, RPL29 is abundantly expressed in all cell lines examined. RPL29K5 methylation was ubiquitously detected, although its levels showed greater variations compared with total RPL29 levels. In general, RPL29K5 methylation levels positively correlated with SET7/9 levels and negatively correlated with LSD1 levels (Fig. 3D). Collectively, these data suggest that the RPL29 methylation levels in cells are mainly determined by the opposing effects of SET7/9 and LSD1.
Rpl29 methylation has no effect on global protein synthesis
To gain insights into the functional relevance of Rpl29 methylation, we first deleted the Rpl29 gene in NIH 3T3 cells using the CRISPR-Cas9 technology (Fig. 4A, Fig. S1) and then re-introduced exogenous WT Rpl29 or the unmethylable K5R mutant (Fig. 4B). Stable clones with expression levels similar to the endogenous Rpl29 level in NIH 3T3 cells were selected for subsequent experiments (Fig. 4B). We confirmed that Rpl29K5 methylation was present in the WT Rpl29 clone, at a similar level as in NIH 3T3 cells, and was absent in the K5R clone (Fig. 4B). Although it has been reported that Rpl29−/− primary MEFs exhibit decreased rates of proliferation (40), deletion of Rpl29 in the immortalized NIH 3T3 cell line, as well as the stable clones expressing exogenous Rpl29 proteins, showed no obvious changes in proliferation, survival, and morphology.
Figure 4.

Rpl29 methylation shows no effect on global protein synthesis. A, Western blotting showing the absence of Rpl29 in Rpl29−/− NIH 3T3 clones generated by CRISPR-Cas9. B, exogenous Rpl29 or the K5R mutant (untagged) was stably expressed in Rpl29−/− NIH 3T3 cells, and the cell lysates were immunoblotted with Rpl29 antibody and D8T9P, respectively. C, SUnSET assay showing global protein synthesis in the indicated cell lines. As a control for protein synthesis inhibition, NIH 3T3 cells were treated with cycloheximide (CHX, 100 μg/ml) for 12 h before the addition of puromycin. D, [35S]methionine labeling assay showing global protein synthesis in the indicated cell lines. Shown are the mean ± S.D. from three independent experiments. The significance of differences was determined with one-way analysis of variance (ANOVA). *, p < 0.05; ns, not significant. E, polysome profiling showing similar fractions of mRNPs and polysomes in all cell lines.
Although Rpl29 is not essential for protein synthesis, it has been shown to enhance translation efficiency in MEFs (40). Using the surface sensing of translation (SUnSET) assay, a method for monitoring global protein synthesis that involves the immunological detection of puromycin-labeled neosynthesized proteins (44), we found that deletion of Rpl29 in NIH 3T3 cells resulted in a slight decrease in global protein synthesis. Re-expression of Rpl29 or the K5R mutant in Rpl29−/− cells led to partial restoration of protein synthesis, with Rpl29 and the K5R mutant showing comparable effects (Fig. 4C). The results were confirmed by metabolic ([35S]methionine) labeling (Fig. 4D), another assay for measuring global protein synthesis (45). Polysome profiling also revealed similar fractions of mRNPs and polysomes in all the cell lines examined (Fig. 4E). Based on these findings, we conclude that Rpl29 methylation has no effect on global protein synthesis in NIH 3T3 cells.
Rpl29 methylation affects its subcellular localization
We next investigated whether Rpl29 methylation alters its subcellular localization by performing immunofluorescence (IF) analysis and confocal microscopy. Consistent with the Rpl29 localization pattern in other cell types (40, 46), NIH 3T3 cells displayed a strong Rpl29 signal in the cytoplasm, with enrichment in perinuclear rough endoplasmic reticulum, and weak signal in the nuclei, with staining in both the nucleoplasm and nucleoli. The Rpl29 signal was specific, as no staining was detected in Rpl29−/− cells. The localization patterns of exogenous Rpl29 and the K5R mutant were generally similar to that of endogenous Rpl29 in NIH 3T3 cells. However, careful examination revealed that, compared with WT Rpl29, the K5R signal was stronger in the cytoplasm, especially in the perinuclear regions, and weaker in the nucleoplasm, which made staining in the nucleoli more prominent in some cells (Fig. 5A).
Figure 5.

Rpl29 methylation facilitates its nuclear enrichment. A and C, the indicated cell lines were immunostained with Rpl29 antibody and counterstained with DAPI. Shown are confocal microscopy images. Scale bars, 20 μm. B, FLAG-tagged Set7/9 or a catalytically inactive mutant (H297A) was stably overexpressed in NIH 3T3 cells, and the cell lysates, as well as that of untransfected (Untrans) cells, were immunoblotted with Set7/9 and β-actin antibodies, respectively. The FLAG-tagged and endogenous (Endo) Set7/9 bands are indicated.
Because the change in localization pattern observed with the unmethylable K5R mutant was subtle, we assessed the effect of enhancing Rpl29 methylation. To this end, FLAG-tagged Set7/9 or the catalytically inactive variant (H297A) was stably transfected in NIH 3T3 cells (Fig. 5B). IF analysis showed a striking alteration of the Rpl29 localization pattern, with a strong signal in the nuclei and weak signal in the cytoplasm, in cells overexpressing Set7/9. The effect depended on Set7/9 activity, as cells overexpressing the H297A mutant showed no change in Rpl29 localization pattern (Fig. 5C). Although we were unable to directly examine the localization pattern of methylated Rpl29 with IF using the dimethyl-RPL29 (Lys-5) (D8T9P) rabbit mAb, our results collectively suggest that Lys-5 methylation facilitates Rpl29 nuclear localization.
Validation of Rpl29 methylation as a cellular biomarker for Set7/9 activity
Set7/9 is a promising therapeutic target for several diseases. Multiple Set7/9 inhibitors have been developed, which exhibit varying degrees of potency and selectivity in biochemical assays in vitro (33–39). However, no direct evidence has been obtained that these compounds have Set7/9 inhibitory effects in vivo, mainly because of the lack of a reliable cellular biomarker for Set7/9 activity. With the identification of Rpl29 as a major substrate of Set7/9, we tested the effect of (R)-PFI-2 on Set7/9 activity in cells. (R)-PFI-2 is a selective Set7/9 inhibitor with nanomolar potency in vitro (36). Treatment of mESCs with 5 μm (R)-PFI-2 resulted in steady decreases of Rpl29K5 methylation, with the signal becoming severely reduced after 14 h and hardly detectable after 24 h (Fig. 6A). At 5 μm, (R)-PFI-2 also led to decreases in Rpl29K5 methylation in NIH 3T3 cells, albeit with less efficiency compared with the effect in mESCs (Fig. 6B), consistent with the observation that NIH 3T3 cells have substantially higher levels of Rpl29K5 methylation (Fig. 3C). Higher doses of (R)-PFI-2 showed a more obvious Set7/9 inhibitory effect in NIH 3T3 cells, with 10 μm being sufficient to largely eliminate Rpl29K5 methylation after 24 h of treatment (Fig. 6C). These data validated Rpl29K5 methylation as a specific cellular biomarker for Set7/9 activity.
Figure 6.
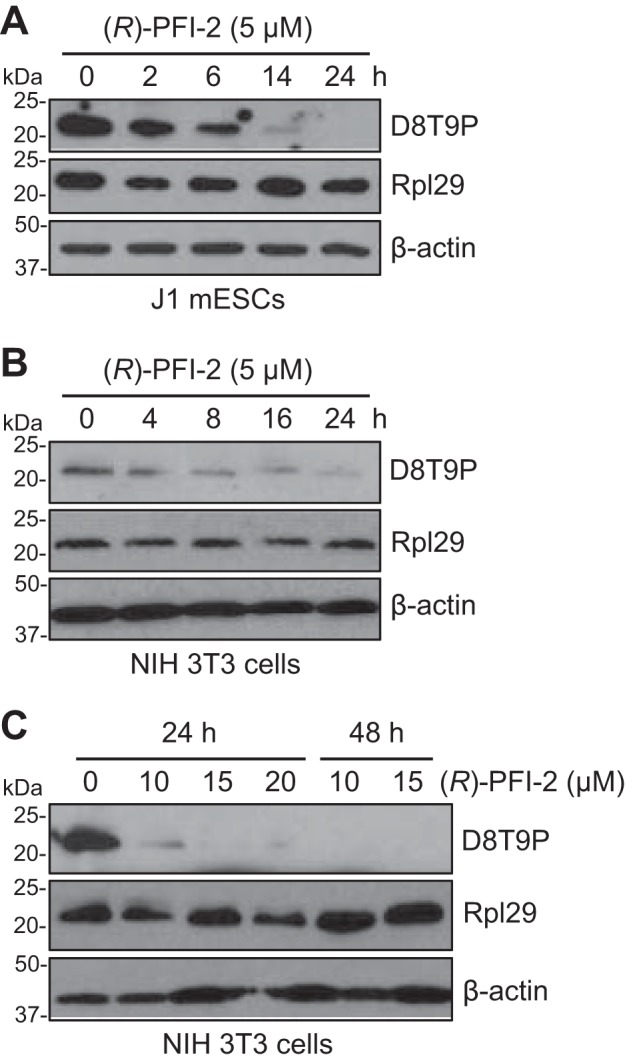
The Set7/9 inhibitor (R)-PFI-2 efficiently inhibits Rpl29 methylation in cells. J1 mESCs (A) or NIH 3T3 cells (B and C) were incubated with (R)-PFI-2 (5–20 μm, as indicated) for various periods of time (2–48 h, as indicated), and the cell lysates were immunoblotted with dimethyl-RPL29 (Lys-5) (D8T9P) rabbit mAb, Rpl29, and β-actin antibodies, respectively.
Discussion
In summary, we demonstrate that Set7/9 specifically methylates Lys-5 in Rpl29, an abundant protein in cells, thus identifying a major Set7/9 substrate. Although Set7/9 is generally considered a monomethyltransferase, it has also been shown to catalyze dimethylation of specific substrates in vitro and in vivo, depending on the sequence contexts of the methylation sites (26). We identified Rpl29K5me2 using antibodies raised against dimethyl peptides. However, in vitro methylation assay suggests that Set7/9 catalyzes both mono- and dimethylation of Rpl29. Indeed, previous work has shown that an antibody against human p53K372me1 (ab16033, Abcam), which presumably recognizes monomethylation, cross-reacts with an unknown Set7/9 substrate with the same molecular weight as Rpl29 (16), further indicating that Rpl29 can be monomethylated.
Rpl29 is conserved in eukaryotes but lacks an ortholog in prokaryotes, suggesting that Rpl29 is not a core component of the ribosome translational machinery and may play a regulatory role in translation. This notion is supported by genetic evidence. Rpl29−/− mice are viable and exhibit general growth deficiencies, and Rpl29−/− MEFs show reduced rates of protein synthesis and proliferation (40). Our data indicate that in NIH 3T3 cells, an immortalized fibroblast cell line with a relatively high level of Rpl29K5 methylation, Rpl29 methylation has no overt effect on global protein synthesis. However, Rpl29 methylation affects its subcellular localization, with the methylated form being enriched in the nuclei, although its functional relevance remains to be determined. It is worth mentioning that Rpl29−/− NIH 3T3 cells, as well as Rpl29−/− mESCs, show no proliferation defects, in contrast to Rpl29−/− MEFs as reported previously (40). We cannot rule out the possibility that Rpl29 and its methylation regulate the synthesis of specific proteins and are functionally more important in special cell types. Nonetheless, the fact that Set7/9−/− mice are viable and fertile with no gross abnormalities (16, 27, 28) suggests that the regulatory role of Rpl29 methylation must be subtle, if any, during normal development and in normal cells.
Most PTMs, including lysine methylation, are reversible, and “correcting” aberrant PTMs by targeting relevant enzymes and regulators (widely known as epigenetic therapy) has become a new frontier for drug discovery. Set7/9, which is not essential for normal development and physiology but regulates cellular pathways involved in various diseases, represents a promising therapeutic target. The identification of Rpl29 as a major Set7/9 substrate provides a specific biomarker for measuring Set7/9 activity in cells. Indeed, we validated the effect of the Set7/9 inhibitor (R)-PFI-2 by showing that it efficiently reduces the Rpl29K5 methylation levels in both mESCs and NIH 3T3 cells.
There are several advantages of using Rpl29 methylation as a biomarker for Set7/9 activity. First, given the functional redundancy of KMTs in histone methylation, it is possible that methylation of non-histone proteins, including some Set7/9 substrates, also involves multiple KMTs. We show that deletion of Set7/9 results in complete elimination of Rpl29 methylation in both mESCs and NIH 3T3 cells, indicating that Rpl29 is methylated exclusively by Set7/9. Second, unlike most known Set7/9 substrates, Rpl29 is abundantly expressed and ubiquitously methylated in all cell lines examined, which makes Set7/9 activity changes readily detectable. Third, methylation of Rpl29 has no overt effects on cell physiology, thus simplifying the interpretation of experimental data, as opposed to the potential complexity associated with methylation events that are functionally more critical. Last, the dimethyl-RPL29 (Lys-5) (D8T9P) rabbit mAb specifically recognizes methylated Rpl29, which would be valuable for testing therapeutics that target Set7/9, as well as for fundamental research on the regulation and functions of Set7/9.
Lsd1, which is frequently overexpressed in cancer (11), is also a promising therapeutic target that has been actively pursued by both academic laboratories and biopharma companies. We show that Lsd1 is a key enzyme that removes Rpl29 methylation, indicating that Rpl29 methylation can be used as a biomarker for Lsd1 activity as well. However, it remains to be determined whether other KDMs, such as Lsd2, are also involved in demethylating Rpl29.
Experimental procedures
Generation of methyl-lysine antibodies
The pan methyllysine antibody D3977 was generated using a peptide library that had a single dimethyl lysine in a peptide library containing degenerate amino acids at surrounding positions. The antibody was an antigen affinity purified polyclonal antibody. The dimethyl-RPL29 (Lys-5) (D8T9P) rabbit mAb was produced by immunizing rabbits with a synthetic peptide corresponding to residues surrounding Lys-5 of Rpl29. As human and murine Rpl29 proteins have identical sequences in their N-terminal regions, the D8T9P antibody recognizes the methylated form of both human and murine Rpl29.
LC-MS/MS analysis
Protein extracts from WT and Set7/9−/− mESC lines were digested with trypsin, and methylated peptides were enriched by immunoaffinity using the polyclonal dimethyl-lysine antibody D3977. Enriched peptides were identified and quantified by LC-MS/MS analysis, as reported previously (47).
Plasmid vectors
The FLAG-Lsd1 expression vector was reported previously (48). The FLAG-tagged Set7/9 and Rpl29 expression constructs were generated by cloning the mouse Set7/9 and Rpl29 cDNAs, respectively, into the pCAG-Flag-IRESblast vector (49). The untagged Rpl29 expression constructs were generated by cloning the cDNAs into the pCAG-IRESblast vector (50). The Set7/9:H297A and Rpl29:K5R point mutations were introduced by PCR-based mutagenesis. The GST-Set7/9 construct was generated by cloning Set7/9 cDNA into the pGEX-6P-1 vector (GE Healthcare). All plasmid constructs were verified by DNA sequencing. The primers used in this study are listed in Table S1.
Cell culture and manipulations
WT (J1) and genetically modified (Set7/9−/− and Rpl29−/−) mESC lines were grown on gelatin-coated Petri dishes and maintained in mESC medium containing 15% fetal bovine serum and 1,000 units/ml of leukemia inhibitory factor (51). Normally, mESCs were cultured in the absence of feeder cells. However, feeder cells were used when mESCs were seeded at low density to derive individual clones for stable expression of exogenous proteins or for disruption of Rpl29 by the CRISPR-Cas9 technology. NIH 3T3 cells and human cancer lines were cultured according to instructions of American Type Culture Collection. Transfection was performed using Lipofectamine 2000 (Invitrogen). Generation of mESC or NIH 3T3 stable clones expressing exogenous proteins was done as previously reported (52). Briefly, cells transfected with the corresponding plasmid vectors were selected with 6 μg/ml of Blasticidin S HCl (Gibco) for 7–10 days, and individual colonies were picked. To inhibit Set7/9, mESCs or NIH 3T3 cells were treated with various doses of (R)-PFI-2 (36) for various periods of time.
Generation of Rpl29−/− cells with CRISPR-Cas9 technology
J1 mESCs or NIH 3T3 cells were transiently co-transfected with the pCAG-Cas9-IRES-GFP vector and two synthesized gBlocks (Integrated DNA Technologies) containing the U6 promoter and Rpl29 targeting sequences in intron 1 and exon 4, respectively (Fig. S1A). 24 h post-transfection, GFP-positive cells were sorted by FACS. Then the cells were seeded at low density, cultured for 7–10 days, and individual colonies were picked. The targeting strategy would delete exons 2 and 3 and create a frameshift (Fig. S1B). The clones were initially screened by PCR (Fig. S1C), and the Rpl29−/− clones were verified by DNA sequencing and Western blotting.
Western blotting and immunoprecipitation
For Western blotting, total proteins from cells were extracted using RIPA buffer (150 mm NaCl, 50 mm Tris-HCl, pH 7.4, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 2 mm EDTA, and 50 mm NaF) containing protease inhibitor mixture (Thermo Scientific 1861279), PMSF, sodium orthovanadate, and β-glycerophosphate. For immunoprecipitation, cells were lysed in lysis buffer (20 mm Tris-HCl, pH 7.9, 150 mm NaCl, 0.1% Nonidet P-40, 1 mm EDTA, 3 mm MgCl2, 10% glycerol, and 1× protease inhibitor mixture), the lysates were incubated with FLAG antibody for 2 h to form immunocomplexes, and immunoprecipitation was performed using Protein A/G UltraLink Resin beads (Thermo Fisher 53133). After denaturation at 100 °C in Laemmli buffer (4% SDS, 20% glycerol, 10% 2-mercaptoethanol, 0.004% bromphenol blue, and 0.125 m Tris HCl, pH 6.8), protein samples were resolved with SDS-PAGE and analyzed by immunoblotting with specific antibodies. The information about all the antibodies used in this study is shown in Table S2.
IF
NIH 3T3 cells grown on glass coverslips were fixed with 4% paraformaldehyde for 7 min and permeabilized with 0.1% Triton X-100 for 10 min. After blocking with 10% goat serum in PBS for 1 h, the coverslips were incubated with Rpl29 mouse polyclonal antibody in PBS containing 0.5% BSA overnight at 4 °C. The signal was detected using anti-mouse Alexa FluorTM 488 IgG (Invitrogen). The coverslips were mounted on slides using ProLongTM Gold antifade reagent with DAPI (Invitrogen). Cells were visualized using an LSM 880 confocal microscope (Zeiss).
In vitro methylation assay
The GST-Set7/9 was expressed and purified as described previously (53). In vitro methylation reactions were performed in a final volume of 30 μl of 50 mm Tris-HCl (pH 8.5), 5 mm MgCl2, 4 mm DTT, and 0.42 μm 3H-labeled S-adenosyl-l-[methyl-3H]methionine (PerkinElmer Life Sciences) (54). Each reaction contains 1 μg of synthesized peptide and 1 μg of recombinant GST-Set7/9. Biotinylated Rpl29 peptides-(2–15) (amino-AKSKNHTTHNQSRK-Biotin) with mono-, di-, trimethylation and unmodified at Lys-5 were supplied by CPC Scientific Inc. Biotinylated H3 peptide-(1–18) was supplied by Keck Biotechnology Resource Laboratory. The reaction was incubated at 30 °C for 1 h and then subjected to fluorography by separation on SDS-PAGE, transferred to a polyvinylidene fluoride membrane, treated with En3HanceTM (PerkinElmer Life Sciences), and exposed to film for 2 days at −80 °C.
Global protein synthesis assays
SUnSET assay was performed as previously described (44). Briefly, cells were incubated with culture medium containing 1 μg/ml of puromycin (Gibco) for 10 min at 37 °C to allow incorporation of puromycin into newly synthesized proteins. After rinsing twice with PBS, cells were cultured in regular medium for an additional 50 min. As a negative control (lack of protein synthesis), cells were treated with cycloheximide (100 μg/ml) for 12 h before the addition of puromycin. Proteins were extracted with CHAPS buffer (150 mm NaCl, 50 mm Tris-HCl, pH 7.4, and 0.5% CHAPS), quantified, and analyzed by immunoblotting with puromycin antibody. Metabolic ([35S]methionine) labeling assay was carried out as previously described (45).
Polysome profiling
Polysome profiling was carried out as previously described (55). Briefly, cells were incubated with cycloheximide (100 μg/ml) in fresh Dulbecco's modified Eagle's medium/F-12 medium for 8 min at room temperature to “freeze” the ribosomes on the translating mRNAs. Next, cell pellets were lysed in 1 ml of hypotonic buffer (5 mm Tris-HCl, 2.5 mm MgCl2, 1.5 mm KCl, and 0.5% Triton X-100) supplemented with EDTA-free protease inhibitor (Roche Applied Science) and RNase inhibitor (Invitrogen). Cell lysates were further processed through a loose Type A glass homogenizer by 10 strokes. Following centrifugation at 16,000 × g for 7 min at 4 °C, the supernatant was collected for ultracentrifugation at 222,228 × g (36,000 rpm) for 2 h at 4 °C using an SW41Ti rotor. 2 ml of sucrose gradient (10%∼50%) was generated through a two-chamber gradient maker system (CBS Scientific). Lysates were fractionated in a fast protein LC (FPLC) system (Bio-Rad), and 200 μl of lysate for each fraction was collected through the fraction collector, whereas the OD at 254 nm was simultaneously monitored by a spectrophotometer. Free ribosome subunits (40S and 60S), monosomes (80S), and polysomes were determined according to their elution.
Author contributions
T. H., A. K. S., and T. C. data curation; T. H., A. K. S., N. V., V. V., M. T. B., and T. C. formal analysis; T. H., A. K. S., N. V., V. V., J. C., S. H., J. B., C. J. F., V. Y., K. A. L., and A. G. investigation; C. J. F., A. G., and C. H. A. resources; V. Y. and K. A. L. software; K. A. L., M. T. B., and T. C. supervision; M. T. B. and T. C. conceptualization; T. C. funding acquisition; T. C. writing-original draft.
Supplementary Material
Acknowledgments
We thank Dr. Ivan Topisirovic (McGill University) for discussion and technical assistance and Dr. Collene Jeter (The University of Texas MD Anderson Cancer Center, MDACC) for training and help with confocal microscopy.
This work was supported by National Institutes of Health NIDDK Grant 1R01DK106418-01 (to T. C.). C. J. F., V. Y., and K. A. L. are employees of Cell Signaling Technology Inc. (CST). A. G. was a former employee of CST and is a current employee of Bluefin Biomedicine, Inc. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Fig. S1 and Tables S1 and S2.
- PTM
- post-translational modification
- KMT
- lysine methyltransferase
- KDM
- lysine demethylase
- Lsd1
- lysine-specific demethylase 1
- MEF
- mouse embryonic fibroblast
- mESC
- mouse embryonic stem cell
- GST
- glutathione S-transferase
- IF
- immunofluorescence.
References
- 1. Wozniak G. G., and Strahl B. D. (2014) Hitting the “mark”: interpreting lysine methylation in the context of active transcription. Biochim. Biophys. Acta 1839, 1353–1361 10.1016/j.bbagrm.2014.03.002 [DOI] [PubMed] [Google Scholar]
- 2. Murray K. (1964) The occurrence of ϵ-N-methyl lysine in histones. Biochemistry 3, 10–15 10.1021/bi00889a003 [DOI] [PubMed] [Google Scholar]
- 3. Allis C. D., and Jenuwein T. (2016) The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 17, 487–500 10.1038/nrg.2016.59 [DOI] [PubMed] [Google Scholar]
- 4. Zhang X., Huang Y., and Shi X. (2015) Emerging roles of lysine methylation on non-histone proteins. Cell Mol. Life Sci. 72, 4257–4272 10.1007/s00018-015-2001-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Carlson S. M., and Gozani O. (2016) Nonhistone lysine methylation in the regulation of cancer pathways. Cold Spring Harb. Perspect. Med. 6, a026435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tschiersch B., Hofmann A., Krauss V., Dorn R., Korge G., and Reuter G. (1994) The protein encoded by the Drosophila position-effect variegation suppressor gene Su(var)3–9 combines domains of antagonistic regulators of homeotic gene complexes. EMBO J. 13, 3822–3831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schubert H. L., Blumenthal R. M., and Cheng X. (2003) Many paths to methyltransfer: a chronicle of convergence. Trends Biochem. Sci. 28, 329–335 10.1016/S0968-0004(03)00090-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shi Y., Lan F., Matson C., Mulligan P., Whetstine J. R., Cole P. A., and Casero R. A. (2004) Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941–953 10.1016/j.cell.2004.12.012 [DOI] [PubMed] [Google Scholar]
- 9. Ciccone D. N., Su H., Hevi S., Gay F., Lei H., Bajko J., Xu G., Li E., and Chen T. (2009) KDM1B is a histone H3K4 demethylase required to establish maternal genomic imprints. Nature 461, 415–418 10.1038/nature08315 [DOI] [PubMed] [Google Scholar]
- 10. Nicholson T. B., and Chen T. (2009) LSD1 demethylates histone and non-histone proteins. Epigenetics 4, 129–132 10.4161/epi.4.3.8443 [DOI] [PubMed] [Google Scholar]
- 11. Zheng Y. C., Ma J., Wang Z., Li J., Jiang B., Zhou W., Shi X., Wang X., Zhao W., and Liu H. M. (2015) A systematic review of histone lysine-specific demethylase 1 and its inhibitors. Med. Res. Rev. 35, 1032–1071 10.1002/med.21350 [DOI] [PubMed] [Google Scholar]
- 12. Klose R. J., Kallin E. M., and Zhang Y. (2006) JmjC-domain-containing proteins and histone demethylation. Nat. Rev. Genet. 7, 715–727 10.1038/nrg1945 [DOI] [PubMed] [Google Scholar]
- 13. Wang H., Cao R., Xia L., Erdjument-Bromage H., Borchers C., Tempst P., and Zhang Y. (2001) Purification and functional characterization of a histone H3-lysine 4-specific methyltransferase. Mol. Cell 8, 1207–1217 10.1016/S1097-2765(01)00405-1 [DOI] [PubMed] [Google Scholar]
- 14. Nishioka K., Chuikov S., Sarma K., Erdjument-Bromage H., Allis C. D., Tempst P., and Reinberg D. (2002) Set9, a novel histone H3 methyltransferase that facilitates transcription by precluding histone tail modifications required for heterochromatin formation. Genes Dev. 16, 479–489 10.1101/gad.967202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chuikov S., Kurash J. K., Wilson J. R., Xiao B., Justin N., Ivanov G. S., McKinney K., Tempst P., Prives C., Gamblin S. J., Barlev N. A., and Reinberg D. (2004) Regulation of p53 activity through lysine methylation. Nature 432, 353–360 10.1038/nature03117 [DOI] [PubMed] [Google Scholar]
- 16. Lehnertz B., Rogalski J. C., Schulze F. M., Yi L., Lin S., Kast J., and Rossi F. M. (2011) p53-dependent transcription and tumor suppression are not affected in Set7/9-deficient mice. Mol. Cell 43, 673–680 10.1016/j.molcel.2011.08.006 [DOI] [PubMed] [Google Scholar]
- 17. Kouskouti A., Scheer E., Staub A., Tora L., and Talianidis I. (2004) Gene-specific modulation of TAF10 function by SET9-mediated methylation. Mol. Cell 14, 175–182 10.1016/S1097-2765(04)00182-0 [DOI] [PubMed] [Google Scholar]
- 18. Subramanian K., Jia D., Kapoor-Vazirani P., Powell D. R., Collins R. E., Sharma D., Peng J., Cheng X., and Vertino P. M. (2008) Regulation of estrogen receptor α by the SET7 lysine methyltransferase. Mol. Cell 30, 336–347 10.1016/j.molcel.2008.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang J., Hevi S., Kurash J. K., Lei H., Gay F., Bajko J., Su H., Sun W., Chang H., Xu G., Gaudet F., Li E., and Chen T. (2009) The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat. Genet. 41, 125–129 10.1038/ng.268 [DOI] [PubMed] [Google Scholar]
- 20. Estève P. O., Chin H. G., Benner J., Feehery G. R., Samaranayake M., Horwitz G. A., Jacobsen S. E., and Pradhan S. (2009) Regulation of DNMT1 stability through SET7-mediated lysine methylation in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 106, 5076–5081 10.1073/pnas.0810362106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang J., Huang J., Dasgupta M., Sears N., Miyagi M., Wang B., Chance M. R., Chen X., Du Y., Wang Y., An L., Wang Q., Lu T., Zhang X., Wang Z., and Stark G. R. (2010) Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc. Natl. Acad. Sci. U.S.A. 107, 21499–21504 10.1073/pnas.1016147107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kontaki H., and Talianidis I. (2010) Lysine methylation regulates E2F1-induced cell death. Mol. Cell 39, 152–160 10.1016/j.molcel.2010.06.006 [DOI] [PubMed] [Google Scholar]
- 23. Munro S., Khaire N., Inche A., Carr S., and La Thangue N. B. (2010) Lysine methylation regulates the pRb tumour suppressor protein. Oncogene 29, 2357–2367 10.1038/onc.2009.511 [DOI] [PubMed] [Google Scholar]
- 24. Maganti A. V., Maier B., Tersey S. A., Sampley M. L., Mosley A. L., Özcan S., Pachaiyappan B., Woster P. M., Hunter C. S., Stein R., and Mirmira R. G. (2015) Transcriptional activity of the islet beta cell factor Pdx1 is augmented by lysine methylation catalyzed by the methyltransferase Set7/9. J. Biol. Chem. 290, 9812–9822 10.1074/jbc.M114.616219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Couture J. F., Collazo E., Hauk G., and Trievel R. C. (2006) Structural basis for the methylation site specificity of SET7/9. Nat. Struct. Mol. Biol. 13, 140–146 10.1038/nsmb1045 [DOI] [PubMed] [Google Scholar]
- 26. Dhayalan A., Kudithipudi S., Rathert P., and Jeltsch A. (2011) Specificity analysis-based identification of new methylation targets of the SET7/9 protein lysine methyltransferase. Chem. Biol. 18, 111–120 10.1016/j.chembiol.2010.11.014 [DOI] [PubMed] [Google Scholar]
- 27. Kurash J. K., Lei H., Shen Q., Marston W. L., Granda B. W., Fan H., Wall D., Li E., and Gaudet F. (2008) Methylation of p53 by Set7/9 mediates p53 acetylation and activity in vivo. Mol. Cell 29, 392–400 10.1016/j.molcel.2007.12.025 [DOI] [PubMed] [Google Scholar]
- 28. Campaner S., Spreafico F., Burgold T., Doni M., Rosato U., Amati B., and Testa G. (2011) The methyltransferase Set7/9 (Setd7) is dispensable for the p53-mediated DNA damage response in vivo. Mol. Cell 43, 681–688 10.1016/j.molcel.2011.08.007 [DOI] [PubMed] [Google Scholar]
- 29. Oudhoff M. J., Freeman S. A., Couzens A. L., Antignano F., Kuznetsova E., Min P. H., Northrop J. P., Lehnertz B., Barsyte-Lovejoy D., Vedadi M., Arrowsmith C. H., Nishina H., Gold M. R., Rossi F. M., Gingras A. C., and Zaph C. (2013) Control of the hippo pathway by Set7-dependent methylation of Yap. Dev. Cell 26, 188–194 10.1016/j.devcel.2013.05.025 [DOI] [PubMed] [Google Scholar]
- 30. Oudhoff M. J., Braam M. J., Freeman S. A., Wong D., Rattray D. G., Wang J., Antignano F., Snyder K., Refaeli I., Hughes M. R., McNagny K. M., Gold M. R., Arrowsmith C. H., Sato T., Rossi F. M., et al. (2016) SETD7 controls intestinal regeneration and tumorigenesis by regulating Wnt/β-catenin and Hippo/YAP signaling. Dev. Cell 37, 47–57 10.1016/j.devcel.2016.03.002 [DOI] [PubMed] [Google Scholar]
- 31. Elkouris M., Kontaki H., Stavropoulos A., Antonoglou A., Nikolaou K. C., Samiotaki M., Szantai E., Saviolaki D., Brown P. J., Sideras P., Panayotou G., and Talianidis I. (2016) SET9-mediated regulation of TGF-β signaling links protein methylation to pulmonary fibrosis. Cell Rep. 15, 2733–2744 10.1016/j.celrep.2016.05.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sasaki K., Doi S., Nakashima A., Irifuku T., Yamada K., Kokoroishi K., Ueno T., Doi T., Hida E., Arihiro K., Kohno N., and Masaki T. (2016) Inhibition of SET domain-containing lysine methyltransferase 7/9 ameliorates renal fibrosis. J. Am. Soc. Nephrol. 27, 203–215 10.1681/ASN.2014090850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mori S., Iwase K., Iwanami N., Tanaka Y., Kagechika H., and Hirano T. (2010) Development of novel bisubstrate-type inhibitors of histone methyltransferase SET7/9. Bioorg. Med. Chem. 18, 8158–8166 10.1016/j.bmc.2010.10.022 [DOI] [PubMed] [Google Scholar]
- 34. Francis N. J., Rowlands M., Workman P., Jones K., and Aherne W. (2012) Small-molecule inhibitors of the protein methyltransferase SET7/9 identified in a high-throughput screen. J. Biomol. Screen 17, 1102–1109 10.1177/1087057112452137 [DOI] [PubMed] [Google Scholar]
- 35. Niwa H., Handa N., Tomabechi Y., Honda K., Toyama M., Ohsawa N., Shirouzu M., Kagechika H., Hirano T., Umehara T., and Yokoyama S. (2013) Structures of histone methyltransferase SET7/9 in complexes with adenosylmethionine derivatives. Acta Crystallogr. D Biol. Crystallogr. 69, 595–602 10.1107/S0907444912052092 [DOI] [PubMed] [Google Scholar]
- 36. Barsyte-Lovejoy D., Li F., Oudhoff M. J., Tatlock J. H., Dong A., Zeng H., Wu H., Freeman S. A., Schapira M., Senisterra G. A., Kuznetsova E., Marcellus R., Allali-Hassani A., Kennedy S., Lambert J. P., et al. (2014) (R)-PFI-2 is a potent and selective inhibitor of SETD7 methyltransferase activity in cells. Proc. Natl. Acad. Sci. U.S.A. 111, 12853–12858 10.1073/pnas.1407358111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Meng F., Cheng S., Ding H., Liu S., Liu Y., Zhu K., Chen S., Lu J., Xie Y., Li L., Liu R., Shi Z., Zhou Y., Liu Y. C., Zheng M., Jiang H., Lu W., Liu H., and Luo C. (2015) Discovery and optimization of novel, selective histone methyltransferase SET7 inhibitors by pharmacophore- and docking-based virtual screening. J. Med. Chem. 58, 8166–8181 10.1021/acs.jmedchem.5b01154 [DOI] [PubMed] [Google Scholar]
- 38. Takemoto Y., Ito A., Niwa H., Okamura M., Fujiwara T., Hirano T., Handa N., Umehara T., Sonoda T., Ogawa K., Tariq M., Nishino N., Dan S., Kagechika H., Yamori T., Yokoyama S., and Yoshida M. (2016) Identification of cyproheptadine as an inhibitor of SET domain containing lysine methyltransferase 7/9 (Set7/9) that regulates estrogen-dependent transcription. J. Med. Chem. 59, 3650–3660 10.1021/acs.jmedchem.5b01732 [DOI] [PubMed] [Google Scholar]
- 39. Fujiwara T., Ohira K., Urushibara K., Ito A., Yoshida M., Kanai M., Tanatani A., Kagechika H., and Hirano T. (2016) Steric structure-activity relationship of cyproheptadine derivatives as inhibitors of histone methyltransferase Set7/9. Bioorg. Med. Chem. 24, 4318–4323 10.1016/j.bmc.2016.07.024 [DOI] [PubMed] [Google Scholar]
- 40. Kirn-Safran C. B., Oristian D. S., Focht R. J., Parker S. G., Vivian J. L., and Carson D. D. (2007) Global growth deficiencies in mice lacking the ribosomal protein HIP/RPL29. Dev. Dyn. 236, 447–460 10.1002/dvdy.21046 [DOI] [PubMed] [Google Scholar]
- 41. Williamson N. A., Raliegh J., Morrice N. A., and Wettenhall R. E. (1997) Post-translational processing of rat ribosomal proteins: ubiquitous methylation of Lys22 within the zinc-finger motif of RL40 (carboxy-terminal extension protein 52) and tissue-specific methylation of Lys4 in RL29. Eur. J. Biochem. 246, 786–793 10.1111/j.1432-1033.1997.00786.x [DOI] [PubMed] [Google Scholar]
- 42. Odintsova T. I., Müller E. C., Ivanov A. V., Egorov T. A., Bienert R., Vladimirov S. N., Kostka S., Otto A., Wittmann-Liebold B., and Karpova G. G. (2003) Characterization and analysis of posttranslational modifications of the human large cytoplasmic ribosomal subunit proteins by mass spectrometry and Edman sequencing. J. Protein Chem. 22, 249–258 10.1023/A:1025068419698 [DOI] [PubMed] [Google Scholar]
- 43. Huang J., Sengupta R., Espejo A. B., Lee M. G., Dorsey J. A., Richter M., Opravil S., Shiekhattar R., Bedford M. T., Jenuwein T., and Berger S. L. (2007) p53 is regulated by the lysine demethylase LSD1. Nature 449, 105–108 10.1038/nature06092 [DOI] [PubMed] [Google Scholar]
- 44. Schmidt E. K., Clavarino G., Ceppi M., and Pierre P. (2009) SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods 6, 275–277 10.1038/nmeth.1314 [DOI] [PubMed] [Google Scholar]
- 45. Gandin V., Masvidal L., Cargnello M., Gyenis L., McLaughlan S., Cai Y., Tenkerian C., Morita M., Balanathan P., Jean-Jean O., Stambolic V., Trost M., Furic L., Larose L., Koromilas A. E., et al. (2016) mTORC1 and CK2 coordinate ternary and eIF4F complex assembly. Nat. Commun. 7, 11127 10.1038/ncomms11127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rohde L. H., Julian J., Babaknia A., and Carson D. D. (1996) Cell surface expression of HIP, a novel heparin/heparan sulfate binding protein, of human uterine epithelial cells and cell lines. J. Biol. Chem. 271, 11824–11830 10.1074/jbc.271.20.11824 [DOI] [PubMed] [Google Scholar]
- 47. Guo A., Gu H., Zhou J., Mulhern D., Wang Y., Lee K. A., Yang V., Aguiar M., Kornhauser J., Jia X., Ren J., Beausoleil S. A., Silva J. C., Vemulapalli V., Bedford M. T., and Comb M. J. (2014) Immunoaffinity enrichment and mass spectrometry analysis of protein methylation. Mol. Cell Proteomics 13, 372–387 10.1074/mcp.O113.027870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nicholson T. B., Singh A. K., Su H., Hevi S., Wang J., Bajko J., Li M., Valdez R., Goetschkes M., Capodieci P., Loureiro J., Cheng X., Li E., Kinzel B., Labow M., and Chen T. (2013) A hypomorphic lsd1 allele results in heart development defects in mice. PLoS ONE 8, e60913 10.1371/journal.pone.0060913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kim S. J., Zhao H., Hardikar S., Singh A. K., Goodell M. A., and Chen T. (2013) A DNMT3A mutation common in AML exhibits dominant-negative effects in murine ES cells. Blood 122, 4086–4089 10.1182/blood-2013-02-483487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen T., Ueda Y., Dodge J. E., Wang Z., and Li E. (2003) Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol. Cell Biol. 23, 5594–5605 10.1128/MCB.23.16.5594-5605.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dan J., Rousseau P., Hardikar S., Veland N., Wong J., Autexier C., and Chen T. (2017) Zscan4 inhibits maintenance DNA methylation to facilitate telomere elongation in mouse embryonic stem cells. Cell Rep. 20, 1936–1949 10.1016/j.celrep.2017.07.070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Veland N., Hardikar S., Zhong Y., Gayatri S., Dan J., Strahl B. D., Rothbart S. B., Bedford M. T., and Chen T. (2017) The arginine methyltransferase PRMT6 regulates DNA methylation and contributes to global DNA hypomethylation in cancer. Cell Rep. 21, 3390–3397 10.1016/j.celrep.2017.11.082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cheng D., Côté J., Shaaban S., and Bedford M. T. (2007) The arginine methyltransferase CARM1 regulates the coupling of transcription and mRNA processing. Mol. Cell 25, 71–83 10.1016/j.molcel.2006.11.019 [DOI] [PubMed] [Google Scholar]
- 54. Nishioka K., Rice J. C., Sarma K., Erdjument-Bromage H., Werner J., Wang Y., Chuikov S., Valenzuela P., Tempst P., Steward R., Lis J. T., Allis C. D., and Reinberg D. (2002) PR-Set7 is a nucleosome-specific methyltransferase that modifies lysine 20 of histone H4 and is associated with silent chromatin. Mol. Cell 9, 1201–1213 10.1016/S1097-2765(02)00548-8 [DOI] [PubMed] [Google Scholar]
- 55. Gandin V., Sikstrom K., Alain T., Morita M., McLaughlan S., Larsson O., and Topisirovic I. (2014) Polysome fractionation and analysis of mammalian translatomes on a genome-wide scale. J. Vis. Exp. 10.3791/51455 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.